

Abstract
Meroterpenes derived from dimethylorsellinic acid (DMOA) and farnesyl pyrophosphate have attracted much biosynthetic attention, yet only recently have synthetic solutions to any family members appeared. A key point of divergence in DMOA-derived meroterpene biosynthesis is the protoaustinoid A carbocation which can be diverted to either the berkeleyone, andrastin, or terretonin structural classes via cyclase-controlled rearrangement pathways. Herein we show that the protoaustinoid bicyclo[3.3.1]nonane nucleus can be reverted to either andrastin or terretonin ring systems under abiotic reaction conditions. The first total syntheses of members of these natural product families are reported as their racemates.
Keywords: total synthesis, meroterpene, biosynthesis, natural product
Graphical abstract
Traversing Biosynthetic Carbocation Landscapes in the Total Synthesis of Andrastin and Terretonin Meroterpenes
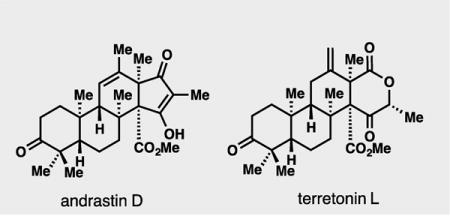
Over 100 structurally fascinating, fungal-derived natural products are assembled from 3,5-dimethylorsellinic acid (DMOA) and farnesyl pyrophosphate (FPP).[1] These meroterpenes, while historically the subject of intense biosynthetic interest,[1,2] have only very recently seen members succumb to total synthesis efforts.[3] The andrastins (see 1–6),[2l,4] terretonins (see 10–15),[2i,5,6] and their downstream congeners in particular, represent a large subset of DMOA-derived members with interesting biological profiles and unique carbocyclic frameworks (Figure 1).[1] To date these compounds have evaded successful chemical synthesis attempts.[7] Herein we report the first total synthesis of members of these families using a strategy that is inspired by nature’s pathway, but uses completely abiotic chemistry. Density functional theory (DFT) calculations have also shed light on the specific molecular interactions responsible for shaping the energetic landscape in the early stages of DMOA-derived meroterpene biosynthesis.[8]
Figure 1.
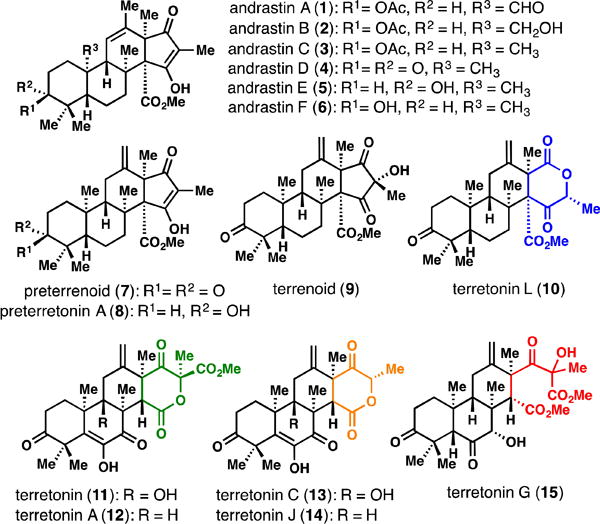
Representative andrastin and terretonin-type meroterpenes.[6]
A key point of divergence in DMOA-derived meroterpene synthesis is the protoaustinoid carbocation (17), which is derived from FPP/DMOA-conjugate 16 via a cyclase-mediated polyene cyclization (Figure 2).[1b] We estimate that over 80% of all DMOA-derived meroterpenes traverse 17 or a related intermediate. Proton loss of Ha affords protoaustinoid A (18) which serves as a precursor to the berkeleyone, austin, and related families.[1b] In Aspergillus nidulans, this process is mediated by the cyclase AusL.[2e] The biosynthesis of the terretonin and andrastin families is generally believed to involve a 1,2-shift pathway prior to proton loss (see 17→19) thus converting the bicyclo[3.3.1]nonane skeleton into a 5,6-fused ring system. In Aspergillus terreus, the Trt1 cyclase mediates selective proton abstraction of Hb forming a trisubstituted alkene,[2i,2j,2m] while in Penicillium chrysogenum, a methyl proton (Hc) is selectively abstracted by AdrI.[2l] Two of our groups (T.R.N and T.J.M.) recently prepared protoaustinoid A (18) en route to distinct total syntheses of berkeleyone A.[3] While these pathways did not directly proceed through carbocation 17, we wondered if 18 (or a suitable synthetic surrogate) could be “reverted” to the andrastin and terretonin ring systems directly in the absence of enzymes, possibly driven by a thermodynamic preference for 5 and/or 8. If feasible, this could potentially open up synthetic access to nearly half of the remaining DMOA-derived members believed to proceed through carbocation 17.
Figure 2.
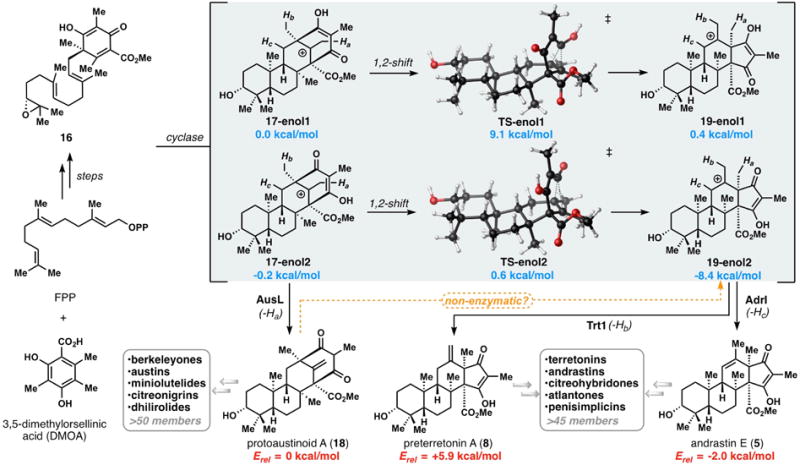
Key steps in the early stages of DMOA-derived meroterpene biosynthesis showing the divergence of cationic vinylogous acid tautomers (17-enol1 and 17-enol2) to form protoaustinoid A (18), preterretonin A (8), and andrastin E (5). Energy values shown are in kcal/mol and were obtained using Gaussian ‘09 at the mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p) level of theory.[9,10]
Although the synthetic strategy described above was computationally predicted to be favorable for the conversion of protoaustinoid A (18) to andrastin E (5) (ΔG = -2.0 kcal/mol, see SI for details),[9,10] DFT calculations indicate that formation of the less stable 1,1-disubstituted alkene-containing preterretonin A (8) is unfavorable by 5.9 kcal/mol. Analysis of the carbocation intermediates believed to be involved in these conversions (i.e. 17 and 19), revealed that the tautomeric form of the vinylogous acid functionality determines whether the protoaustinoid or andrastin framework is energetically favored. For one pair of regioisomers (17-enol1 and 19-enol1), the two different ring systems are comparable in energy (ΔG = +0.4 kcal/mol) and kinetically accessible (ΔG‡ = +9.1 kcal/mol). However, the regioisomeric vinylogous acid 17-enol2 presents an exergonic pathway to the andrastin 5,6-fused bicyclic framework, and 19-enol2 (ΔG = -8.4 kcal/mol), by a nearly barrierless pathway (ΔG‡ = +0.8 kcal/mol for TS-enol2). The basis for the disparate stability of these isomers is derived from greater stabilization of the carbocation by the neighboring carbonyl oxygen lone-pair in 19-enol2 than the more distal carbonyl in 19-enol1 (see SI for details). Intriguingly, these theoretical findings suggest that controlling tautomeric state may be an effective molecular approach by which biosynthetic machinery can influence the chemical fate of the proposed cationic intermediates.
While these calculations indicate the facility of the rearrangement, surprisingly we have had difficulty in forming either preterretonin (8) or andrastin E (5) via treatment of protoaustinoid A (18) (or its corresponding A-ring ketone) with acid, wherein tautomerization between enol isomers is expected to occur. Under a variety of acidic conditions, only decomposition or complex, unidentified product mixtures have resulted. We initially attributed these failures to the difficulty in protonating the exocyclic olefin over several more basic functional groups combined with the sensitivity of the 1,3-diketone. Seeking alternative chemoselective methods for carbocation generation, we were drawn to recent work by Shigehisa and co-workers who reported an exceedingly mild, Co-catalyzed hydroalkoxylation reaction via an oxidative variant of the classic Mukaiyama hydration process.[11,12] Substrate 21 was prepared in one-step from tetracyclic building block 20, an intermediate we have prepared in gram quantities en route to the synthesis of berkeleyone A (Scheme 1).[3a] Subjecting 21 to modified Shigehisa conditions (cat. Co(II), PhSiH3, N-fluoropyridinium salt (F+) oxidant) yet omitting the alcohol trapping agent cleanly promoted the desired rearrangement.[13] Notably, we have been unable to elicit this bond rearrangement on 21 using a variety of Brønsted acids, although the corresponding regioisomeric vinylogous ester of 21 gives detectable quantities of rearranged product.[14] When one equivalent of phenylsilane and N-fluoropyridinium salt were employed, the andrastin framework was favored over the exocyclic, terretonin-type alkene system (~3:1 ratio of 22:23 along with recovered 21). Increasing the equivalents of F+ oxidant and phenylsilane to 2.5 allowed for very clean formation of 22 as essentially a single olefin isomer in 90% isolated yield. The structure of the newly minted 5,6-fused ring system was confirmed by single crystal X-ray diffraction and following Krapcho-type demethylation (LiCl, DMSO, Δ), (±)-andrastin D (4) was furnished in 77% yield thus completing the first total synthesis of a member of this meroterpene class.
Scheme 1.
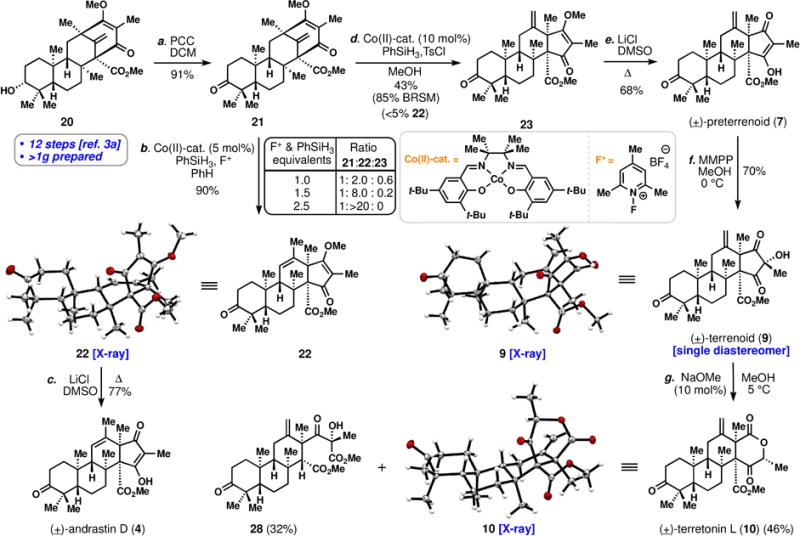
Total syntheses of andrastin D (4), preterrenoid (7), terrenoid (9), and terretonin L (10). Reagents and conditions: a) PCC (4 equiv), DCM, 25 °C, 2 h, 91%; b) Co(II)-cat. (5 mol %), PhSiH3 (2.5 equiv), F+ (2.5 equiv), PhH, rt, 2 h, 90%; c) LiCl (40 equiv), DMSO, 120 °C, 2 h, 77%; d) Co(II)-cat. (10 mol %), PhSiH3 (2.5 equiv), TsCl (1.5 equiv), MeOH, rt, 3 h, 43% (85% BRSM); e) LiCl (40 equiv), DMSO, 120 °C, 1.5 h, 68%; f) MMPP (2 equiv), MeOH, 0 °C, 2 h, 70%; g) NaOMe (0.1 equiv), MeOH, 5 °C, 24 h, 46% + 32% of 24; PCC = pyridinum chlorochromate, MMPP = magnesium monoperoxyphthalate.
While this strategy offers high yielding, selective entry into the andrastins, it presents a challenge for the synthesis of terretonins as the thermodynamic product is obtained. Cognizant of the disparate alkene patterns produced by titanocene-mediated epoxide opening cascades, as compared to their acid-mediated counterparts,[15] we wondered if a purely radical-based homoallyl-type rearrangement/HAT process could be used to forge 23 preferentially (Figure 3).[16] After significant exploration, we discovered that when 21 was subjected to conditions developed by Carreira for radical olefin hydrochlorination (cat. Co(II), PhSiH3, TsCl, MeOH),[17] we observed formation of 23 in 43% isolated yield with only trace amounts of 22 (<5%). The remaining mass balance was largely recovered starting material and we did not observe the expected tertiary chloride product.[18] We suspect that the steric encumbrance surrounding radical intermediates 24–26 precludes efficient trapping with TsCl and a final hydrogen atom transfer (HAT) from the less-hindered methyl position ensues preferentially forming 23.[15,19,20] We also assume that under the more powerfully oxidizing Co(II)/F+ conditions, radical 26 is converted into carbocation 27 which is ultimately funneled into 22 via loss of a proton.[11]
Figure 3.
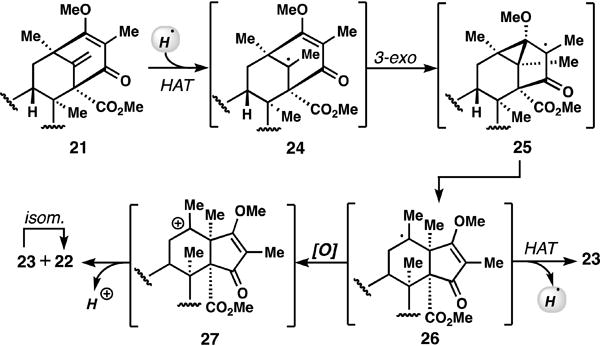
Abiotic radical-based HAT isomerization process
Demethylation of 23 (LiCl, Δ) afforded preterrenoid (7), thus allowing us to explore late-stage oxidative chemistry potentially leading to the hallmark terretonin lactone ring systems which are found in several permutations (see colored rings, Figure 1).[2i] Stereoselective oxidation of 7 with magnesium monoperoxyphthalate (MMPP) afforded terrenoid (9) as a single diastereomer whose structure was confirmed by single crystal X-ray diffraction. Treating this key biosynthetic precursor with catalytic quantities of NaOMe in MeOH afforded (±)-terretonin L (10) as the major product (46%), thus formally accomplishing the same retro-Claisen/esterification cascade as the putative hydrolase Trt14 in Aspergillus oryzae.[2i] Notably a single diastereomer was obtained, likely due to a preference for the newly-formed methyl stereocenter to reside in the pseudoequatorial position away from the top face of the congested polycyclic ring system. Interestingly, a ring-opened ester assigned as 28 was also isolated in 32% yield; this structure would correspond to 6,7-deoxyterretonin G (see 15, Figure 1). Abe and co-workers did not observe a product of this type when reacting terrenoid (9) with Trt14, indicating the chemoselectivity of this enzyme.
In summary, we have examined early, fundamental steps in DMOA-derived meroterpene synthesis both experimentally and computationally. By initially analyzing the energetic landscape of the system, we realized that a biomimetic 1,2-shift should be feasible in the absence biosynthetic machinery and by investigating abiotic reaction conditions, we demonstrate that selective formation of either andrastin or terretonin scaffolds are possible from protoaustinoid bicyclo[3.3.1]nonane skeletons. With a better understanding of the key skeletal forming chemistry in DMOA-derived meroterpene biosynthesis, efforts can now focus on exploring the synthesis of more highly oxidized congeners. Finally, in-depth studies on how the aforementioned cyclase enzymes manipulate these cationic intermediates, particularly how they prevent energetically favorable rearrangements, may lead to new insights in meroterpene enzymology.
Supplementary Material
Acknowledgments
T.J.M acknowledges the NSF (CAREER Award# 155454) for funding. T.J.M. is a Cottrell Scholar. D. J. T acknowledges support from NSF grants CHE-1565933 and CHE-030089 [XSEDE program]. G. X. thanks the Swiss National Science Foundation (SNSF) for a postdoctoral mobility fellowship (P2BEP2_162076). We thank Dr. Hasan Celik for NMR spectroscopic assistance. Dr. Antonio DiPasquale and Mr. Nick Settineri are acknowledged for X-ray crystallographic analysis wherein support from NIH Shared Instrument Grant (S10-RR027172) is also acknowledged. This work was supported by the High Performance Computing facilities operated by, and the staff of, the Yale Center for Research Computing.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Geris R, Simpson TJ. Nat Prod Rep. 2009;26:1063. doi: 10.1039/b820413f. [DOI] [PubMed] [Google Scholar]; b) Matsuda Y, Abe I. Nat Prod Rep. 2016;33:26. doi: 10.1039/c5np00090d. [DOI] [PubMed] [Google Scholar]; c) Matsuda Y, Awakawa T, Mori T, Abe I. Curr Opin Chem Biol. 2016;31:1. doi: 10.1016/j.cbpa.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 2.a) Cox R. Nat Prod Rep. 2014;31:1045. doi: 10.1039/c4np00059e. [DOI] [PubMed] [Google Scholar]; b) Simpson TJ, Stenzel DJ. J Chem Soc Chem Commun. 1981:1042. [Google Scholar]; c) Scott FE, Simpson TJ, Trimble LA, Vederas JC. J Chem Soc Chem Commun. 1986;214 [Google Scholar]; d) Matsuda Y, Awakawa T, Wakimoto T, Abe I. J Am Chem Soc. 2013;135:10962. doi: 10.1021/ja405518u. [DOI] [PubMed] [Google Scholar]; e) Lo HC, Entwistle R, Guo CJ, Ahuja M, Szewczyk E, Hung JH, Chiang YM, Oakley BR, Wang CCC. J Am Chem Soc. 2012;134:4709. doi: 10.1021/ja209809t. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Matsuda Y, Iwabuchi T, Fujimoto T, Awakawa T, Nakashima Y, Mori T, Zhang H, Hayashi F, Abe I. J Am Chem Soc. 2016;138:12671. doi: 10.1021/jacs.6b08424. [DOI] [PubMed] [Google Scholar]; g) Uchida R. J Antibiot. 1996;49:418. doi: 10.7164/antibiotics.49.418. [DOI] [PubMed] [Google Scholar]; h) McIntyre CR, Simpson TJ. J Chem Soc Chem Commun. 1981;1043 [Google Scholar]; i) Matsuda Y, Iwabuchi T, Wakimoto T, Awakawa T, Abe I. J Am Chem Soc. 2015;137:3393. doi: 10.1021/jacs.5b00570. [DOI] [PubMed] [Google Scholar]; j) Matsuda Y, Awakawa T, Itoh T, Wakimoto T, Kushiro T, Fujii I, Ibizuka Y, Abe I. ChemBioChem. 2012;13:1738. doi: 10.1002/cbic.201200369. [DOI] [PubMed] [Google Scholar]; k) Matsuda Y, Quan Z, Mitsuhashi T, Li C, Abe I. Org Lett. 2016;18:296. doi: 10.1021/acs.orglett.5b03465. [DOI] [PubMed] [Google Scholar]; l) Matsuda Y, Awakawa T, Abe I. Tetrahedron. 2013;69:8199. [Google Scholar]; m) Guo CJ, Knox BP, Chiang Y-M, Lo HC, Sanchez JF, Lee KH, Oakley BR, Bruno KS, Wang CCC. Org Lett. 2012;14:5684. doi: 10.1021/ol302682z. [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Matsuda Y, Wakimoto T, Mori T, Awakawa T, Abe I. J Am Chem Soc. 2014;135:15326. doi: 10.1021/ja508127q. [DOI] [PubMed] [Google Scholar]; o) Valiante V, Mattern DJ, Schüffler A, Horn F, Walther G, Scherlach K, Petzke L, Dickhaut J, Guthke R, Hertweck C, Nett M, Thines E, Brakhage AA. ACS Chem Biol. 2017;article ASAP doi: 10.1021/acschembio.7b00003. [DOI] [PubMed] [Google Scholar]
- 3.a) Ting CP, Xu G, Zeng X, Maimone TJ. J Am Chem Soc. 2016;138:14868. doi: 10.1021/jacs.6b10397. [DOI] [PubMed] [Google Scholar]; b) Elkin M, Szewczyk SM, Scruse AC, Newhouse TR. J Am Chem Soc. 2017;139:1790. doi: 10.1021/jacs.6b12914. [DOI] [PubMed] [Google Scholar]
- 4.Uchida R, Shiomi K, Inokoshi J, Sunazuka T, Tanaka H, Iwai Y, Takayanagi H, Ōmura S. J Antibiot. 1996;49:418. doi: 10.7164/antibiotics.49.418. [DOI] [PubMed] [Google Scholar]
- 5.a) Springer JP, Dorner JW, Cole RJ, Cox RH. J Org Chem. 1979;44:4852. [Google Scholar]; b) Li GY, Li BG, Yang T, Yin JH, Qi HY, Liu GY, Zhang GL. J Nat Prod. 2005;68:1243. doi: 10.1021/np0501738. [DOI] [PubMed] [Google Scholar]; c) Liu XH, Miao FP, Qiao MF, Cichewicz RH, Ji NY. RSC Advances. 2013;3:588. [Google Scholar]; d) Fukuda T, Kurihara Y, Kanamoto A, Tomoda H. J Antibiot. 2014;67:593. doi: 10.1038/ja.2014.46. [DOI] [PubMed] [Google Scholar]
- 6.For a complete list of terretonin structures isolated to date, see the Supporting Information.
- 7.Okamota R, Takeda K, Tokuyama H, Ihara M, Toyota M. J Org Chem. 2013;78:93. doi: 10.1021/jo301948h. [DOI] [PubMed] [Google Scholar]
- 8.For a review of DFT calculations on terpene-forming carbocation rearrangements, see:; Tantillo DJ. Nat Prod Rep. 2011;28:1035. doi: 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- 9.Frisch MJ, et al. Gaussian ’09, revision D.01. Gaussian, Inc; Wallingford, CT: 2013. [Google Scholar]
- 10.For systematic evaluation and recommendation of using B3LYP geometries and mPW1PW91 single point energies, see:; Matsuda SPT, Wilson WK, Xiong Q. Org Biomol Chem. 2006;4:530. doi: 10.1039/b513599k. [DOI] [PubMed] [Google Scholar]
- 11.Shigehisa H, Aoki T, Yamaguchi S, Shimizu N, Hiroya K. J Am Chem Soc. 2013;135:10306. doi: 10.1021/ja405219f. [DOI] [PubMed] [Google Scholar]
- 12.For a recent review on metal-mediated radical olefin hydrofunctionalization, see:; Crossley SWM, Obradors C, Martinez RM, Shenvi RA. Chem Rev. 2016;116:8912. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The corresponding free vinylogous acid of 21 was not compatible with the reaction conditions of this transformation as the N-fluoropyridinium salt led to significant byproduct formation even in the absence of the Co(II) catalyst.
- 14.Demethylation and remethylation of 21 yielded an inseperable ~1:1 mixture of 21 and its corresponding vinylogous ester regioisomer (iso-21). When this mixture was heated with p-TsOH in PhH we detected small amounts of the ring-shifted product, but only from iso-21. The published synthetic pathway to 20 however, cannot be used to make iso-21 selectively since this regioisomer does not undergo a key bridgehead deprotonation reaction (see ref. 3a). Thus this was not considered to be a viable option for the synthesis of andrastins.
- 15.Justicia J, Álvarez de Cienfuegos L, Campaña AG, Miguel D, Jakoby V, Gansäuer A, Cuerva JM. Chem Soc Rev. 2011;40:3525. doi: 10.1039/c0cs00220h. [DOI] [PubMed] [Google Scholar]
- 16.For recent examples of 3-exo radical cyclizations initiated by a metal-catalyzed HAT process, see:; Lo JC, et al. J Am Chem Soc. 2017;139:2484. doi: 10.1021/jacs.6b13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaspar B, Carreira EM. Angew Chem Int Ed. 2008;47:5758. doi: 10.1002/anie.200801760. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;120:5842. [Google Scholar]
- 18.Similar results are obtained by replacing TsCl with tert-Butyl hydroperoxide (TBHP). Presumably these reagents oxidize the Co(II) precatalyst to Co(III) which is required for the intial HAT.
- 19.Crosley SWM, Barabé F, Shenvi RA. J Am Chem Soc. 2014;136:16788. doi: 10.1021/ja5105602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DFT studies on a simplified model system indicate that the radical rearrangment is both facile and significantly exergonic. The conversion of model radical 24 to 26 is downhill by 8 kcal/mol with activaton barriers of 11.6 kcal/mol for 24 → 25 and 4.9 kcal/mol for 25 → 26 (see Supporting Information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.