

Abstract
One ultimate goal of synthetic chemistry is to install or manipulate any functional group at any position of a molecule. This Account discusses the potential and possible approaches to use catalysis to enable a reaction to occur at one of many C–H bonds or at one of several nearly identical functional groups.
Graphical Abstract
Twenty years ago, one-third of the articles in the “Holy Grail” issue of Accounts of Chemical Research focused on catalysis,1,3 and spectacular advances have been made in catalysis since that time. Homogeneous catalysis has changed from a collection of reactions for special purposes (albeit some producing billions of pounds of product per year) to mainstream chemistry that has revolutionized all types of organic synthesis. The number and scope of new catalytic processes that have been discovered during these two decades is staggering, ranging from reactions with general utility to reactions that seem merely curious and hard to envision using, but triggering new ideas.
These catalytic reactions occur in many cases with remarkable selectivity for one functional group over another, including selective reactions at a functional group that is typically less reactive toward most reagents in the absence of a catalyst. Perhaps best known is the selectivity of ruthenium complexes for reaction at an alkene to cleave the C–C bond during catalytic olefin metathesis in the presence of ketones, esters, amides, and alcohols.4 Although catalysts have changed the order of reactivity of functional groups toward many reagents, even the most spectacular reactions occur, in many cases, where the molecules dictate, not where chemists and their catalysts dictate. Reactions occur at the less hindered alkene, ketone, or alcohol over the more hindered ones, at a ketone over an ester, or at an ester over an amide. They occur at the more acidic C–H bonds over the less acidic C–H bond, at the weaker C–H bond over the stronger C–H bond, or at the more electron-rich C–H bond over the more electron-poor C–H bond.
No doubt, some catalysts violate these trends in site selectivity. For example, many catalysts react at stronger aryl C–H bonds over weaker alkyl C–H bonds.5 But what catalyst reacts at the more electron-rich, more hindered aryl C–H bond or the stronger and more electron-rich of the alkyl C–H bonds? Or what catalyst reacts selectively at one of two ketones or one of two alcohols or one of two C–H bonds that have equivalent steric and electronic properties but are chemically nonequivalent due to the remaining structure of the molecule. And what catalyst reacts at the C–C bonds in the framework of a molecule, rather than the periphery? No catalyst reacts with unstrained versions of such C–C bonds to restructure it from within.
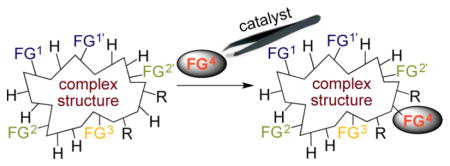
Thus, we are far from achieving the most general goal of synthetic chemistry that could be achieved with catalysts–conducting a reaction at any bond of a molecule to install any atom or group of atoms by selection of the proper catalyst and reagent (Figure 1). Obviously no single catalyst could achieve this general task. However, one can imagine creating a suite of catalysts that a well-versed synthetic chemist can use to induce reactions at positions dictated by the catalyst, rather than positions dictated by the local environment of a C–H bond, C–C bond, or functional group. Progress has been made toward this goal, as described in the next few sections. Yet, we remain far from achieving it; therefore, this Account also describes potential new paths to achieve this general goal of synthetic chemistry.
Figure 1.
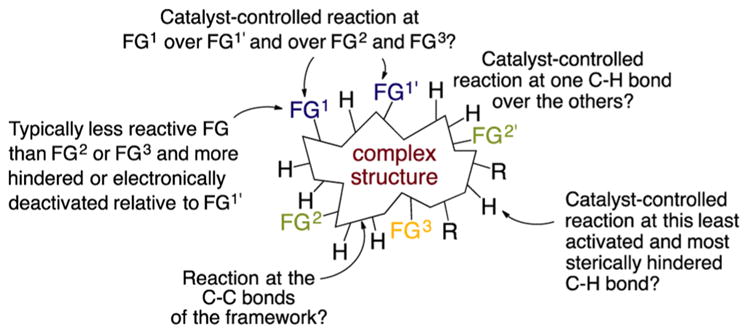
Challenge of controlling site-selectivity for one functional group over other similar or more reactive functional groups, for one C–H bond over another, and for C–C bonds.
SELECTIVITY FOR ONE FUNCTIONAL GROUP OVER ANOTHER
Discoveries of catalysts over the past 20 years have given chemists in the field of homogeneous catalysis confidence that one can conduct reactions at a functional group that is typically less reactive toward classical reagents in the presence of a functional group that is typically more reactive toward such reagents and even at a single C–H bond over common functional groups in the molecule (Figure 2). As just noted, olefin metathesis occurs at a nonpolar C = C bond in preference to alternative processes at more polar and, therefore, typically more reactive C=O or O–H bonds. Palladium-catalyzed cross-coupling reactions are exquisitely selective for an aryl halide over many other functional groups, as exemplified by Suzuki couplings used in the synthesis of vancomycin (Figure 2A),6 whereas iridium catalysts (at least for the borylation and silylation of C–H bonds) are exquisitely selective for aryl C–H bonds over aryl C–X bonds in which X is any of the halogens.7,8 This selectivity was recently exploited in the 100 kg preparation of doravirine for phase III clinical trials (Figure 2B).9 Likewise, many palladium-catalyzed reactions occurring through oxidation states of palladium higher than Pd(0) occur at aryl C–H bonds over aryl carbon–halogen bonds.10
Figure 2.

(A) Suzuki coupling in the synthesis of vancomycin showing the remarkable selectivity of the palladium catalyst for aryl bromide bonds over other potentially reactive bonds. (B) C–H borylation in the synthesis of doravirine showing the remarkable selectivity of the iridium catalyst for an aryl C–H bond over aryl halide bonds.
However, many molecules contain multiple functional groups of the same class. They often contain multiple alkenes or carbonyl groups or alcohols or amines. The selective reaction at one of these functional groups over the other with one catalyst but selective reaction at another of these functional groups with a different catalyst remains a challenge. For example, the catalytic hydrogenation of an ester or amide over a ketone is rare if not unknown.
Although this challenge of conducting a reaction at one type of functional group over another or at one of multiple functional groups of the same type constantly arises during multistep syntheses, the selectivity for reaction at one C–H bond over another is the most striking challenge of site selectivity. This challenge arises because many molecules have many C–H bonds that are similar to each other. The need to achieve selective reactions at one C–H bond over another highlights the limitations of our capabilities to achieve site-selective reactions, but this need has led to progress suggesting that the general goal of catalyst controlled site selectivity can be achieved.
Twenty years ago, Bergman wrote that selectively activating a C–H bond in the presence of functional groups was one holy grail of chemistry.3 Now, perhaps due to articulating this challenge, chemists have discovered many catalysts that achieve this task. In our own work, the borylation of aryl C–H bonds occurs in the presence of ketones, esters, amides, nitriles, amines, all four halides bound to an arene, basic and protic nitrogens of heterocycles, and even phenols (Figure 3A).8,11 The reactions occur with unusually reliable site selectivity in arenes controlled by steric effects and directing effects. Metal-catalyzed functionalizations of aryl C–H bonds occur ortho to a wide range of substituents, first ortho to a ketone12 and now ortho to many groups including carboxylic acid units located varied distances from the arene (Figure 3B).13 Thus, a suite of catalysts provides remarkable, although far from absolute, control of the position at which a reaction occurs on aromatic C–H bonds.
Figure 3.

Several approaches to install functionality at specific C–H bonds. (A) General scheme for the combination of borylation of an aryl C–H bond and functionalization of the intermediate arylboronate. (B) Generic scheme for palladium-catalyzed directed functionalization of aryl C–H bonds under oxidative conditions. (C) Chemoenzymatic methods for fluorination of specific C–H bonds and different positions controlled by the site-selectivity of mutant P450 enzymes.
We are much further from creating a suite of catalysts that control the site of reaction at aliphatic C–H bonds. Many functionalizations of alkyl C–H bonds are oxidative processes, and the cleavage of the C–H bond occurs with an electron-poor site on a metal complex or reagent. As a result, these reactions occur at the most electron-rich, typically weak C–H bond.14 In other words, these types of functionalizations occur at tertiary C–H bonds and electron-rich secondary C–H bonds. Although chemical catalysts for the functionalization of primary C–H bonds are now known,15 such catalysts that functionalize primary C–H bonds in a practical way without a functional group to direct the catalyst to the primary C–H bond are not known. Likewise, catalysts that functionalize the more electron-poor C–H bond of a set of nonacidic methylene C–H bonds are not known, and catalysts that react at the more hindered C–H bond among a set of electronically similar C–H bonds are not known. How one would create a small-molecule catalyst that reacts at the more hindered C–H bond of a set of C–H bonds having similar bond strengths and electronic properties is hard to imagine.
Even if a catalyst exists that functionalizes a C–H bond selectively at a specific position, the range of functional groups or substituents that chemists would like to install at such a position is large. Thus, it might seem that dozens of catalysts must be discovered to install dozens of functional groups at each C–H bond. However, one strategy followed by our group and others to avoid this requirement is to discover reactions that create versatile synthetic intermediates. By doing so, one can diversify the intermediate to create a series of products with various functional groups or substituents at a specific position or use one system for C–H bond functionalization to address a wide range of synthetic needs. We did so by discovering complexes that catalyze the borylation and silylation of aromatic and heteroaromatic C–H bonds. The products of these reactions can be converted to those containing a new C–C, C–N, C–O, or C–halogen bond in biaryls, alkylarenes, aminoarenes, arylamine derivatives, aryl ethers, phenols, or aryl halides, as shown in Figure 3A.16–18
Others19–22 have outlined a similar strategy using enzymatic oxidation of C–H bonds. By this strategy, the combination of P450-catalyzed hydroxylation of an alkyl C–H bond, followed by deoxyfluorination (Figure 3C) or by acylation or alkylation of the alcohol is used to create a set of molecules with functional groups introduced at a specific aliphatic C–H bond.22 Thus, a programmed method to install groups that can be readily derivatized at one position over others would be a significant step toward enabling the installation any functional group at any C–H bond in a molecule.
INSPIRATIONS FOR FUTURE DIRECTIONS
Given these current limitations, but recent progress, how can we create additional catalysts that ultimately lead to complete control over site selectivity? Classically, control over site selectivity has been achieved with protective groups. Protection of the more reactive alcohol or ketone as an ether or acetal renders it less reactive, and this protection allows a reaction to occur at the less reactive alcohol, ketone, or carbonyl group. Although successful in many cases, an important goal of selective catalysis is to eliminate our reliance on protective groups that add steps to the sequence used to prepare complex molecules and that is impractical for the synthesis of bulk chemicals. The potential use of biomass feedstocks, which possess multiple functional groups, illustrates the challenge of conducting a reaction at one functional group among many as part of potential production of bulk chemicals.
The functionalization of aromatic C–H bonds suggests that site-selective reactions can be achieved more generally. Recently, researchers at Merck23 used a few examples of medicinally active molecules to illustrate how one can functionalize a C–H bond in many positions selectively with current methods by choosing the proper reagent and catalyst. Of course, one cannot place any group at any position, but many positions of the molecule are accessible. An adaption of their analysis of the ability to functionalize C–H bonds in clopidogrel is provided in Figure 4 and shows that one type of C–H bond functionalization or another allows a reaction to occur selectively at almost any of the C–H bonds in this molecule. However, a similar depiction of their analysis of the potential to functionalize quinine shows that some positions can be functionalized by current methods, but a similar number of positions of the quinine scaflold cannot be functionalized site-selectively.
Figure 4.

Depiction of an analysis by researchers at Merck23 of the potential to functionalize specific C–H bonds in two medicinally active compounds.
How can one develop catalytic reactions at positions that are inaccessible by current methods? Certainly the discovery of new modes of reactivity that lead to reactions at typically unreactive positions can address this issue. However, approaches to use existing reactivity but to change the site at which these reactions occur can provide an additional approach to this synthetic problem.
Studies using the combination of supramolecular chemistry and catalysis to redirect a catalyst to one functional group or one C–H bond over another is being pursued in several ways. As noted in the introduction, the selective reaction at one hydroxyl group over another, particularly in a carbohydrate or polyol natural product, is a synthetic challenge that lies at the heart of biosynthetic pathways to important carbohydrate biopolymers, peptide natural products, or macrolide natural products. Although we do not have enzymes that catalyze a reaction at every individual hydroxyl group selectively in any carbohydrate, work with peptide catalysts points toward a path to create such catalysts. Libraries of peptides with varying structures have been created that recognize portions of organic substrates and catalyze the selective acetoxylation, sulfonation, or phosphorylation of one hydroxyl group over others.24 With access to a library of peptides, a catalyst of this type can be chosen that reacts at a specific hydroxyl group that is not the inherently most reactive hydroxyl group in complex molecules, such as erythromycin A, teicoplanin (Figure 5), or D-myoinositol.24–26 Likewise, libraries of peptide catalysts have been created for the halogenation of arenes, and members of this peptide library catalyze the halogenation of one arene over other arenes in complex structures, like vancomycin, due to their ability to recognize the overall structure of the molecule.27
Figure 5.

Use of a peptide catalyst to functionalize one of many alcohols of teicoplanin.
Nature attacks the problem of site selectivity by evolution (and accompanying lengthy time scales). Perhaps the most striking examples are the selective hydroxylations of C–H bonds in largely hydrocarbyl reactants, including steroids, fatty acids, and alkanes. However, additional classes of enzymes react at aryl C–H bonds with high selectivity. For example, a series of halogenases react at the 5, 6, or 7 position of the indole in tryptophan exclusively, and studies are being conducted to improve activity and broaden the scope of substrates that react with these enzymes.28 Of course, glycosyltransferases also catalyze reactions at one hydroxyl group over the many other hydroxyl groups in carbohydrates as part of the synthesis of complex carbohydrates and glycoproteins.29
Although many enzymes catalyze many reactions selectively, there are positions of molecules at which no known enzyme causes a reaction to occur. Thus, chemists, biochemists, and engineers have developed approaches to access these positions of a molecule by evolving the catalyst in the laboratory. Again, research on this topic has been conducted intensively with enzymes that catalyze hydroxylations of C–H bonds. The methods of directed evolution have been used to create artificial P450s that catalyze the hydroxylation of steroids at positions where natural enzymes do not react (Figure 6A) and with light hydrocarbons that hardly react at all with natural P450s.30–32
Figure 6.

(A) Directed evolution of P450 enzymes leading to non-native site selectivity for hydroxylation of steroids. (B) Directed evolution of P450 analogs containing iridium in the porphyrin of the active site catalyze enantioselectively.
Although these methods for evolution of enzymes in the laboratory are powerful, these enzymes undergo a limited range of reactions. The promiscuous reactivity of a heme active site has been shown to encompass the additions of carbenes33,34 and nitrenes35 to alkenes, as well as insertions of carbenes into Si–H bonds36 and nitrenes into C–H bonds.37,38 However, the scope of these reactions is severely limited and chemical synthesis requires the installation of a wide range of functional groups, such as amino groups, cyano groups, fluorine atoms, trifluoromethyl groups, substituted carbonyl groups, or entire substituted aryl or heteroaryl groups. Enzymes are not known that catalyze the installation of these structural units onto an organic scaflold. Thus, several groups, including ours, are seeking methods to combine protein architectures with computational design and directed evolution to create artificial enzymes,39 including artificial metalloenzymes,40 that catalyze the reactions of chemists, instead of the reactions of Nature. To do so, active sites are needed that catalyze the installation or interconversion of functional groups at or near room temperature with a tolerance for water and with ligands that comprise the side chains of natural amino acids or that can be installed as unnatural amino acids or as cofactors bound to the protein by noncovalent interactions. Clearly, the community is far from achieving this goal in a general way, but some progress has been made in this direction.
In our own group, we have developed approaches to replace, in a formal sense, the iron in heme proteins with metals that catalyze reactions that are different from those catalyzed by iron–porphyrin complexes (Figure 6B).41,42 Although such systems are far from catalyzing multiple reactions at programmed sites, they do catalyze reactions that are not induced by natural enzymes and that can be evolved to react with high selectivity. To do so, we expressed native and mutant myoglobins and cytochromes P450 that lack the heme unit and then loaded these apo proteins with native porphyrins containing non-native metals, such as rhodium, ruthenium, iridium, silver, cobalt, etc. We found that the metalloprotein containing a methyliridium unit in place of the iron catalyzes a wide range of atom-transfer processes that are not catalyzed by iron. For example, these systems catalyze the stereoselective insertion of a carbene into a C–H bond and the stereoselective cyclopropanation of unactivated alkenes,42,43 in some cases with kinetics that rival those of natural enzymes in biosynthetic pathways.42
Over the past decade, others have pioneered the development of systems that combine streptavidin and organometallic compounds containing a tether to biotin to create artificial metalloenzymes that catalyze stereoselective reactions characteristic of organometallic systems, such as hydrogenation, allylic substitution, annulation by C–H bond activation, and olefin metathesis.40 These studies collectively show that the ability to use mutagenesis to change the environment around such catalysts complements typical variations of ligands in transition metal chemistry. This approach to the generation of artificial metalloenzymes enables the generation of libraries of catalysts that can be stereoselective and, perhaps, ultimately site selective for reaction at one C–H bond or one functional group of a molecule over another to install the functional groups of chemists and not just those of Nature.
Most of the discussion in this Account has focused on conducting reactions at bonds or groups of atoms at the periphery of molecules, such as C–H bonds, O–H bonds, and C=O bonds of carbonyl groups. As noted in the introduction, perhaps the most challenging transformations are those at carbon–carbon bonds in the cyclic framework of organic molecules, as well as specific carbon–carbon bonds in a chain. Catalysts are being developed that cleave C–C bonds in strained rings, such as cyclopropanes and cyclobutanes.44,45 However, little progress has been made on methods to restructure complex molecules by cleaving C–C bonds in the unstrained rings of the core. Of course the driving force for such a process must stem from the reagent or from forming another strong bond in an accompanying step if C–C bonds in stable structures are to be cleaved.
Synthetic chemistry is a complex web of chemical reactions that is rapidly increasing in size and number of connections between the radii of this web. Thus, there will be no single development that will allow chemists to catalyze a reaction at any position they desire. However, novel reactivity, in conjunction with novel approaches to control regioselectivity discussed in this Account, should make it possible to conduct reactions at positions of molecules previously inaccessible, whether inaccessible due to the lack of reactivity of these positions or due to the presence of positions in the same molecule that are inherently more reactive toward most reagents in the absence of a catalyst.
Acknowledgments
The author thanks the NIH (Grant GM-115812) for current support of our work on C–H bond silylation and borylation discussed in this Account, the NIH (Grant GM-55382) for support of our ongoing work on selective functionalization of C–H bonds to form C–X bonds and for the selective functionalization of O–H bonds, and the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 for support of our current work on artificial metalloenzymes discussed in this Account.
Footnotes
Notes
The author declares no competing financial interest.
References
- 1.Bard AJ, Fox MA. Artificial Photosynthesis: Solar Splitting of Water to Hydrogen and Oxygen. Acc Chem Res. 1995;28:141–145. [Google Scholar]
- 2.Breslow R. Biomimetic Chemistry and Artificial Enzymes: Catalysis by Design. Acc Chem Res. 1995;28:146–153. [Google Scholar]
- 3.Arndtsen BA, Bergman RG, Mobley TA, Peterson TH. Selective Intermolecular Carbon-Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc Chem Res. 1995;28:154–162. [Google Scholar]
- 4.Vougioukalakis GC, Grubbs RH. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]
- 5.Hartwig JF. Organotransition Metal Chemistry. Chapter 18 University Science Books; Sausalito, CA: 2010. Catalytic C-H Bond Activation and Functionalization. [Google Scholar]
- 6.Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. Total synthesis of the vancomycin aglycon. J Am Chem Soc. 1999;121:10004. [Google Scholar]
- 7.Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J Am Chem Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- 8.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. C-H Activation for the Construction of C-B Bonds. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 9.Campeau LC, Chen Q, Gauvreau D, Girardin M, Belyk K, Maligres P, Zhou G, Gu C, Zhang W, Tan L, O’Shea PD. A Robust Kilo-Scale Synthesis of Doravirine. Org Process Res Dev. 2016;20:1476–1481. [Google Scholar]
- 10.Yoo EJ, Ma S, Mei TS, Chan KSL, Yu JQ. Pd-Catalyzed Intermolecular C–H Amination with Alkylamines. J Am Chem Soc. 2011;133:7652–7655. doi: 10.1021/ja202563w. [DOI] [PubMed] [Google Scholar]
- 11.Hartwig JF. Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Acc Chem Res. 2012;45:864–873. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
- 12.Murai S, Kakiuchi F, Sekine S, Tanaka Y, Kamatani A, Sonoda M, Chatani N. Efficient Catalytic Addition of Aromatic Carbon-Hydrogen Bonds to Olefins. Nature. 1993;366:529–531. [Google Scholar]
- 13.Engle KM, Mei TS, Wasa M, Yu JQ. Weak Coordination as a Powerful Means for Developing Broadly Useful C–H Functionalization Reactions. Acc Chem Res. 2012;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Newhouse T, Baran PS. If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew Chem, Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Schlecht S, Semple TC, Hartwig JF. Thermal, Catalytic, Regiospecific Functionalization of Alkanes. Science. 2000;287:1995–1997. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]
- 16.Murphy JM, Liao X, Hartwig JF. Meta Halogenation of 1,3-Disubstituted Arenes via Iridium-Catalyzed Arene Borylation. J Am Chem Soc. 2007;129:15434–15435. doi: 10.1021/ja076498n. [DOI] [PubMed] [Google Scholar]
- 17.Murphy JM, Tzschucke CC, Hartwig JF. One-pot synthesis of arylboronic acids and aryl trifluoroborates by Ir-catalyzed borylation of arenes. Org Lett. 2007;9:757–760. doi: 10.1021/ol062903o. [DOI] [PubMed] [Google Scholar]
- 18.Tzschucke CC, Murphy JM, Hartwig JF. Arenes to anilines and aryl ethers by sequential iridium-catalyzed borylation and copper-catalyzed coupling. Org Lett. 2007;9:761–764. doi: 10.1021/ol062902w. [DOI] [PubMed] [Google Scholar]
- 19.Rentmeister A, Arnold FH, Fasan R. Chemo-enzymatic fluorination of unactivated organic compounds. Nat Chem Biol. 2009;5:26–28. doi: 10.1038/nchembio.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang K, Shafer BM, Demars MD, Stern HA, Fasan R. Controlled Oxidation of Remote sp3 C–H Bonds in Artemisinin via P450 Catalysts with Fine-Tuned Regio- and Stereoselectivity. J Am Chem Soc. 2012;134:18695–18704. doi: 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolev JN, O’Dwyer KM, Jordan CT, Fasan R. Discovery of Potent Parthenolide-Based Antileukemic Agents Enabled by Late-Stage P450-Mediated C-H Functionalization. ACS Chem Biol. 2014;9:164–173. doi: 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyagi V, Alwaseem H, O’Dwyer KM, Ponder J, Li QY, Jordan CT, Fasan R. Chemoenzymatic synthesis and antileukemic activity of novel C9-and C14-functionalized parthenolide analogs. Bioorg Med Chem. 2016;24:3876–3886. doi: 10.1016/j.bmc.2016.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem Soc Rev. 2016;45:546–576. doi: 10.1039/c5cs00628g. [DOI] [PubMed] [Google Scholar]
- 24.Miller SJ. In search of peptide-based catalysts for asymmetric organic synthesis. Acc Chem Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- 25.Lewis CA, Miller SJ. Site-selective derivatization and remodeling of erythromycin A by using simple peptide-based chiral catalysts. Angew Chem, Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 26.Han S, Miller SJ. Asymmetric Catalysis at a Distance: Catalytic, Site-Selective Phosphorylation of Teicoplanin. J Am Chem Soc. 2013;135:12414–12421. doi: 10.1021/ja406067v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pathak TP, Miller SJ. Site-Selective Bromination of Vancomycin. J Am Chem Soc. 2012;134:6120–6123. doi: 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Payne JT, Andorfer MC, Lewis JC. Regioselective Arene Halogenation using the FAD-Dependent Halogenase RebH. Angew Chem, Int Ed. 2013;52:5271–5274. doi: 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taniguchi N, Honke K, Fukuda M, Clausen H, Furukawa K, Hart GW, Kannagi R, Kawasaki T, Kinoshita T, Muramatsu T, Saito M, Shaper JH, Sugahara K, Tabak LA, Van den Eijnden DH, Yanagishita M, Dennis JW, Furukawa K, Hirabayashi Y, Kawakita M, Kimata K, Lindahl U, Narimatsu H, Schachter H, Stanley P, Suzuki A, Tsuji H, Yamashita K, editors. Handbook of Glycosyltransferases and Related Genes. Springer; Japan: Tokyo: 2002. [Google Scholar]
- 30.Kille S, Zilly FE, Acevedo JP, Reetz MT. Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat Chem. 2011;3:738–743. doi: 10.1038/nchem.1113. [DOI] [PubMed] [Google Scholar]
- 31.Roiban GD, Reetz MT. Expanding the toolbox of organic chemists: directed evolution of P450 monooxygenases as catalysts in regio- and stereoselective oxidative hydroxylation. Chem Commun. 2015;51:2208–2224. doi: 10.1039/c4cc09218j. [DOI] [PubMed] [Google Scholar]
- 32.Jung ST, Lauchli R, Arnold FH. Cytochrome P450: taming a wild type enzyme. Curr Opin Biotechnol. 2011;22:809–817. doi: 10.1016/j.copbio.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coelho PS, Brustad EM, Kannan A, Arnold FH. Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- 34.Bordeaux M, Tyagi V, Fasan R. Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts. Angew Chem, Int Ed. 2015;54:1744–1748. doi: 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farwell CC, Zhang RK, McIntosh JA, Hyster TK, Arnold FH. Enantioselective Enzyme-Catalyzed Aziridination Enabled by Active-Site Evolution of a Cytochrome P450. ACS Cent Sci. 2015;1:89–93. doi: 10.1021/acscentsci.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kan SBJ, Lewis RD, Chen K, Arnold FH. Directed evolution of cytochrome c for carbon-silicon bond formation: Bringing silicon to life. Science. 2016;354:1048–1051. doi: 10.1126/science.aah6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, Arnold FH. Enantioselective Intramolecular C-H Amination Catalyzed by Engineered Cytochrome P450 Enzymes In Vitro and In Vivo. Angew Chem, Int Ed. 2013;52:9309–9312. doi: 10.1002/anie.201304401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh R, Bordeaux M, Fasan R. P450-Catalyzed Intra-molecular sp(3) C-H Amination with Arylsulfonyl Azide Substrates. ACS Catal. 2014;4:546–552. doi: 10.1021/cs400893n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kries H, Blomberg R, Hilvert D. De novo enzymes by computational design. Curr Opin Chem Biol. 2013;17:221–228. doi: 10.1016/j.cbpa.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 40.Durrenberger M, Ward TR. Recent achievements in the design and engineering of artificial metalloenzymes. Curr Opin Chem Biol. 2014;19:99–106. doi: 10.1016/j.cbpa.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 41.Key HM, Dydio P, Clark DS, Hartwig JF. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature. 2016;534:534–537. doi: 10.1038/nature17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dydio P, Key HM, Nazarenko A, Rha JYE, Seyedkazemi V, Clark DS, Hartwig JF. An artificial metalloenzyme with the kinetics of native enzymes. Science. 2016;354:102–106. doi: 10.1126/science.aah4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Key HM, Dydio P, Clark DS, Hartwig JF. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature. 2016;534:534–537. doi: 10.1038/nature17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murakami M, Ishida N. Potential of Metal-Catalyzed C–C Single Bond Cleavage for Organic Synthesis. J Am Chem Soc. 2016;138:13759–13769. doi: 10.1021/jacs.6b01656. [DOI] [PubMed] [Google Scholar]
- 45.Ko HM, Dong G. Cooperative activation of cyclobutanones and olefins leads to bridged ring systems by a catalytic [4 + 2] coupling. Nat Chem. 2014;6:739–744. doi: 10.1038/nchem.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]