

Abstract
Hypoglycemia is the most common complication of diabetes, causing morbidity and death. Recurrent hypoglycemia alters the cascade of physiological and behavioral responses that maintain euglycemia. The extent to which these responses are normally triggered by decreased whole-brain cerebral glucose metabolism (CMRglc) has not been resolved by previous studies. We measured plasma counterregulatory hormonal responses and whole-brain CMRglc (along with blood-to-brain glucose transport rates and brain glucose concentrations) with 1-[11C]-d-glucose positron emission tomography during hyperinsulinemic glucose clamps at nominal plasma glucose concentrations of 90, 75, 60, and 45 mg/dL (5.0, 4.2, 3.3, and 2.5 mmol/L) in 18 healthy young adults. Clear evidence of hypoglycemic physiological counterregulation was first demonstrated between 75 mg/dL (4.2 mmol/L) and 60 mg/dL (3.3 mmol/L) with increases in both plasma epinephrine (P = 0.01) and glucagon (P = 0.01). In contrast, there was no statistically significant change in CMRglc (P = 1.0) between 75 mg/dL (4.2 mmol/L) and 60 mg/dL (3.3 mmol/L), whereas CMRglc significantly decreased (P = 0.02) between 60 mg/dL (3.3 mmol/L) and 45 mg/dL (2.5 mmol/L). Therefore, the increased epinephrine and glucagon secretion with declining plasma glucose concentrations is not in response to a decrease in whole-brain CMRglc.
Introduction
The human brain cannot synthesize glucose and can only store glycogen and other substrates to maintain normal brain function for only a few minutes if the blood-borne supply of glucose is lost. Thus, tight regulation of glucose metabolism and homeostasis is critical for brain function and development (1). Hypoglycemia, a rare event in people without diabetes, is the most common complication in those living with diabetes, causing morbidity and death (2). In diabetes, it is the result of therapeutic insulin excess and impaired glucagon and epinephrine responses against falling plasma glucose concentrations. This syndrome of defective glucose counterregulation is associated with a 25-fold or greater increased risk of severe iatrogenic hypoglycemia during intensive glycemic therapy. Thus, better understanding of the mechanisms underpinning the development of counterregulation is key to developing novel strategies to prevent recurrent hypoglycemia.
When plasma glucose concentrations fall, signals to and from the brain lead to a cascade of physiological and behavioral responses that normally maintain euglycemia (1). The extent to which these responses are triggered by decreased cerebral glucose metabolism (CMRglc) is unclear. Two previous studies have not resolved this issue. Using the Kety-Schmidt cerebral blood flow (CBF) measurement technique with arterial-venous sampling and four successive plasma glucose steps of 85 mg/dL (4.7 mmol/L), 75 mg/dL (4.2 mmol/L), 65 mg/dL (3.6 mmol/L), and 55 mg/dL (3.1 mmol/L), Boyle et al. (3) calculated whole-brain CMRglc (brain glucose uptake) by the Fick principle (CBF × arteriovenous difference) and reported that the first increase in glucose counterregulatory hormones coincided with the first decrease at the same plasma concentration of 65 mg/dL (3.6 mmol/L). However, in a two-step study conducted at 90 mg/dL (5.0 mmol/L) and then at 54 mg/dL (3.0 mmol/L) using [13C]-magnetic resonance spectroscopy of the occipital lobes and [13C]-glucose infusions, van de Ven et al. (4) found an increase in counterregulatory hormones at 54 mg/dL (3.0 mmol/L) with no change in tricarboxylic acid (TCA) cycle rates. The TCA cycle accounts for 90–95% of CMRglc at normoglycemia, and this relative contribution remains stable at 54 mg/dL (3.0 mmol/L), so changes in TCA cycle rates should reflect changes in overall CMRglc (5–7). To reconcile these findings and determine if physiological counterregulation responses to hypoglycemia occur before or are coincident with a reduction in CMRglc, we measured plasma counterregulatory hormonal responses and whole-brain CMRglc. In addition, we measured blood-to-brain glucose transport rates (CTXglc) and brain free glucose concentrations to place this work in broader methodological and physiological contexts. We made measurements with 1-[11C]-d-glucose positron emission tomography (PET) during hyperinsulinemic glucose clamps at nominal plasma glucose concentrations of 90, 75, 60, and 45 mg/dL (5.0, 4.2, 3.3, and 2.5 mmol/L) in 18 healthy young adults.
Research Design and Methods
Study Participants
The study was approved by the Human Research Protection Office and Institutional Review Board at the Washington University School of Medicine in St. Louis (approval 201011750) for compliance with the ethical standards of the Declaration of Helsinki of 1975 (revised 1983) and conducted at the Clinical Research Unit (CRU) of the Institute for Clinical and Translational Sciences and at the Neurological-Neurosurgical Intensive Care Unit (NNICU) PET Research Facility (Washington University in St. Louis). Eighteen healthy young adults gave their written informed consent to participate in this study. All subjects had negative medical histories, normal physical examinations, and normal fasting plasma glucose concentrations, blood counts, plasma electrolytes, liver and renal function tests, and electrocardiograms. No subject was on any medications or had a personal history or first-degree relative with a history of diabetes, heart disease, or psychiatric or neurological conditions.
Subjects were assigned by block randomization to one of two groups where they underwent a stepped hyperinsulinemic-euglycemic-hypoglycemic clamp. Two groups were needed in order to study glucose counterregulatory responses and cerebral glucose metabolism rates at four different glycemic levels and to avoid exceeding the maximum amount of radiation exposure allowed per year in each subject. Ten subjects (six females/four males, four African American/six Caucasian, age 28.9 ± 6.9 years, BMI 24.4 ± 3.6 kg/m2) were assigned to a stepped hyperinsulinemic glucose clamp at 90 mg/dL (5.0 mmol/L) and 60 mg/dL (3.3 mmol/L). The other eight subjects (two females/six males, three African American/five Caucasian, age 27.6 ± 8.2 years, BMI 24.0 ± 2.8 kg/m2) were assigned to a stepped hyperinsulinemic glucose clamp at 75 mg/dL (4.2 mmol/L) and 45 mg/dL (2.5 mmol/L).
Experimental Design
Subjects were admitted to the CRU in the morning after a 10-h overnight fast. Two intravenous catheters were placed, one in an antecubital vein for infusions of insulin, glucose, and potassium chloride, and the other in a contralateral antecubital vein for injection of the radionuclide 1-[11C]-d-glucose. A radial arterial line was placed with local anesthetic, after Allen testing, for blood sampling and measurement of time-radioactivity curves. Patients were then transported to the NNICU PET and positioned in an ECAT EXACT HR+ PET scanner (Siemens/CTI, Knoxville, TN) with their heads centered in the gantry field of view.
After placement of all lines and after 30 min of supine rest, regular human insulin (Novo Nordisk, Bagsværd, Denmark) was infused at 2.0 mU/kg/min (12 pmol/kg/min). In group 1, plasma glucose was clamped at 90 mg/dL (5.0 mmol/L) for 2 h and 60 mg/dL (3.3 mmol/L) for 2 h. In group 2, plasma glucose was clamped at 75 mg/dL (4.2 mmol/L) for 2 h and 45 mg/dL (2.5 mmol/L) for 2 h (Fig. 1). To maintain plasma glucose concentrations at target levels, 20% glucose was infused at variable rates, based on bedside plasma glucose measurements obtained every 5 min. Electrocardiograms and vital signs (Table 1) were continuously monitored on a Phillips IntelliVue MP70. Every 30 min, heart rate, blood pressure, and symptom scores were recorded and arterial samples were obtained for measurement of epinephrine, norepinephrine, insulin, C-peptide, glucagon, cortisol, fatty acids, β-hydroxybutyrate, and lactate concentrations. Subjects remained supine throughout the study.
Figure 1.

Diagram of the experimental protocol. HR, heart rate; BP, blood pressure; Sx, symptoms; D 20%, 20 g/100 mL dextrose monohydrate infusion.
Table 1.
Vital signs measured for 10 subjects clamped to nominal plasma glucose concentrations of 90 and then 60 mg/dL and 8 subjects clamped to 75 and then 45 mg/dL
| Nominal glucose (mg/dL) |
||||
|---|---|---|---|---|
| 90 | 75 | 60 | 45 | |
| Heart rate (bpm) | 71 ± 2 | 68 ± 4 | 82 ± 4 | 71 ± 3 |
| Systolic blood pressure (mmHg) | 134 ± 4 | 135 ± 5 | 127 ± 4 | 125 ± 6 |
| Diastolic blood pressure (mmHg) | 71 ± 2 | 69 ± 2 | 64 ± 2 | 58 ± 3 |
| Mean arterial pressure (mmHg) | 92 ± 2 | 91 ± 3 | 85 ± 3 | 80 ± 3 |
For each subject at each clamped glycemic level, five measurements were made at 30-min intervals during 2 h of PET scanning. Averages for all collections are shown. To convert glucose to mmol/L, multiply by 0.05551.
After 20 min of stabilizing the glycemic clamp, attenuation measurements were made with [68Ge]-[68Ga]-rotating rod sources. Regional cerebral blood volume (CBV) was then measured for 5 min beginning 2 min after brief inhalation of 22 ± 4 SD mCi of C[15O] (8,9). Regional CBF was measured with a 60-s emission scan after rapid intravenous injection of 19 ± 2 SD mCi of H2[15O] in saline (8,10). Thirty minutes later, 10 ± 3 SD mCi of 1-[11C]-d-glucose was administered through the antecubital vein, and dynamic PET acquisition was performed for 60 min. Forty-four separate frames were collected: 16 for 30 s, 8 for 60 s, 16 for 120 s, and 4 for 180 s. All PET emission data were collected in three dimensions by retraction of interslice septa. For quantitative processing of H2[15O] and C[15O], arterial blood was automatically sampled simultaneously with scanning at 5 mL/min from the radial artery to a scintillation detector. For 1-[11C]-d-glucose, 0.1–0.2 mL radial arterial blood was sampled manually every 10–15 s for 3 min and then every 10–15 min for 60 min. All data collection, procedures, and PET imaging were identically performed during each glycemic level (Fig. 1). At the conclusion of the second 1-[11C]-d-glucose scan, insulin infusion was discontinued, a meal was served, and glucose infusions were adjusted until euglycemia was achieved. All radiopharmaceuticals were produced in the Cyclotron Facility of the Washington University Mallinckrodt Institute of Radiology.
Subsequently, each subject was scanned using MRI on a Magnetom Trio-TIM at 3T (Siemens, Erlangen, Germany). Magnetization-prepared rapid gradient echo series (MP-RAGE) were obtained with 2,400 ms TR, 3.16 ms TE, and 1,000 ms TI in a 256 × 256 × 176 field of view at 1 mm3 isotropic resolution. All imaging equipment operators and PET data processing personnel were blinded to experimental conditions and interventions.
Analytical Methods
Glucose metabolism and cerebral hemodynamics were estimated from quantitative measurements of 1-[11C]-d-glucose, H2[15O], and C[15O] in arterial blood and from cerebral regions as detected by PET using methods well validated against the Kety-Schmidt technique and arterial-venous sampling (11). Arterial bedside plasma glucose was measured with a glucose oxidase method (Yellow Springs Glucose Analyzer 2; Yellow Springs Instruments, Yellow Springs, OH). Arterial samples for plasma insulin, C-peptide (12), glucagon (13), and cortisol (14) were measured with radioimmunoassay. Plasma epinephrine and norepinephrine (15) were measured with a radioenzymatic single-isotope derivative method. Blood nonesterified fatty acids (16), β-hydroxybutyrate (17), and lactate (18) were measured with enzymatic methods. Subjects were asked to score hypoglycemic symptoms from 0 (none) to 6 (severe) according to previously published methods (19).
Image Analysis
MP-RAGE images were coregistered onto motion-corrected PET images using the Functional MRI of the Brain Software Library (20). Masks for brain parenchyma, which excluded cerebellum, brain stem, dural venous sinuses, and cerebral spinal fluid, were created using anatomical parcellations and segmentations from the Destrieux atlas (21). Whole-brain time-resolved PET radioactivity data were generated by masking and summation of voxels without blurring operations.
CBF was estimated using an adaptation of the Kety autoradiography model (10) for positron emissions from H2[15O] in brain tissue, ψ(t), and from the radial artery, ψa(t):
 |
The permeability surface area product of the model is PS. The convolution operation is ⊗. The tissue partition coefficient was assumed to be λ = 0.95. Emissions ψa(t) were measured by automated sampling of blood from the radial artery through a catheter tubing, connected to a plastic scintillator and photomultiplier for detecting 511-keV positron annihilations, and a peristaltic pump (22). The delay and dispersion intrinsic to the catheter tubing were measured using a phantom, which delivered radiolabeled whole blood as a Heaviside impulse to the automated sampling apparatus; ψa(t) was corrected using the measured impulse response.
The four-compartment model for cerebral glucose kinetics (Fig. 2) has been previously described (11) using coupled differential equations for the quantities of intravascular glucose (q1); intracellular glucose (q2); intracellular metabolites, including metabolic pools such as lactate, glutamate, and glutamine (q3); and intravascular metabolites (q4). Steady-state kinetic coefficients, kmn, describe flux from compartment qn to compartment qm; k0n describe elimination from the model field of view via compartment qn. CBV was measured using previously described methods (9). Arterial positron emissions from 1-[11C]-d-glucose, φa(t), were measured by periodic manual radial-artery sampling. The model defines k04 = CBF/CBV, the reciprocal mean transit time, and
 |
 |
 |
CTXglc, CMRglc, and free brain glucose described in this work are defined by
 |
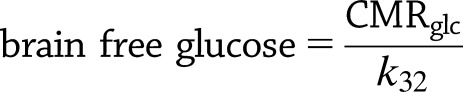 |
Arterial whole-blood concentration of total glucose (Cb) was estimated from assays of arterial plasma glucose concentration (Ca) and the hematocrit (Hct) by assuming rapid exchange between plasma and intraerythrocytic glucose and Cb = Ca(1 – 0.30 Hct) (23–25). The brain net extraction fraction for glucose (Enet) may be calculated using model rate constants, independently from arterial glucose concentrations, for our model of 1-[11C]-d-glucose PET, to be
 |
estimating the balance of glucose metabolic demand and glucose supplied by CBF (11). Enet also equals the fractional arterial-venous difference of glucose concentrations for the whole brain as used by the Fick principle to calculate CMRglc.
Bayesian parameter estimation was performed using Markov chain Monte Carlo with simulated annealing and Metropolis-Hastings importance sampling using all available measured quantities (26). For CBF, prior probabilities were set using values previously reported by Herscovitch et al. (27) for humans. For cerebral glucose kinetics, prior probabilities for adjustable model parameters were set using values previously reported by Powers et al. (11,28) for macaque monkeys and human neonates.
Figure 2.

Four-compartment model used for parameter estimations by Bayesian analysis. Compartment 1 is the arterial and capillary vascular space that exchanges 1-[11C]-d-glucose with brain tissue. Rate constant k12 replaces back-exchange to compartment 1 with egress from the field of view. Compartment 2 represents intracellular free 1-[11C]-d-glucose. Compartment 3 represents intracellular [11C]-metabolites. Compartment 4 is the vascular space to which the metabolites exit. Venous washout was set to the measured reciprocal mean transit time (CBF divided by CBV).
Statistics
Data were pooled across the two groups of subjects because they were comparable in age, sex, race, and BMI. Data were analyzed by general linear model repeated-measures ANOVA using SAS software version 9.3 (SAS Institute, Cary, NC). Mixed-effects analysis methods were implemented to assess the effect of glycemic levels (90, 75, 60, and 45 mg/dL or 5.0, 4.2, 3.3, and 2.5 mmol/L) on all collected data except for demographics and vital signs. All data measurements obtained at each glycemic level were averaged across subjects. Measurements at each glycemic level were compared pairwise to 90 mg/dL nominal glucose, and P values were adjusted for multiple comparisons by the Tukey method. P < 0.05 was considered statistically significant. Data are expressed as means ± SEM, except where otherwise specified. Based on our previous data (29,30), a sample size of 10 subjects provides 80% power to detect a difference of 20% and >95% power to detect a difference of 30% in CMRglc. Sample sizes were based on power calculations (two sided, α = 0.05) from the means and variances of the primary statistical end points from our previous data.
Results
Plasma glucose targets were met during the hyperinsulinemic-euglycemic-hypoglycemic clamps (Fig. 3A). The SEM of measured clamped arterial plasma glucose concentrations was <1 mg/dL. Consistent with the experimental design and clamping methodology, plasma insulin concentrations were raised from baseline and sustained throughout the study (Fig. 3B) and C-peptide concentrations were suppressed even during nominal 75 mg/dL (4.2 mmol/L) clamps (Table 2).
Figure 3.

Clamped arterial plasma glucose (A), insulin (B), epinephrine (C), norepinephrine (D), and glucagon (E) concentrations and total symptom scores (F) at nominal target glucose concentrations of 90, 75, 60, and 45 mg/dL. Data are means ± SEM collected at each nominal glucose for all interval sample collections throughout scanning. The base of each bar plot lists means, numbers of subjects, and nominal glucose concentrations for clamping. P values <0.05 describe contrasts from measurements at 90 mg/dL. To convert glucose to mmol/L, multiply by 0.05551. For insulin in pmol/L, multiply by 6.945. For epinephrine in pmol/L, multiply by 5.485.
Table 2.
Biochemistries for 10 subjects clamped to 90 and then 60 mg/dL and 8 subjects clamped to 75 and then 45 mg/dL
| Nominal glucose (mg/dL) |
||||
|---|---|---|---|---|
| 90 | 75 | 60 | 45 | |
| C-peptide (ng/mL) | 0.931 ± 0.085 | 0.263 ± 0.030† | 0.124 ± 0.008† | 0.100 ± 0.0† |
| Cortisol (μg/dL) | 11.6 ± 1.3 | 8.7 ± 0.8 | 15.5 ± 1.5 | 20.9 ± 2.1‡ |
| Free fatty acids (μmol/L) | 192 ± 28 | 105 ± 15 | 64 ± 13 | 107 ± 36 |
| β-hydroxybutyrate (μmol/L) | 262 ± 37 | 225 ± 51 | 238 ± 48 | 194 ± 21 |
| Lactate (μmol/L) | 1,451 ± 119 | 1,363 ± 83 | 1,443 ± 133 | 1,844 ± 141 |
For each subject at each clamped glycemic level, blood samples were collected every 30 min for five total collections. Averages for all collections are shown. To convert glucose to mmol/L, multiply by 0.05551. For C-peptide in nmol/L, multiply by 0.3323. For cortisol in nmol/L, multiply by 27.59. For norepinephrine in nmol/L, multiply by 0.005911. P values compare biochemical measurements to euglycemic measurements made at 90 mg/dL.
†P value <0.0001.
‡P value <0.002.
Counterregulatory and Symptom Responses to Hypoglycemia
Clear evidence of hypoglycemic counterregulation was first demonstrated between 75 mg/dL (4.2 mmol/L) and 60 mg/dL (3.3 mmol/L) (Fig. 3C–F and Table 2). Plasma epinephrine (P = 0.01) and glucagon (P = 0.01) concentrations both increased at the nominal glucose clamp level of 60 mg/dL (3.3 mmol/L) (Fig. 3C and E). Symptom scores first increased (P = 0.008) at the nominal glucose clamp level of 45 mg/dL (2.5 mmol/L) (Fig. 3F). As expected, under hyperinsulinemic conditions, plasma free fatty acids and β-hydroxybutyrate levels were low (Table 2). Plasma lactate levels tended to increase only at the lowest glucose clamp level (Table 2).
Whole-Brain Glucose Metabolism and Transport
In contrast to the significant changes in counterregulatory hormones, there were no statistically significant changes in CMRglc (P = 1.0) between 75 mg/dL (4.2 mmol/L) and 60 mg/dL (3.3 mmol/L). However, between 60 mg/dL (3.3 mmol/L) and 45 mg/dL (2.5 mmol/L), CMRglc significantly decreased (P = 0.02) (Fig. 4C).
Figure 4.

CTXglc (A), brain free glucose (B), CMRglc (C), CBF (D), CBV (E), and Enet (F) at nominal target glucose concentrations of 90, 75, 60, and 45 mg/dL. Data are means ± SEM collected at each nominal glucose throughout scanning.
CTXglc fell as arterial plasma glucose decreased (P < 0.05 for 75, 60, and 45 compared with 90) (Fig. 4A), limiting CMRglc at a plasma glucose concentration of 50 mg/dL (2.8 mmol/L) to 45 mg/dL (2.5 mmol/L), as shown in Fig. 5. Brain free glucose concentrations also progressively decreased as plasma glucose levels were reduced (Fig. 4B). CTXglc, CMRglc, and brain free glucose were all significantly decreased at the lowest arterial plasma glucose concentration. The brain net extraction fraction for glucose increased as plasma glucose fell (Fig. 4F). CBF was unchanged throughout but CBV increased (P = 0.02) at the lowest nominal glucose clamp level (Fig. 4D and E).
Figure 5.

CTXglc and CMRglc for each subject continuously plotted against arterial plasma glucose directly measured during clamping and scanning. For each subject, at each scan session, circles mark CTXglc and crosses mark CMRglc. The separation between CTXglc and CMRglc decreases markedly at lower arterial plasma glucose.
Discussion
The mechanism for the attenuated sympathoadrenal response to hypoglycemia in diabetes is unknown. It is a functional disorder distinct from classic diabetic autonomic neuropathy. Unlike the loss of the insulin and glucagon responses that reside at the islet level, the alteration resulting in attenuated sympathoadrenal responses has been postulated to reside within the central nervous system (1). It had been hypothesized that hypoglycemia causes a decrease in the rate of cerebral glucose metabolism, which in turn causes an increase in sympathoadrenal activity and a decrease in cognitive function. However, using 1-[11C]-d-glucose PET, this study documents that the sympathoadrenal response to hypoglycemia is not a response to a decrease in whole-brain glucose metabolism, since the hormonal counterregulatory responses to hypoglycemia occur at higher plasma glucose than necessary to produce a reduction in CMRglc. We found an increase in counterregulatory hormones with no change in whole-brain CMRglc at 60 mg/dL (3.3 mmol/dL) and found a reduction in whole-brain CMRglc at 45 mg/dL (2.5 mmol/L), at which level CTXglc became rate limiting. Therefore, the increase in counterregulatory hormones is not a response to a reduction in whole-brain cerebral glucose metabolism. Our data are congruent with those reported by van de Ven et al. (4) in which a reduction in plasma glucose from 90 mg/dL (5.0 mmol/L) to 54 mg/dL (3.0 mmol/L) produced an increase in counterregulatory hormones and no change in occipital lobe TCA flux, which is proportionately representative of CMRglc at these plasma glucose levels (7). Indeed, given the scatter in the nadir glucose concentrations in the latter study and the present findings, it would appear that the glycemic threshold for a decrease in whole-brain CMRglc during hypoglycemia is in the range of 50 mg/dL (2.8 mmol/L) or less. Taken together, these data indicate that the counterregulatory responses to falling plasma glucose concentrations are not triggered by decreases in whole-brain glucose metabolism. However, we cannot categorically exclude the possibility that these neuroendocrine responses are triggered by reductions in local glucose metabolism in one or more critical brain regions (11,31,32). Measurement of CMRglc in small brain regions in human subjects is technically difficult and limited by the direct proportionality of region volumes with the signal-to-noise ratio of measurements. Image-based PET methods based on the relative distribution of [18F]-fluorodeoxyglucose radioactivity can be used to provide data for small regions, but these are still subject to signal-to-noise limitations and, furthermore, explicitly must assume that the rate constants and the lumped constant are the same throughout the brain without a means to determine the validity of these assumptions (31,32).
The overall trends observed in our data in humans are in agreement with the trends observed in anesthetized macaques using the identical four-compartment model (11). In that study, Enet derived from simultaneous arterial-venous glucose sampling by vascular cannulation showed good agreement with 1-[11C]-d-glucose PET estimates of Enet during hypoglycemia, providing methodological validation of the PET measurements corresponding to the Fick principle. Our data, similar to the data in macaques (11), demonstrate characteristic nonlinear responses of blood to brain glucose transport and cerebral metabolic rate of glucose to arterial plasma glucose clamping from normoglycemia to hypoglycemia. Our data indicate that blood-to-brain glucose transport becomes limiting to whole-brain glucose metabolism at plasma glucose concentrations of 45–50 mg/dL (2.5–2.8 mmol/L). This is consistent with data from van de Ven et al. (4), who reported no reduction in CMRglc at 54 mg/dL (3.0 mmol/L), but very different from Boyle et al. (3), who reported reduced CMRglc at 65 mg/dL (3.6 mmol/L). Our methodology and that used by Boyle et al. are very similar. Boyle et al. used the Fick principle to measure CMRglc, and we have validated our PET CMRglc method against arteriovenous differences. Even so, after thorough consideration of the experimental techniques used by us and by Boyle et al., we were unable to find a clear methodological or physiological explanation for this discrepancy.
Published data in humans show no differences of whole-brain blood-to-brain glucose transport or whole-brain glucose metabolism between healthy subjects and patients with uncomplicated, reasonably well-controlled type 1 diabetes during hypoglycemia (30,33,34) or even after prolonged interprandial hypoglycemia (29). Thus, the findings of this study can be extended to people with endogenous insulin-deficient diabetes who require treatment with insulin and often suffer episodes of iatrogenic hypoglycemia (1). Moreover, published data in humans demonstrate that recently antecedent hypoglycemia can shift the glycemic threshold for counterregulatory responses and symptoms to lower plasma glucose concentrations without altering whole-brain blood-to-brain glucose transport or whole-brain glucose metabolism (30). Identifying the glycemic threshold for true changes in blood-to-brain glucose transport and brain glucose metabolism is consequential for understanding the pathophysiology of iatrogenic hypoglycemia, a central problem for people with diabetes requiring insulin (1). The present data suggest that the shifts in glycemic thresholds for sympathoadrenal and other responses to hypoglycemia to lower glucose concentrations after recent antecedent hypoglycemia (1) are not the result of a shift in the glycemic threshold for a decrease in whole-brain CMRglc.
Article Information
Acknowledgments. The authors acknowledge the assistance of the staff of the Washington University Clinical Research Unit in the performance of this study; the technical assistance of Hussain Jafri (Mallinckrodt Institute of Radiology, Washington University in St. Louis), Matthew Beach (Department of Pediatrics, Washington University in St. Louis), and Krishan Jethi (Department of Medicine, Washington University in St. Louis); the staff at Washington University Core Laboratory for Clinical Studies in the performance of the laboratory assays; and the Washington University Center for Clinical Imaging Research.
Funding. This work was supported in part by the American Diabetes Association (ADA) (Clinical/Translational Research Award 1-11-CT-14), National Institutes of Health (NIH) (DK101440), Washington University Diabetes Research Center Immunoassay Core (P30 DK020579), and Washington University Institute of Clinical and Translational Sciences (grant UL1 TR000448 from the National Center for Advancing Translational Sciences of the NIH). J.S.S. was supported in part by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the NIH (U54 HD087011 to the Intellectual and Developmental Disabilities Research Center at Washington University in St. Louis). A.M.A. was supported in part by the Robert Wood Johnson Foundation (Harold Amos Medical Faculty Development Program [HAMFDP]).
The content of this article is solely the responsibility of the authors and does not necessarily represent the official view of the NIH, HAMFDP, or ADA.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. J.J.L. helped with data processing, analyzed the data, and edited the manuscript. N.K. and A.M.S. performed the studies. S.N. and H.H. helped with the statistical analyses. J.A.A.-D. and J.S.S. helped with data processing and data analysis. K.S. helped revise the manuscript. W.J.P. helped design the study, analyzed and interpreted the data, and revised the manuscript. P.E.C. helped design the study, interpreted the data, and revised the manuscript. A.M.A. designed and performed the study, analyzed and interpreted the data, and wrote and edited the manuscript. A.M.A. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. This study was previously presented and published in abstract form at the 73rd Scientific Sessions of the American Diabetes Association, Chicago, IL, 21–25 June 2013, and at the 2016 Pediatric Diabetes Research Consortium Second Annual Research Symposium, St. Louis, MO, 7 April 2016.
References
- 1.Cryer PE. Hypoglycemia in Diabetes: Pathophysiology, Prevalence and Prevention. Alexandria, VA, American Diabetes Association, Inc., 2016 [Google Scholar]
- 2.Sprague JE, Arbeláez AM. Glucose counterregulatory responses to hypoglycemia. Pediatr Endocrinol Rev 2011;9:463–473; quiz 474–475 [PMC free article] [PubMed] [Google Scholar]
- 3.Boyle PJ, Nagy RJ, O’Connor AM, Kempers SF, Yeo RA, Qualls C. Adaptation in brain glucose uptake following recurrent hypoglycemia. Proc Natl Acad Sci U S A 1994;91:9352–9356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van de Ven KC, de Galan BE, van der Graaf M, et al. Effect of acute hypoglycemia on human cerebral glucose metabolism measured by 13C magnetic resonance spectroscopy. Diabetes 2011;60:1467–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen PJ, Alexander SC, Smith TC, Reivich M, Wollman H. Effects of hypoxia and normocarbia on cerebral blood flow and metabolism in conscious man. J Appl Physiol 1967;23:183–189 [DOI] [PubMed]
- 6.Gottstein U, Bernsmeier A, Sedlmeyer I. The carbohydrate metabolism of the human brain. I. Studies with substrate-specific enzymatic methods in normal brain circulation. Klin Wochenschr 1963;41:943–948 [in German] [DOI] [PubMed] [Google Scholar]
- 7.Lewis LD, Ljunggren B, Norberg K, Siesjö BK. Changes in carbohydrate substrates, amino acids and ammonia in the brain during insulin-induced hypoglycemia. J Neurochem 1974;23:659–671 [DOI] [PubMed] [Google Scholar]
- 8.Videen TO, Perlmutter JS, Herscovitch P, Raichle ME. Brain blood volume, flow, and oxygen utilization measured with 15O radiotracers and positron emission tomography: revised metabolic computations. J Cereb Blood Flow Metab 1987;7:513–516 [DOI] [PubMed] [Google Scholar]
- 9.Martin WR, Powers WJ, Raichle ME. Cerebral blood volume measured with inhaled C15O and positron emission tomography. J Cereb Blood Flow Metab 1987;7:421–426 [DOI] [PubMed] [Google Scholar]
- 10.Raichle ME, Martin WR, Herscovitch P, Mintun MA, Markham J. Brain blood flow measured with intravenous H2(15)O. II. Implementation and validation. J Nucl Med 1983;24:790–798 [PubMed] [Google Scholar]
- 11.Powers WJ, Dagogo-Jack S, Markham J, Larson KB, Dence CS. Cerebral transport and metabolism of 1-11C-D-glucose during stepped hypoglycemia. Ann Neurol 1995;38:599–609 [DOI] [PubMed] [Google Scholar]
- 12.Kuzuya H, Blix PM, Horwitz DL, Steiner DF, Rubenstein AH. Determination of free and total insulin and C-peptide in insulin-treated diabetics. Diabetes 1977;26:22–29 [DOI] [PubMed] [Google Scholar]
- 13.Ensinck J. Immunoassays for glucagon. In Handbook of Experimental Pharmacology Lefebvre PJ, Ed. New York, Springer-Verlag, 1983, p. 203–221 [Google Scholar]
- 14.Farmer RW, Pierce CE. Plasma cortisol determination: radioimmunoassay and competitive protein binding compared. Clin Chem 1974;20:411–414 [PubMed] [Google Scholar]
- 15.Shah SD, Clutter WE, Cryer PE. External and internal standards in the single-isotope derivative (radioenzymatic) measurement of plasma norepinephrine and epinephrine. J Lab Clin Med 1985;106:624–629 [PubMed] [Google Scholar]
- 16.Hosaka K, Kikuchi T, Mitsuhida N, Kawaguchi A. A new colorimetric method for the determination of free fatty acids with acyl-CoA synthetase and acyl-CoA oxidase. J Biochem 1981;89:1799–1803 [DOI] [PubMed] [Google Scholar]
- 17.Pinter JK, Hayashi JA, Watson JA. Enzymic assay of glycerol, dihydroxyacetone, and glyceraldehyde. Arch Biochem Biophys 1967;121:404–414 [DOI] [PubMed] [Google Scholar]
- 18.Lowry OH, Passonneau JV, Hasselberger FX, Schulz DW. Effect of ischemia on known substrates and cofactors of the glycolytic pathway in brain. J Biol Chem 1964;239:18–30 [PubMed] [Google Scholar]
- 19.Towler DA, Havlin CE, Craft S, Cryer P. Mechanism of awareness of hypoglycemia. Perception of neurogenic (predominantly cholinergic) rather than neuroglycopenic symptoms. Diabetes 1993;42:1791–1798 [DOI] [PubMed] [Google Scholar]
- 20.Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 2004;23(Suppl. 1):S208–S219 [DOI] [PubMed] [Google Scholar]
- 21.Destrieux C, Fischl B, Dale A, Halgren E. Automatic parcellation of human cortical gyri and sulci using standard anatomical nomenclature. Neuroimage 2010;53:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutchins GD, Hichwa RD, Koeppe RA. A continuous flow input function detector for H2[15O] blood flow studies in positron emission tomography. IEEE Trans Nucl Sci 1986;33:546–549 [Google Scholar]
- 23.Dillon RS. Importance of the hematocrit in interpretation of blood sugar. Diabetes 1965;14:672–674 [DOI] [PubMed] [Google Scholar]
- 24.Murphy JR. Erythrocyte metabolism. I. The equilibration of glucose-C14 between serum and erythrocytes. J Lab Clin Med 1960;55:281–285 [PubMed] [Google Scholar]
- 25.Lefevre PG, McGinniss GF. Tracer exchange vs. net uptake of glucose through human red cell surface. New evidence for carrier-mediated diffusion. J Gen Physiol 1960;44:87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JJ, Bretthorst GL, Derdeyn CP, et al. Dynamic susceptibility contrast MRI with localized arterial input functions. Magn Reson Med 2010;63:1305–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herscovitch P, Raichle ME, Kilbourn MR, Welch MJ. Positron emission tomographic measurement of cerebral blood flow and permeability-surface area product of water using [15O]water and [11C]butanol. J Cereb Blood Flow Metab 1987;7:527–542 [DOI] [PubMed] [Google Scholar]
- 28.Powers WJ, Rosenbaum JL, Dence CS, Markham J, Videen TO. Cerebral glucose transport and metabolism in preterm human infants. J Cereb Blood Flow Metab 1998;18:632–638 [DOI] [PubMed] [Google Scholar]
- 29.Fanelli CG, Dence CS, Markham J, et al. Blood-to-brain glucose transport and cerebral glucose metabolism are not reduced in poorly controlled type 1 diabetes. Diabetes 1998;47:1444–1450 [DOI] [PubMed] [Google Scholar]
- 30.Segel SA, Fanelli CG, Dence CS, et al. Blood-to-brain glucose transport, cerebral glucose metabolism, and cerebral blood flow are not increased after hypoglycemia. Diabetes 2001;50:1911–1917 [DOI] [PubMed] [Google Scholar]
- 31.Cranston I, Reed LJ, Marsden PK, Amiel SA. Changes in regional brain (18)F-fluorodeoxyglucose uptake at hypoglycemia in type 1 diabetic men associated with hypoglycemia unawareness and counter-regulatory failure. Diabetes 2001;50:2329–2336 [DOI] [PubMed] [Google Scholar]
- 32.Powers WJ, Videen TO, Markham J, et al. Selective defect of in vivo glycolysis in early Huntington’s disease striatum. Proc Natl Acad Sci U S A 2007;104:2945–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Criego AB, Tkac I, Kumar A, Thomas W, Gruetter R, Seaquist ER. Brain glucose concentrations in healthy humans subjected to recurrent hypoglycemia. J Neurosci Res 2005;82:525–530 [DOI] [PubMed] [Google Scholar]
- 34.van de Ven KC, van der Graaf M, Tack CJ, Heerschap A, de Galan BE. Steady-state brain glucose concentrations during hypoglycemia in healthy humans and patients with type 1 diabetes. Diabetes 2012;61:1974–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]