

Abstract
The preparation of all possible stereo-isomers of a given chiral molecule bearing multiple stereocenters by a simple and unified method is a significant challenge in asymmetric catalysis. We report stereodivergent allylic substitutions with aryl acetic acid esters catalyzed synergistically by a metallacyclic iridium complex and benzotetramisole. Through permutations of the enantiomers of the two chiral catalysts, all four stereoisomers of the products bearing two adjacent stereocenters are accessible with high diastereoselectivity and enantioselectivity. The resulting chiral activated ester products can be converted readily to enantioenriched amides, unactivated esters, and carboxylic acids in a one-pot manner.
Transition-metal-catalyzed allylic substitutions are useful methods for the enantioselective construction of carbon–carbon bonds.1 If both the nucleophiles and electrophiles of the allylation reactions are prochiral, synthetically useful adducts that contain two contiguous stereocenters can be constructed in one step. However, most reported reactions of this type that afford products enantioselectively and diastereoselectively form one out of two possible relative configurations (anti vs syn).2 Few methods provide stereodivergent access to all four possible stereoisomers of the products with either the anti or syn configuration. Recently, Carreira and co-workers reported the allylation of aldehydes in a stereodivergent fashion by the synergistic reactivity of iridium and amine catalysts under acidic conditions.3 Zhang and co-workers reported the combination of iridium and zinc catalysts for the related allylation of α-hydroxy phenones.4 An approach to the stereodivergent allylation of carbonyl compounds in the carboxylic acid oxidation state has not been published.5
Mechanistic studies6 have revealed that metallacyclic iridium complexes7 developed in our group govern the geometry, facial selectivity, and regioselectivity of the allyl moiety in allylation reactions (Scheme 1, A). Lewis basic chiral tertiary amines are known to react with acyl precursors to form C1-ammonium enolates that have a well-defined geometry and that react with high facial selectivity (Scheme 1, B).8 The metallacyclic iridium catalyst for allylic substitution that we discovered7 operates under basic conditions. Thus, a system with a Lewis basic catalyst displacing an alkoxide or phenoxide anion to generate the enolate would be compatible with our iridium catalysts. We envisioned that the allylation reaction between A as an electrophile and B as a nucleophile would be highly regio-, diastereo-, and enantioselective. Furthermore, the iridium complex and the Lewis base (LB) could dictate the configurations of the two stereogenic centers of the product arising from the electrophile (marked blue) and the nucleophile (marked red), respectively. Thus, our proposed allylation method could access all four possible stereoisomers of the product by simple permutations of the enantiomers of the two catalysts (IrR + LBR, IrS + LBR, IrR + LBS, IrS + LBS).9
Scheme 1.
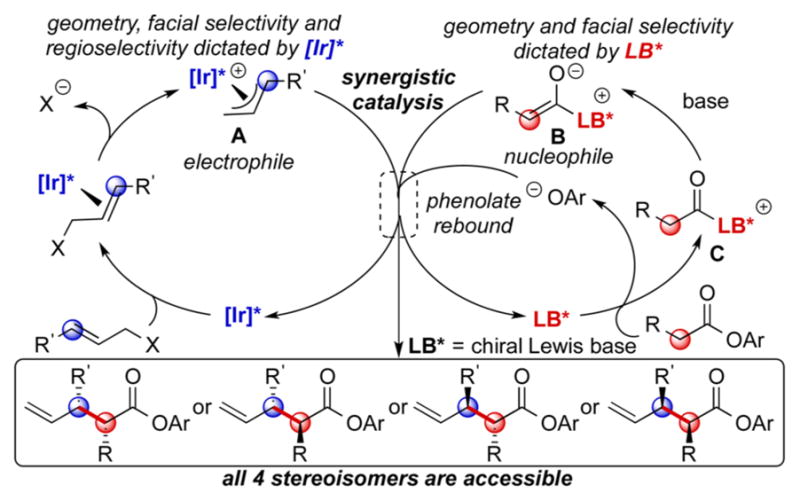
Proposed Mechanism for Synergistic Catalysis
A critical concern that underlies our proposed transformation is the turnover of the Lewis base catalyst. Regeneration of this catalyst typically requires an intramolecular acyl transfer to a proximal nucleophile on the acylammonium intermediate. In this case, only lactones and lactams are accessible as the products.8c,d,10 Although external nucleophiles can be employed as acyl acceptors,11 this external nucleophile can react with intermediate C before allylation occurs. In addition, the direct allylation of an external nucleophile can compete or override the allylation of the enolate.
We considered that the “rebound” strategy disclosed recently by Scheidt,12 Smith,13 and Snaddon14 could be followed to regenerate the Lewis base. In this scenario, the electron-deficient phenolate (Scheme 1, OAr−) substituted by the Lewis base catalyst serves as an acyl acceptor after α-functionalization of the ester. The low concentration and low nucleophilicity of the electron-deficient phenolate would prevent the direct allylation of the phenolate.
Herein we report stereodivergent allylic substitutions with aryl acetic acid esters catalyzed synergistically by a metallacyclic iridium complex and a chiral Lewis base. By variation of the combination of enantiomers of the two catalysts, all four stereoisomers of the products are formed with high diastereoselectivity and enantioselectivity. The resulting chiral activated esters are readily converted to enantioenriched amides, unactivated esters, and carboxylic acids in a one-pot manner.
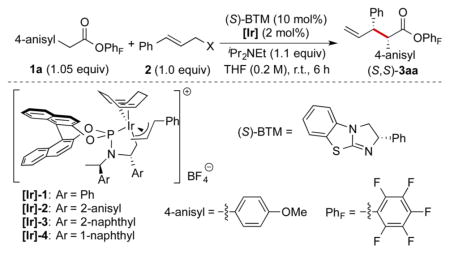
To develop a stereodivergent allylation of aryl acetic acid esters, we treated 1a with 2a (X = OBoc) in the presence of iPr2NEt as the base, iridium catalyst [Ir]-1, and a range of Lewis base catalysts (Table S1). These studies revealed that benzotetramisole (BTM)15 is compatible with our proposed synergistic catalysis (>99% yield, >20:1 dr; see the Supporting Information (SI) for details). Reactions conducted with other Lewis bases, such as tetramisole and quinine, delivered the product in lower yields (<40%) or with lower diastereoselectivity (<4:1). The use of the pentafluorophenyl ester as the nucleophile precursor was critical; reactions conducted with esters derived from other electron-deficient phenols, such as 4-nitrophenol and 2,4,6-tricholorophenol, gave the corresponding products in low yield and dr (Table S2; see the SI for details).
To test the effect of the leaving group on the allyl moiety in this reaction, various cinnamyl alcohol derivatives were subjected to the reaction conditions (Table 1). The reaction conducted with tert-butyl cinnamyl carbonate (2a) gave (S,S)-3aa in the highest yield (97%) with >20:1 dr and >99% ee (entry 7). Only the branched product was observed. A similar result was obtained in the absence of iPr2NEt, indicating that the tert-butoxide generated from oxidative addition of 2a and subsequent decarboxylation acted as a base to deprotonate the acyl–BTM adduct (entry 8).
Table 1.
Evaluation of the Reaction Conditions for the Allylation of 1aa
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | [lr] | X | b/lb | drb | ee/%c | yield/%d |
| 1 | [lr]-1 | OAc | n.d. | n.d. | n.d. | 13 |
| 2 | OBz | >99:1 | >20:1 | n.d. | 59 | |
| 3 | OPiv | n.d. | n.d. | n.d. | 8 | |
| 4 | OPO(OEt)2 | n.d. | 6:1 | n.d. | 38 | |
| 5 | OCOOMe | >99:1 | 5:1 | n.d. | 43 | |
| 6 | OTroc | >99:1 | 9:1 | n.d. | 90 | |
| 7 | OBoc | >99:1 | >20:1 | >99 | >99 (97) | |
| 8e | [Ir]-1 | OBoc | >99:1 | >20:1 | >99 | 99 (97) |
| 9e | [lr]-2 | >99:1 | 11:1 | n.d. | 71 | |
| 10e | [lr]-3 | >99:1 | >20:1 | >99 | >99 (>99) | |
| 11e | [lr]-4 | >99:1 | >20:1 | >99 | 99 (99) | |
| 12e,f | [lr]-1 | 10:1 | <1:20 | >99 | 98 (97) | |
| 13e,f | [lr]-2 | >20:1 | 1:13 | n.d. | 57 | |
| 14e,f | [lr]-3 | 10:1 | <1:20 | >99 | 99 (>99) | |
| 15e,f | [lr]-4 | >20:1 | 1:13 | >99 | 99 (>99) | |
The absolute configuration of (S,S)-3aa was assigned by analogy.
Determined by 1H NMR analysis of the crude reaction mixtures.
Determined by chiral supercritical fluid chromatography of the major isomers.
Combined yields of the two diastereomers of the branched product and the linear product, determined by 1H NMR analysis with mesitylene as an internal standard. Yields in parentheses are for all isomers isolated.
Without iPr2NEt.
(R)-BTM was used instead of (S)-BTM.
Metallacyclic iridium catalysts with different aryl substituents on the phosphoramidite ligands were evaluated. Reactions conducted with [Ir]-3 and [Ir]-4 bearing naphthyl substituents on the ligands afforded (S,S)-3aa quantitatively with excellent diastereoselectivity and enantioselectivity (>20:1 dr, >99% ee; entries 10 and 11). However, the reaction conducted with [Ir]-2 bearing 2-methoxyphenyl substituents on the ligand gave (S,S)-3aa in a lower yield of 71% with a lower dr of 11:1 (entry 9).
When the reaction was conducted with (R)-BTM as the Lewis base catalyst instead of (S)-BTM, the diastereoselectivity was completely reversed; (R,S)-3aa was obtained instead of (S,S)-3aa in 97% yield with >20:1 dr and >99% ee (entry 12). A small amount of linear product was observed when the reaction was conducted with the catalyst combination of [Ir]-1 and (R)-BTM. However, employing [Ir]-4 and (R)-BTM as the catalysts suppressed the formation of the linear product while maintaining high dr and ee (entry 15).
To examine the stereodivergence of our allylation method, 1a and 2a were treated with four different combinations of the enantiomers of the two catalysts under otherwise identical conditions (Scheme 2). As a result, all four stereoisomers of 3aa were obtained individually in high yield with excellent diastereo-and enantioselectivity, indicating nearly complete control of the configurations at the allyl electrophile and the enolate nucleophile by the metallacyclic Ir complex and the BTM base, respectively, and the dominance of catalyst control of these configurations over potential substrate control. The absolute configurations of the products are consistent with a stereo-chemical model based on previous mechanistic studies of the Ir6 and BTM16 catalysts (Scheme S1), rendering the stereochemical outcome of our allylation method predictable.
Scheme 2.
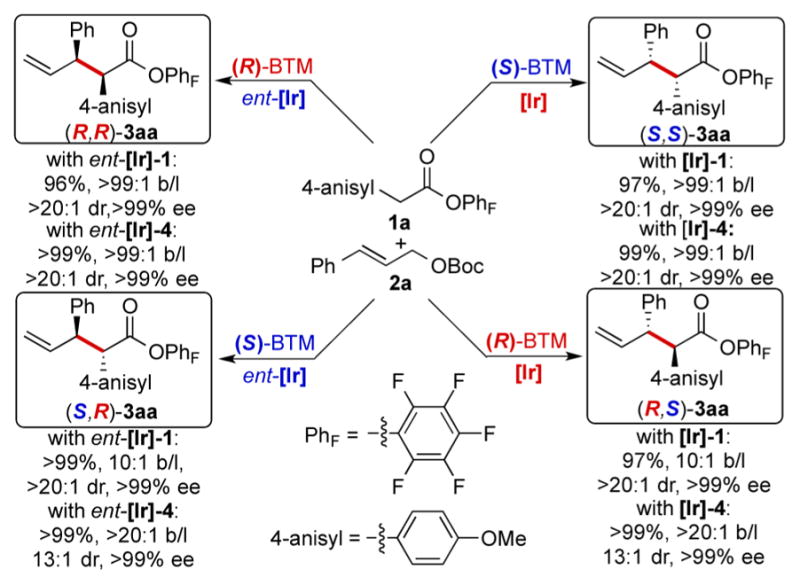
Synthesis of All Four Stereoisomers of 3aa
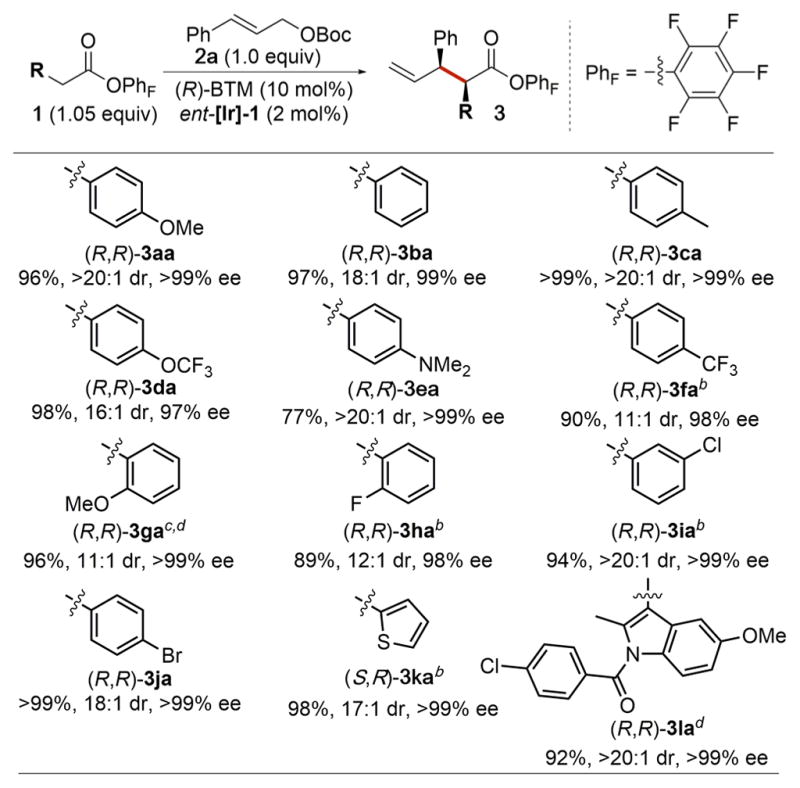
The scope of aryl acetic acid esters that underwent the stereodivergent allylic substitutions is summarized in Table 2. Various para-substituted phenyl acetic acid esters were suitable for this transformation. Electron-donating (3aa, 3da, 3ea), electron-neutral (3ba, 3ca) and electron-withdrawing (3fa) functional groups on the phenyl ring of the phenyl acetic acid ester were tolerated in this reaction, furnishing the corresponding products in high yields (≥77%) with high dr (≥11:1) and excellent ee (≥97%). The reaction with 4-(methylsulfonyl)-phenyl acetic acid ester (1m), a substrate bearing a readily enolizable position as a result of the strong electron-withdrawing effect of the sulfonyl group, formed the product 3ma in high yield (88%) but with modest dr (3.8:1). Further investigations showed that the low diastereoselectivity resulted from the competing reaction of 1m with 2a occurring without participation of BTM, not from racemization of the product (Table S4).
Table 2.
Scope of Esters for the Allylationa
|
Combined yields of the two isolated diastereomers are reported. The branched products were obtained exclusively.
20 mol % (R)-BTM was used.
The reaction time was extended to 9 h.
1.1 equiv of iPr2NEt was added.
Substitutions at the ortho (3ga, 3ha) or meta (3ia) position of the phenyl ring of the phenyl acetic acid ester had little effect on the allylation reaction; the corresponding products were all obtained in ≥89% yield with ≥11:1 dr and ≥98% ee. The allylation also occurred with heteroaryl acetic acid esters. For example, 1l, which is derived from the nonsteroidal anti-inflammatory drug indomethacin, was allylated in 92% yield with >20:1 dr and >99% ee. In the cases of 3ga and 3la, addition of 1.1 equiv of iPr2NEt was necessary to reach full conversion of the starting allylic carbonates within 9 h, presumably by accelerating the enolization of the acyl–BTM intermediate.
The scope of allylic carbonates that underwent the stereo-divergent allylic substitutions with aryl acetic acid esters is summarized in Table 3.17 Various substituents on the phenyl ring of the cinnamyl carbonate were tolerated, giving the corresponding products in ≥90% yield with ≥17:1 dr and ≥98% ee (3aa–ah). The allylic substitutions also occurred with allylic carbonates containing heteroaryl and alkenyl substituents. Allylic carbonates bearing a thiazole ring (2j) or a pyrimidine ring (2k) reacted to form the products 3aj and 3ak, respectively, with high diastereoselectivity and enantioselectivity (>20:1 dr, >99% ee). The reaction with tert-butyl sorbyl carbonate proceeded smoothly, furnishing 3al in 90% yield with 17:1 dr and >99% ee.
Table 3.
Scope of Allylic Carbonates for the Allylationa
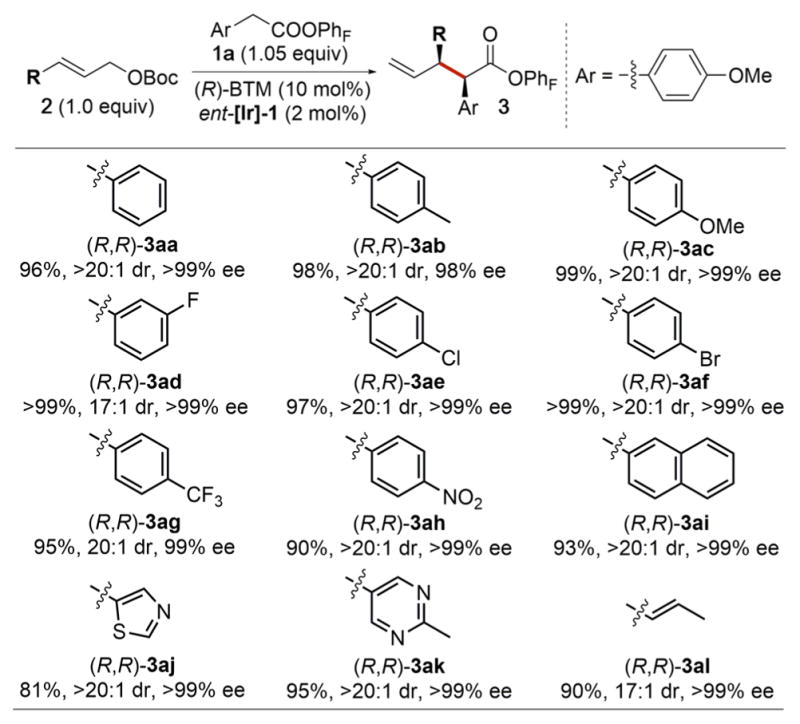
|
Combined yields of the two isolated diastereomers are reported. The branched products were obtained exclusively.
To demonstrate the stereodivergence of this allylation reaction further, the reactions in Table 4 were conducted. In these reactions, the same enantiomer of the iridium catalyst was used with the two enantiomers of the Lewis basic catalyst BTM. Both diastereomers of 3ca, 3ea, 3ja, 3ac, 3ad, and 3ak were isolated in high yield with high regio-, diastereo-, and enantioselectivity.
Table 4.
Examples of Stereodivergencea
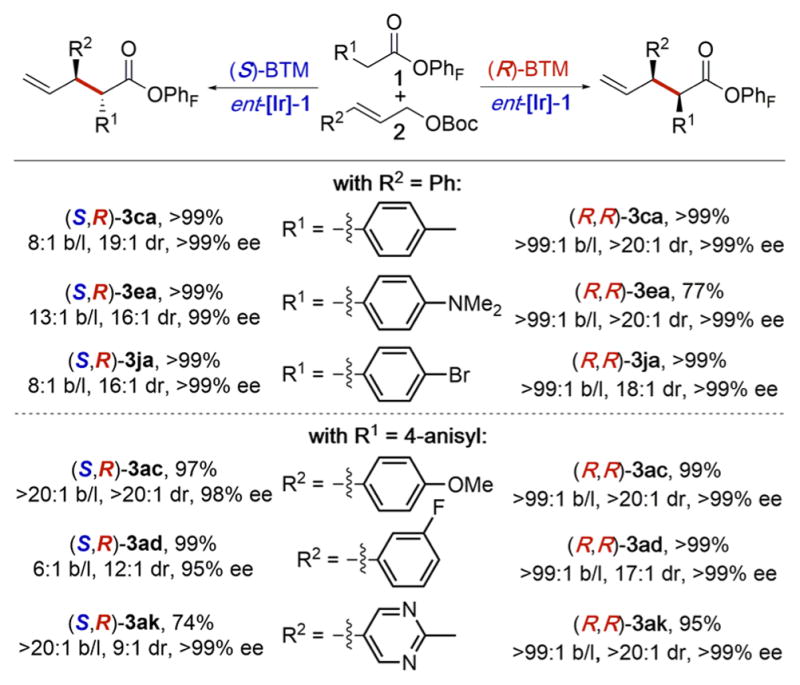
|
See the SI for experimental details.
The pentafluorophenyl ester products generated from this allylation are readily elaborated under mild conditions (Scheme 3). Addition of benzylamine and iPr2NEt to the reaction mixture at the end of the allylation reaction generated amide 4aa in a one-pot manner (98% yield with >20:1 dr and 98% ee). Similarly, one-pot syntheses of methyl ester 5aa and carboxylic acid 6aa were realized with 4-dimethylaminopyridine (DMAP) as the catalyst for methanolysis and hydrolysis of 3aa. Finally, primary alcohol 7aa was obtained through reduction of 3aa in 98% yield with >20:1 dr and >99% ee.
Scheme 3. Derivatizations of (R,R)-3aaa.
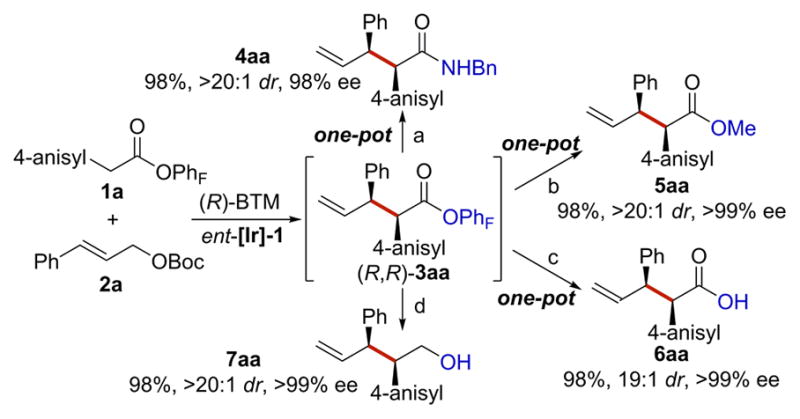
aConditions: (a) iPr2NEt (1.5 equiv), BnNH2 (1.3 equiv), r.t., 12 h; (b) DMAP (0.2 equiv), Et3N (5.0 equiv), MeOH/THF, 65 °C, 12 h; (c) DMAP (0.2 equiv), Et3N (5.0 equiv), H2O/THF, 65 °C, 12 h; (d) LiAlH4 (1.5 equiv), THF, r.t., 12 h.
In summary, we have shown that the combination of a metallacyclic iridium complex and a chiral Lewis base catalyzes stereodivergent allylic substitutions with aryl acetic acid esters. All four possible stereoisomers of the resulting products containing two contiguous stereocenters are accessible by simple permutations of the enantiomers of the two catalysts. The activated pentafluorophenyl esters as nucleophile precursors in this reaction allowed regeneration of the Lewis base catalyst through a “rebound” strategy while simultaneously allowing the resulting allylation products to be converted readily to enantioenriched amides, unactivated esters, and carboxylic acids. Studies to expand the scope with respect to general aliphatic carboxylic acid derivatives are underway.
Supplementary Material
Acknowledgments
Dr. Antonio DiPasquale is acknowledged for X-ray crystallography and support from NIH Shared Instrumentation Grant S10-RR027172. We thank the NIH (GM-55382 to J.F.H. and F32GM106641 to J.J.B.) for support.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b11692.
Experimental procedures and spectra (PDF)
Crystallographic data for ent-3aj (CIF)
References
- 1.For selected reviews, see: Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804.Helmchen G, Dahnz A, Dubon P, Schelwies M, Weihofen R. Chem Commun. 2007:675. doi: 10.1039/b614169b.Lu Z, Ma S. Angew Chem, Int Ed. 2008;47:258. doi: 10.1002/anie.200605113.Hartwig JF, Stanley LM. Acc Chem Res. 2010;43:1461. doi: 10.1021/ar100047x.Tosatti P, Nelson A, Marsden SP. Org Biomol Chem. 2012;10:3147. doi: 10.1039/c2ob07086c.Oliver S, Evans PA. Synthesis. 2013;45:3179.Liu Y, Han SJ, Liu WB, Stoltz BM. Acc Chem Res. 2015;48:740. doi: 10.1021/ar5004658.
- 2.For selected recent publications, see: Trost BM, Miller JR, Hoffman CM. J Am Chem Soc. 2011;133:8165. doi: 10.1021/ja2029602.Chen W, Hartwig JF. J Am Chem Soc. 2013;135:2068. doi: 10.1021/ja311363a.Chen W, Hartwig JF. J Am Chem Soc. 2014;136:377. doi: 10.1021/ja410650e.Liu WB, Reeves CM, Stoltz BM. J Am Chem Soc. 2013;135:17298. doi: 10.1021/ja4097829.Liu WB, Reeves CM, Virgil SC, Stoltz BM. J Am Chem Soc. 2013;135:10626. doi: 10.1021/ja4052075.Chen W, Chen M, Hartwig JF. J Am Chem Soc. 2014;136:15825. doi: 10.1021/ja506500u.Grassi D, Alexakis A. Chem Sci. 2014;5:3803.Zhang X, Liu WB, Tu HF, You SL. Chem Sci. 2015;6:4525. doi: 10.1039/c5sc01772f.Zhuo CX, Cheng Q, Liu WB, Zhao Q, You SL. Angew Chem, Int Ed. 2015;54:8475. doi: 10.1002/anie.201502259.Jiang X, Chen W, Hartwig JF. Angew Chem, Int Ed. 2016;55:5819. doi: 10.1002/anie.201600235.Liu WB, Okamoto N, Alexy EJ, Hong AY, Tran K, Stoltz BM. J Am Chem Soc. 2016;138:5234. doi: 10.1021/jacs.6b02153.Wu QF, Zheng C, Zhuo CX, You SL. Chem Sci. 2016;7:4453. doi: 10.1039/c6sc00176a.
- 3.(a) Krautwald S, Sarlah D, Schafroth MA, Carreira EM. Science. 2013;340:1065. doi: 10.1126/science.1237068. [DOI] [PubMed] [Google Scholar]; (b) Krautwald S, Schafroth MA, Sarlah D, Carreira EM. J Am Chem Soc. 2014;136:3020. doi: 10.1021/ja5003247. [DOI] [PubMed] [Google Scholar]; (c) Næsborg L, Halskov KS, Tur F, Mønsted SMN, Jørgensen KA. Angew Chem, Int Ed. 2015;54:10193. doi: 10.1002/anie.201504749. [DOI] [PubMed] [Google Scholar]; (d) Sandmeier T, Krautwald S, Zipfel HF, Carreira EM. Angew Chem, Int Ed. 2015;54:14363. doi: 10.1002/anie.201506933. [DOI] [PubMed] [Google Scholar]
- 4.Huo X, He R, Zhang X, Zhang W. J Am Chem Soc. 2016;138:11093. doi: 10.1021/jacs.6b06156. [DOI] [PubMed] [Google Scholar]
- 5.For selected publications that give access to all stereoisomers of the products, see: Huang Y, Walji AM, Larsen CH, MacMillan DWC. J Am Chem Soc. 2005;127:15051. doi: 10.1021/ja055545d.Lee EC, Hodous BL, Bergin E, Shih C, Fu GC. J Am Chem Soc. 2005;127:11586. doi: 10.1021/ja052058p.Yan XX, Peng Q, Li Q, Zhang K, Yao J, Hou XL, Wu YD. J Am Chem Soc. 2008;130:14362. doi: 10.1021/ja804527r.Nojiri A, Kumagai N, Shibasaki M. J Am Chem Soc. 2009;131:3779. doi: 10.1021/ja900084k.Simmons B, Walji AM, MacMillan DWC. Angew Chem, Int Ed. 2009;48:4349. doi: 10.1002/anie.200900220.McInturff EL, Yamaguchi E, Krische MJ. J Am Chem Soc. 2012;134:20628. doi: 10.1021/ja311208a.Shi SL, Wong ZL, Buchwald SL. Nature. 2016;532:353. doi: 10.1038/nature17191.
- 6.(a) Madrahimov ST, Markovic D, Hartwig JF. J Am Chem Soc. 2009;131:7228. doi: 10.1021/ja902609g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Madrahimov ST, Hartwig JF. J Am Chem Soc. 2012;134:8136. doi: 10.1021/ja212217j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen M, Hartwig JF. J Am Chem Soc. 2015;137:13972. doi: 10.1021/jacs.5b09980. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Madrahimov ST, Li Q, Sharma A, Hartwig JF. J Am Chem Soc. 2015;137:14968. doi: 10.1021/jacs.5b08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ohmura T, Hartwig JF. J Am Chem Soc. 2002;124:15164. doi: 10.1021/ja028614m. [DOI] [PubMed] [Google Scholar]; (b) Kiener CA, Shu C, Incarvito C, Hartwig JF. J Am Chem Soc. 2003;125:14272. doi: 10.1021/ja038319h. [DOI] [PubMed] [Google Scholar]
- 8.(a) Gaunt MJ, Johansson CCC. Chem Rev. 2007;107:5596. doi: 10.1021/cr0683764. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Beutner GL. Angew Chem, Int Ed. 2008;47:1560. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]; (c) Paull DH, Weatherwax A, Lectka T. Tetrahedron. 2009;65:6771. doi: 10.1016/j.tet.2009.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Morrill LC, Smith AD. Chem Soc Rev. 2014;43:6214. doi: 10.1039/c4cs00042k. [DOI] [PubMed] [Google Scholar]
- 9.For selected reviews of synergistic catalysis, see: Zhong C, Shi X. Eur J Org Chem. 2010;2010:2999.Allen AE, MacMillan DWC. Chem Sci. 2012;3:633. doi: 10.1039/C2SC00907B.Du Z, Shao Z. Chem Soc Rev. 2013;42:1337. doi: 10.1039/c2cs35258c.
- 10.For selected publications, see: Bekele T, Shah MH, Wolfer J, Abraham CJ, Weatherwax A, Lectka T. J Am Chem Soc. 2006;128:1810. doi: 10.1021/ja058077g.Liu G, Shirley ME, Van KN, McFarlin RL, Romo D. Nat Chem. 2013;5:1049. doi: 10.1038/nchem.1788.Fugard AJ, Thompson BK, Slawin AMZ, Taylor JE, Smith AD. Org Lett. 2015;17:5824. doi: 10.1021/acs.orglett.5b02997.
- 11.Lee SY, Neufeind S, Fu GC. J Am Chem Soc. 2014;136:8899. doi: 10.1021/ja5044209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawanaka Y, Phillips EM, Scheidt KA. J Am Chem Soc. 2009;131:18028. doi: 10.1021/ja9094044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.West TH, Daniels DSB, Slawin AMZ, Smith AD. J Am Chem Soc. 2014;136:4476. doi: 10.1021/ja500758n. [DOI] [PubMed] [Google Scholar]
- 14.Schwarz KJ, Amos JL, Klein JC, Do DT, Snaddon TN. J Am Chem Soc. 2016;138:5214. doi: 10.1021/jacs.6b01694. [DOI] [PubMed] [Google Scholar]
- 15.Birman VB, Li X. Org Lett. 2006;8:1351. doi: 10.1021/ol060065s. [DOI] [PubMed] [Google Scholar]
- 16.(a) Liu P, Yang X, Birman VB, Houk KN. Org Lett. 2012;14:3288. doi: 10.1021/ol301243f. [DOI] [PubMed] [Google Scholar]; (b) Robinson ERT, Walden DM, Fallan C, Greenhalgh MD, Cheong PHY, Smith AD. Chem Sci. 2016;7:6919. doi: 10.1039/c6sc00940a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.In the isolated products, a small amount of (R,R)-3aa impurity derived from the catalyst ent-[Ir]-1 was observed by GC.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.