

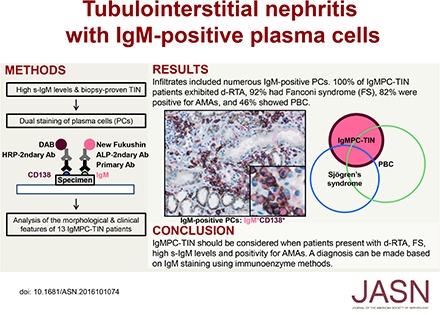
Abstract
Infiltration by IgG-positive plasma cells is a common finding in tubulointerstitial nephritis. Indeed, it has been thought that CD138-positive mature plasma cells secrete mainly IgG, and the occurrence of tubulointerstitial nephritis with CD138-positive plasma cells secreting IgM has rarely been reported. Routine immunofluorescence of fresh frozen sections is considered the gold standard for detection of immune deposits. However, the immunoenzyme method with formalin-fixed, paraffin-embedded sections is superior for detecting IgM- or IgG-positive cells within the renal interstitium, thus histologic variants may often go undetected. We recently discovered a case of tubulointerstitial nephritis showing IgM-positive plasma cell accumulation within the interstitium. To further explore the morphologic and clinical features of such cases, we performed a nationwide search for patients with biopsy-proven tubulointerstitial nephritis and high serum IgM levels. We identified 13 patients with tubulointerstitial nephritis and IgM-positive plasma cell infiltration confirmed with the immunoenzyme method. The clinical findings for these patients included a high prevalence of distal renal tubular acidosis (100%), Fanconi syndrome (92%), and anti-mitochondrial antibodies (82%). The pathologic findings were interstitial nephritis with diffusely distributed CD3-positive T lymphocytes and colocalized IgM-positive plasma cells, as well as tubulitis with CD3-positive T lymphocytes in the proximal tubules and collecting ducts. Additionally, levels of H+-ATPase, H+, K+-ATPase, and the HCO3−-Cl− anion exchanger were markedly decreased in the collecting ducts. We propose to designate this group of cases, which have a common histologic and clinical form, as IgM-positive plasma cell–tubulointerstitial nephritis.
Keywords: IgM, plasma cell, tubulointerstitial nephritis (TIN), Fanconi syndrome, renal tubular acidosis (RTA)
Interstitial nephritis presents with histologic interstitial abnormalities that reflect infiltration by various inflammatory cells, including lymphocytes, plasma cells, and macrophages,1,2 and is often accompanied by tubulitis, resulting in a condition called tubulointerstitial nephritis (TIN). The causes of TIN vary and include drug reactions and autoimmune diseases such as Sjögren syndrome or IgG4-related disease.3 Infiltration of IgG-positive plasma cells is a common finding in TIN,4 whereas infiltration of IgM-positive plasmacytoid large lymphocytes is rare, as we previously reported.5 CD138 is a specific marker of plasma cells6 and has recently been used in the diagnosis of IgG4-related disease.7 It had been thought that CD138-positive mature plasma cells secrete IgG, but not IgM. In one recent study, however, it was observed that IgM-positive plasmablasts developed into IgM-positive mature plasma cells.8
Primary biliary cirrhosis/cholangitis (PBC) is a chronic autoimmune cholestatic liver disease characterized by nonsuppurative destruction of interlobular bile ducts. PBC is accompanied by the presence of serum anti-mitochondrial antibodies (AMAs) and elevated serum concentrations of IgM.9 Distal renal tubular acidosis (d-RTA) is the main feature of the functional renal complications in patients with PBC,10 and is found in up to 33% of these patients. In addition, concomitant Fanconi syndrome has also been reported,11–15 and in most past reports the renal involvement was histologically diagnosed as TIN.5,11,13–21
In this paper, we report the novel clinical and morphologic features of 13 patients diagnosed with TIN and IgM-positive plasma cell infiltration, which we term “IgM-positive plasma cell–tubulointerstitial nephritis” (IgMPC-TIN).
Results
Clinical Features
We examined the clinical data from a total of 21,786 patients from whom renal biopsy samples were collected and identified 13 patients whose renal biopsies revealed IgMPC-TIN (13 of 21786, 0.06%). The clinical details for these 13 patients at the time of renal biopsy are summarized in Table 1. The patients were predominantly women (12 of 13, 92%) with an average age of 51±9 years (range 38–66) at the time of renal biopsy. They all had elevated serum IgM levels (889±435 mg/dl) (range 369–2045 mg/dl) and a normal serum and urinary protein electrophoresis pattern, and immunofixation revealed they were negative for paraprotein. These patients were all diagnosed with d-RTA, and 92% were also diagnosed with Fanconi syndrome and found to be positive for glycosuria (although glycosuria was not detected in patient 6 at the time of biopsy, glycosuria appeared over the clinical course). In addition, 82% of patients were found to be positive for either serum AMA or anti-mitochondrial M2 antibodies. However, only six patients (46%) satisfied the PBC criteria. All patients had mild-to-moderate renal insufficiency (eGFR: 37.1±12.1 ml/min) (range 15.2–60.3 ml/min). Increased serum alkaline phosphatase and γ-glutamyltransferase levels were found in eight and six patients, respectively. All patients had some level of proteinuria (1.8±1.9 g/g creatinine, range 0.22–6.60 g/g creatinine). Elevated levels of urinary β2-microglobulin were a common abnormal finding. There were four patients (31%) with Sjögren syndrome and two patients (15%) with hypocomplementemia.
Table 1.
Clinical and immunohistologic data of IgMPC-TIN at the time of renal biopsy
| Variable | Patient No. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
| Age/sex | 38/F | 63/F | 40/F | 38/F | 53/F | 60/M | 55/F | 48/F | 55/F | 66/F | 52/F | 44/F | 56/F |
| s-IgM, mg/dl (33–269) | 1040 | 860 | 838 | 1152 | 708 | 682 | 405 | 369 | 844 | 663 | 665 | 1284 | 2045 |
| s-IgG, mg/dl (870–1700) | 1490 | 2610 | 1242 | 1409 | 897 | 1387 | 1041 | 1194 | 994 | 1450 | 1232 | 1549 | 981 |
| s-IgA, mg/dl (110–410) | 158 | 179 | 107 | 270 | 168 | 168 | 151 | 193 | 599 | 262 | 262 | 264 | 126 |
| s-IEP | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal | ND | Normal | Normal | Normal | Normal |
| u-IEP | Normal | Normal | Normal | Normal | ND | Normal | Normal | Normal | ND | Normal | Normal | Normal | Normal |
| RTA | Distal | Distal | Distal | Distal | Distal | Distal | Distal | ND | Distal | Distal | Distal | Distal | Distal |
| Fanconi syndrome | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes |
| Glycosuria | Yes | Yes | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| AMA, (fold) (<20) | 160 | 320 | <20 | 160 | 160 | <20 | 20 | ND | ND | 80 | 320 | ND | <20 |
| Anti-M2 antibody, (index) (<7.0) | 30.9 | 24.4 | <7.0 | 131 | 76.8 | ND | 112 | ND | ND | 178 | 157 | 562 | 10.3 |
| PBC | Yes | Yes | No | No | No | No | No | No | No | Yes | Yes | Yes | Yes |
| (Biopsy-proven) | (Biopsy-proven) | (Biopsy-proven) | (Biopsy-proven) | (Biopsy-proven) | |||||||||
| s-Cr, mg/dl (M: 0.65–1.07, F: 0.46–0.79) | 0.85 | 1.14 | 0.92 | 1.84 | 1.13 | 1.88 | 1.14 | 1.09 | 1.00 | 1.50 | 1.50 | 1.38 | 2.71 |
| eGFR, ml/min | 60.3 | 37.8 | 54.5 | 26.5 | 40.1 | 30.0 | 39.3 | 43.0 | 45.4 | 27.6 | 29.6 | 34.0 | 15.2 |
| ALP, IU/L (107–322) | 398 | 668 | 244 | 1227 | 309 | 259 | 323 | 139 | 505 | 290 | 812 | 1559 | 806 |
| γ-GT, IU/L (M: 13–64, F: 9–32) | 149 | 99 | 263 | 21 | 43 | 34 | 17 | 30 | 28 | 13 | 24 | 56 | 178 |
| U-P, g/gCr | 0.41 | 0.93 | 0.52 | 5.11 | 0.22 | 0.30 | 0.78 | 1.39 | 0.60 | 6.60 | 2.14 | 3.09 | 1.50 |
| U-β2m, μg/L (<200) | 16,744 | 78,200 | 22,103 | 42,700 | 8010 | 9702 | 72,311 | 2661 | 20,000 | 84,400 | >60,000 | 69,359 | 66,987 |
| Sjӧgren syndrome | Yes | Yes | No | No | Yes | Yes | No | No | No | No | No | No | No |
| Hypocomplementemia | Yes | No | No | No | No | No | No | No | No | No | No | Yes | No |
| IgM-positive PCs, cells per hpf | 123 | 20 | 94 | 113 | 41 | 31 | 16 | 52 | 57 | 30 | 67 | 47 | 55 |
| IgM-positive PCs/total PCs, % | 77.4 | 58.6 | 86.5 | 66.7 | 77.7 | 70.0 | 87.5 | 72.3 | 69.2 | 58.3 | 80.3 | 63.5 | 89.2 |
| IgM-positive PCs/IgM-positive cells, % | 60.6 | 67.8 | 89.9 | 90.0 | 84.7 | 66.7 | 44.6 | 90.9 | 83.8 | 69.5 | 78.7 | 77.6 | 86.4 |
F, female; M, male; s-IgM, serum immunoglobulin M; s-IgG, serum immunoglobulin G; s-IgA, serum immunoglobulin A; s-IEP, serum immunofixation electrophoresis; u-IEP, urinary immunofixation electrophoresis; RTA, renal tubular acidosis; ND, not determined; s-Cr, serum creatinine; ALP, alkaline phosphatase; γ-GT, γ-glutamyltransferase; U-P, urinary protein; gCr, g creatinine; U-β2m, urinary β2-microglobulin; PCs, plasma cells.
The clinical data at the time of follow-up are summarized in Table 2. Eleven patients (85%) were treated with an intermediate dose of glucocorticoid (prednisolone 29±5.3 mg, range 20–40 mg). Nearly all patients receiving a glucocorticoid responded to the therapy, and their renal function was preserved with a significant decrease in urinary protein (P<0.01) and serum IgM levels (P<0.001) at the last follow-up (0.2–15.0 years) (Supplemental Figure 1). However, eight out of 11 patients continued to exhibit renal insufficiency (eGFR<60 ml/min).
Table 2.
Clinical data of IgMPC-TIN at the time of follow-up
| Variable | Patient No. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
| Tx | PSL, CyA, UDCA | UDCA | PSL, UDCA | PSL, Citrates | PSL, Citrates | PSL | None | PSL | PSL | PSL, UDCA | PSL, NaHCO3 | PSL, UDCA | PSL, UDCA, NaHCO3 |
| PSL, mg | 30 | 0 | 30 | 20 | 30 | 40 | 0 | 30 | 30 | 30 | 30 | 20 | 30 |
| Follow-up period, yr | 7.1 | 5.4 | 1.2 | 0.3 | 0.4 | 1.6 | 0.9 | 2.8 | 2.2 | 15.0 | 6.2 | 0.8 | 0.2 |
| Follow-up s-Cr, mg/dl | 0.73 | 1.31 | 0.81 | 1.59 | 0.95 | 1.52 | 1.04 | 0.78 | 1.25 | 2.30 | 1.28 | 1.40 | 1.98 |
| Follow-up eGFR, ml/min | 67.8 | 31.8 | 62.2 | 30.4 | 48.5 | 37.9 | 32.7 | 61.2 | 35.2 | 16.3 | 34.1 | 33.5 | 21.4 |
| Follow-up s-IgM, mg/dl | 342 | 654 | 216 | 749 | 206 | 183 | 483 | 32 | ND | 350 | ND | 616 | 1064 |
| Follow-up ALP, IU/L | 177 | 447 | 309 | 569 | 165 | 210 | 353 | 68 | 784 | 285 | 242 | 457 | 306 |
| Follow-up γ-GT, IU/L | 17 | 35 | 68 | 37 | 28 | 34 | 22 | 22 | 36 | 17 | 36 | 35 | 136 |
| Follow-up U-P, g/gCr | 0.16 | 0.86 | 0.03 | 2.17 | 0.09 | 0.06 | 0.69 | 0.16 | ND | 1.30 | 0.65 | 0.54 | 1.23 |
| Follow-up U-β2m, μg/L | 3446 | 83,900 | 1606 | 109,600 | 816 | 840 | 60,392 | 1420 | 17,067 | 28,698 | 17,830 | 35,650 | 32,309 |
| Follow-up glycosuria | No | Yes | No | Yes | No | No | Yes | ND | Yes | Yes | Yes | Yes | Yes |
Tx, treatment; PSL, prednisolone; CyA, cyclosporine A; UDCA, ursodeoxycholic acid; s-Cr, serum creatinine; s-IgM, serum immunoglobulin M; ND, not determined; ALP, alkaline phosphatase; γ-GT, γ-glutamyltransferase; U-P, urinary protein; gCr, g creatinine; U-β2m, urinary β2-microglobulin
Morphologic Features
In all cases, the morphologic findings were similar. Light microscopy showed minor glomerular abnormalities or mild mesangial proliferation with extensive infiltration of lymphocytes and plasma cells into the interstitium (interstitial nephritis) and mild-to-moderate infiltration of lymphocytes into the tubules (tubulitis) (Figure 1). Although focal areas of tubular atrophy and mild interstitial fibrosis were also observed, characteristic signs of IgG4-related kidney disease, including storiform fibrosis, eosinophil infiltration, and obliterative phlebitis, were not observed (Figure 1). Vascular changes were nearly absent. In four patients, immunofluorescence microscopy revealed scanty glomerular staining for IgM along the capillary walls. In patient 8, mesangial IgA and C3c staining was observed, which means this patient was incidentally complicated by IgA nephropathy. No staining was observed in the tubular basement membrane and IgM-positive cells in the interstitium could not be clearly detected by conventional immunofluorescent staining. Immunohistochemical staining of formalin-fixed, paraffin-embedded renal biopsy specimens after antigen retrieval revealed the presence of both IgM- and IgG-positive cells within the interstitium (Figure 2, A, B, D, and E). The infiltrates included numerous diffusely distributed IgM-positive cells and CD138-positive plasma cells (Figure 2, A, C, D, and F), with a few IgG-positive cells (Figure 2, B and E) and with a small number of IgG4-positive cells (Supplemental Figure 2E). These plasma cells stained positively with both anti-λ and anti-κ light chain antibodies (Supplemental Figure 3), but no monoclonality was confirmed. The averaged number of infiltrating IgM-positive plasma cells per high-power field (hpf) of renal interstitium from patients with IgMPC-TIN was markedly higher than from patients with other forms of TIN chosen as controls for staining (15 TIN with Sjögren syndrome, 12 with IgG4-related kidney disease, three with TIN with ANCA-related vasculitis, three with granulomatous TIN, four with drug-related TIN, one with antibody-mediated rejection, two with chronic pyelonephritis, and four with unknown TIN) (see Concise Methods section for details) (Figure 3, Supplemental Figure 2I). Receiver operating characteristic (ROC) curve analysis revealed that optimal predictive cutoff number for infiltrating IgM-positive plasma cells was 13 per hpf, with an area under the ROC curve of 0.99 (95% confidence interval [95% CI], 0.979 to 1.007; P<0.001) (Supplemental Figure 4). The sensitivity and specificity were 100% (95% CI, 75.3% to 100%) and 93.2% (95% CI, 81.3% to 98.6%), respectively. Moreover, immunohistochemical dual staining showed that many IgM-positive cells were also CD138-positive. The IgM-positive plasma cell fraction among total CD138-positive cells was 73.5%±10.5% (range 58.3%–89.2%), which confirms that the IgM-positive cells were plasma cells (Figures 2, G and H, and 4, Table 1).
Figure 1.

Light microscopy from IgMPC-TIN patients showed interstitial nephritis and tubulitis. (A) Representative photomicrograph showing interstitial nephritis with an area of tubular atrophy and mild fibrosis (patient 1). However, storiform fibrosis, eosinophil infiltration, and obliterative phlebitis, which are characteristic findings of IgG4-related disease, were not observed (periodic acid–methenamine silver staining). Bar = 100 µm. (B) Higher magnification image showing infiltration by lymphocytes and plasma cells into the interstitium (periodic acid–Schiff staining). Bar = 20 µm. (C) Highest magnification image showing infiltration by lymphocytes and plasma cells (arrowheads) into the interstitium. Bar = 20 µm.
Figure 2.

Immunohistochemical staining revealed the presence of many IgM-positive plasma cells within the renal interstitium. Representative images of renal (A–H) and hepatologic (I) immunohistochemical staining of infiltrating cells in patients 3 (A–F) and 1 (G and H) with IgMPC-TIN and a patient with PBC (I). Light photomicrographs show IgM-positive cells (A and D), IgG-positive cells (B and E), and CD138-positive plasma cells (C and F) within the renal interstitium. The interstitial cellular infiltrates were composed of diffusely spread IgM-positive cells. Many IgM-positive cells infiltrating the renal interstitium (G and H) and portal tract (I) were also CD138-positive (red: IgM; brown: CD138). (A–C): Bar =100 µm; (D–G, and I): bar =50 µm; (H): bar =20 µm.
Figure 3.

The averaged number of infiltrating IgM-positive plasma cells per high-power field of renal interstitium from patients (n=13) with IgMPC-TIN was significantly higher than from patients (n=44) with other forms of TIN chosen as controls for staining. *P<0.001.
Figure 4.

The IgM-positive plasma cell fraction in the renal interstitium of patients with IgMPC-TIN (n=13) was significantly higher than in the portal tract of patients with PBC (n=28). The IgM-positive plasma cell fraction among CD138-positive cells was calculated as the percentage of dual-positive IgM-CD138 cells among the total CD138-positive cells. *P<0.001.
IgM-CD138 dual-positive cells (IgM-positive plasma cells) were also observed in the portal tract of the liver in 28 patients clinically diagnosed with PBC (the IgM-positive plasma cell fraction among CD138-positive cells was 42.6%±19.4%, range 8.3%–68.1%) (Figures 2I and 4). Furthermore, in two patients with IgMPC-TIN from whom both kidney and liver specimens were obtained (patients 12 and 13), IgM-positive plasma cells were confirmed in both the portal tract of the liver and the interstitium of the kidney (data not shown). The IgM-positive plasma cell fraction in the renal interstitium of patients with IgMPC-TIN was significantly higher than in the portal tract of patients with PBC (P<0.001) (Figure 4).
The dual staining of CD3 and aquaporin 2 (AQP2) in Figure 5 shows that infiltrating CD3-positive T lymphocytes were diffusely distributed in the proximal tubular epithelium (proximal tubulitis) and the AQP2-positive epithelium of the collecting ducts (collecting duct tubulitis). The proximal tubulitis was more pronounced than the collecting duct tubulitis. By contrast, CD20-positive B lymphocytes were focally distributed within the interstitium without tubulitis (data not shown). In nearly all patients, CD3-positive T lymphocytes and IgM-positive plasma cells predominantly infiltrated the same lesions. In addition, levels of H+-ATPase, H+, K+-ATPase, and HCO3−-Cl− anion exchanger (AE-1) in the cortical collecting ducts (CCDs) were significantly lower in IgMPC-TIN patients than in control patients with TIN without d-RTA (Figure 6).
Figure 5.

The dual staining of CD3 and AQP2 shows tubulitis in the proximal tubules and collecting ducts. Representative light photomicrographs (patient 13) show that infiltrating CD3-positive T lymphocytes were diffusely distributed in the renal interstitium (A and B), among proximal tubular epithelial cells (proximal tubulitis) (arrowheads) (C), and among collecting duct epithelial cells (collecting duct tubulitis) (arrow) (D) (red: AQP2, brown: CD3). (A): Bar = 100 μm; (B): bar = 50 μm; (C and D): bar = 20 μm.
Figure 6.

The levels of proton pumps and AE-1 in CCDs were significantly lower in patients with IgMPC-TIN than in control patients with TIN without d-RTA. The photomicrographs show the greatly diminished levels or absence of H+-ATPase (A), H+, K+-ATPase (B), and AE-1 (C) in CCDs from patient 13 with IgMPC-TIN, and the mildly decreased levels (arrowheads) of H+-ATPase (D), H+, K+-ATPase (E), and AE-1 (F) in CCDs from a patient with TIN without d-RTA. Bar = 20 μm. In quantitative image analysis of renal immunohistochemical staining, the levels of H+-ATPase (G), H+, K+-ATPase (H), and AE-1 (I) in CCDs from patients with IgMPC-TIN (n=13) were significantly lower than those in CCDs from control TIN patients (n=10) without d-RTA. *P<0.05; **P<0.01; ***P<0.001.
Discussion
In this study, we proposed a novel histologic entity exhibiting TIN with IgM-positive plasma cell infiltration (IgMPC-TIN) and reported its clinical features, included d-RTA, Fanconi syndrome, high serum IgM levels, and AMA positivity. This unique histologic entity may have been overlooked until now due to the use of conventional immunofluorescent staining with biopsy specimens from patients with TIN. The immunoenzyme method is essential for histologic diagnosis of IgMPC-TIN. Furthermore, we also determined that IgM-CD138 dual-positive plasma cells infiltrated the portal tract of the liver in patients with PBC as well as the renal interstitium in patients with IgMPC-TIN.
The clinical courses of these IgMPC-TIN patients enabled us to make three important observations, which will be further discussed below: (1) IgMPC-TIN is coincident with high serum IgM levels and AMA positivity, d-RTA, and Fanconi syndrome; (2) TIN presents with IgM-positive plasma cells; and (3) intermediate-dose glucocorticoid therapy preserves kidney function in these patients for prolonged periods.
AMA and/or anti-mitochondrial M2 antibodies are detected in >95% of patients with PBC,22 whereas their prevalence in the general population is between 0.16% and 1%.23 The AMA-positive population is thus much larger than the PBC population, which has a prevalence of 0.02%–0.04%, including asymptomatic PBC.24,25 The occurrence of AMA and/or anti-mitochondrial M2 antibodies may be prerequisite for a diagnosis of IgMPC-TIN, but this does not mean IgMPC-TIN is involved in PBC. Of our 13 patients, less than half the patients (46%) have been diagnosed with PBC. In addition, the IgM-positive plasma cell fraction among the total CD138-postive cells is much higher in IgMPC-TIN than in PBC. We therefore propose that IgMPC-TIN is not the renal involvement of PBC, although they have features in common like the presence of IgM-positive plasma cells.
Although d-RTA is often accompanied by Sjögren syndrome,26 only 31% of our patients were diagnosed with Sjögren syndrome. This implies that the d-RTA seen with IgMPC-TIN may be caused by factors other than Sjögren syndrome. Previous reports suggest that d-RTA with Sjögren syndrome likely reflects the complete absence of H+-ATPase27 and/or AE-128 from the CCDs. In all of our patients, we immunohistochemically confirmed diminished levels of H+-ATPase, H+, K+-ATPase, and AE-1 in the CCDs, although the use of a single method of immunohistologic staining for the evaluation is a limitation. In patients with PBC and Sjögren syndrome, dysfunction of anion exchanger 2 (AE-2)29 and Na+/H+ exchanger 1 (NHE1)30 in bile duct epithelial cells and dysfunction of the anion exchanger in the salivary gland31 have been reported. In our series, AE-1 levels in CCD epithelial cells may be decreased due to the presence of unknown humoral factors (e.g., autoantibodies) commonly observed in autoimmune conditions such as PBC and Sjögren syndrome.32 Similarly, CCD tubulitis with CD3-positive T lymphocytes may also cause pump failure. However, the precise mechanism underlying d-RTA in patients with IgMPC-TIN is unknown.
Glucosuria is a potential symptom in Fanconi syndrome due to proximal tubular dysfunction. This proximal tubular dysfunction may be caused by proximal tubulitis with CD3-positive T lymphocytes, which is the same mechanism that underlies the aforementioned CCD dysfunction. Because the bicarbonate-loading test was not performed in all of our patients, we can only speculate about the potential occurrence of concomitant proximal-RTA.
The possibility that AMAs lead to Fanconi syndrome/TIN in patients with PBC due to mitochondrial cytopathy was previously discussed.11 However, additional in vitro studies will be required to determine whether AMAs can be a direct cause of mitochondrial cytopathy, because AMAs likely have no pathogenic role in patients with PBC.33 At present, we are assuming the relations among IgMPC-TIN, PBC, and Sjögren syndrome are as shown in Figure 7.
Figure 7.

Relations among IgMPC-TIN (red) and overlapping diseases. Only 46% of patients with IgMPC-TIN satisfied the criteria for PBC, whereas 31% satisfied the criteria for Sjӧgren syndrome.
TIN presented with IgM-positive plasma cells. Infiltration of IgG-positive plasma cells is a common finding in TIN, including IgG4-related kidney disease.4 By contrast, infiltration of IgM-positive plasmacytoid lymphocytes is very rare.5 Although CD138 is a better and more specific marker of plasma cells6 than CD38, plasmablasts, immature plasma cells, and mature plasma cells all stained positively for CD138.34 Generally, antigen-activated B cells with T cell help undergo affinity maturation within germinal centers and persist as long-lived IgG plasma cells in the bone marrow. Thus, CD138-positive mature plasma cells secrete IgG, but not IgM. However, it was recently reported that short-lived IgM-positive plasmablasts develop into long-lived IgM-positive mature plasma cells.8 We observed that many IgM-positive cells also stained positively for CD138, which suggests that IgM-secreting cells in the renal interstitium are long-lived mature plasma cells or short-lived plasmablasts. Long-lived IgM-positive plasma cells were not antigen-selected, and IgM secreted from these cells could potentially provide protective host immunity against lethal conditions.8 On the other hand, IgM-positive plasmablasts reportedly exert inhibitory effects (regulatory B cell) via IL-10 production,35 and infiltrating IgM-positive cells may act to protect against interstitial inflammation. Precisely distinguishing between IgM-positive plamablasts and mature plasma cells is difficult due to the commonality of these cell markers (B lymphocyte–induced maturation protein-1: Blimp-1; interferon-regulatory factor 4: IFR4; and X-box–binding protein 1: XBP1).34
In patients with PBC, immunohistochemical examination has clarified that the cells infiltrating portal tracts are IgM-positive.36–38 The environmental niche of long-lived CD38-positive plasma cells and their effect on inflammation may have important implications for PBC.39 In our study, we confirmed that cells infiltrating the portal tract in patients with PBC are IgM-positive and stain positively for CD138, which is also the case for cells infiltrating the renal interstitium in patients with IgMPC-TIN. Although the precise mechanisms underlying the increased numbers of IgM-positive cells in the portal tract are unclear, previous reports indicate that T lymphocytes from patients with PBC have an enhanced ability to produce lymphokines that promote the proliferation and differentiation of B lymphocytes in vitro40 and in vivo.37 We suggest that a common pathway to autoimmune inflammation is also present in the kidney, leading to the proliferation of IgM-positive plasma cells in the renal interstitium. Given that the cells infiltrating the tubular epithelium were mainly CD3-postive T lymphocytes, we suppose autoreactive T lymphocytes, not IgM-positive plasma cells, most likely caused the proximal and CCD tubulitis.
Finally, intermediate dose of glucocorticoid therapy preserved kidney function for long periods in these patients. The use of glucocorticoid for the general treatment of patients with TIN has been controversial.41 In nearly all of our patients, glucocorticoid improved the laboratory findings and relieved the clinical symptoms (Supplemental Figure 1, Table 2). Unfortunately, however, the patients’ status remained at what is considered CKD. A prospective study gathering additional similar patients will be needed to confirm the ideal duration and dose of glucocorticoid for treatment of patients with IgMPC-TIN.
In conclusion, we described the clinical and morphologic features of 13 patients diagnosed with IgMPC-TIN. This novel histologic variant of TIN should be considered when patients present with d-RTA, Fanconi syndrome, high serum IgM levels, and positivity for AMA. IgMPC-TIN should ultimately be diagnosed after IgM staining using the immunoenzyme method, not immunofluorescence.
Concise Methods
Patients with IgMPC-TIN
Patients exhibiting both increased serum IgM levels (>270 mg/dl) and histologic TIN were identified from among 21,786 patients using samples from renal biopsies performed at our university and collaborating hospitals and universities. Thirteen patients histologically confirmed to have dominant IgM-positive plasma cell infiltration of the renal interstitium (IgMPC-TIN) became the subjects for analysis. In addition, liver biopsy specimens from two patients with IgMPC-TIN were immunohistochemically characterized and compared with those from 28 patients with definite PBC (disease control). Clinical and laboratory data were obtained from each patient at the time of renal biopsy. Follow-up data were obtained from all patients as much as possible. d-RTA was defined as normal anion gap metabolic acidosis with urinary pH persistently >5.5.42 Proximal-RTA could not be diagnosed due to the absence of bicarbonate-loading tests. Fanconi syndrome was defined by the coexistence of hypokalemia, hypophosphatemia, normoglycemic glycosuria, and pan-aminoaciduria.11 Sjögren syndrome was defined on the basis of the diagnostic criteria.43 PBC was diagnosed on the basis of the presence of at least three of the following criteria: alkaline phosphatase or γ-glutamyltransferase levels greater than the upper limit of normal; positive AMAs at a titer of 1:20; increased IgM levels; absence of biliary obstruction on ultrasonography, computed tomography, or cholangiography; or compatible liver biopsy.9,11 All data were collected in accordance with protocols approved by each institution’s internal review board.
Control TIN Specimens
A database search of our university and collaborating hospitals’ renal biopsies was performed for all patients between 1993 and 2017 and with a final diagnosis of TIN, including some biopsies with associated glomerular disease. The cause of TIN in each patient was determined on the basis of laboratory and clinical data. Ultimately, 44 patients (15 TIN with Sjögren syndrome, 12 with IgG4-related kidney disease, three with TIN with ANCA-related vasculitis, three with granulomatous TIN, four with drug related TIN, one with antibody-mediated rejection, two with chronic pyelonephritis, and four with unknown TIN) were selected, and their renal specimens were stained for IgM and CD138 and analyzed as controls using the same methods used for IgMPC-TIN. In addition, among the 44 patients, renal specimens from ten TIN patients without d-RTA were stained for proton pumps and anion exchangers and were analyzed as TIN controls without d-RTA.
Light Microscopy
Kidney and liver tissue samples were fixed in 10% formalin or Bouin, embedded in paraffin, and cut into 2-μm-thick sections. Sections were stained with periodic acid–Schiff, hematoxylin-eosin, or periodic acid–methenamine silver.
Immunohistochemistry
Rabbit anti-human IgG polyclonal antibody (Dako, Glostrup, Denmark), rabbit anti-human IgM polyclonal antibody (Dako), mouse anti-human CD138 monoclonal antibody (Dako), anti-human IgG4 monoclonal antibody (Zymed Laboratories, San Francisco, CA), anti-human κ monoclonal antibody (Nichrei, Tokyo, Japan), and anti-human λ monoclonal antibody (Nichrei) for staining plasma cells; rabbit anti-human CD3 monoclonal antibody (Nichrei) for staining T lymphocytes; mouse anti-human CD20 monoclonal antibody (Dako) for staining B lymphocytes; rabbit anti-human AQP2 polyclonal antibody (Atlas antibodies, Stockholm, Sweden) for staining CCD epithelial cells; rabbit anti-human ATP6V1E1 polyclonal antibody (Sigma-Aldrich, St. Louis, MO) for staining H+-ATPase in CCDs; mouse anti-human band 3 monoclonal antibody (Atlas antibodies) for staining AE-1 in CCDs; and rabbit anti-human ATP12A polyclonal antibody (Atlas antibodies) for staining H+, K+-ATPase in CCDs were used as primary antibodies. Formalin-fixed, paraffin embedded sections were dewaxed, rehydrated, and subjected to microwave heating for 15 minutes. For immunoglobulin staining, sections were digested with protease from Bacillus amyloliquefaciens (P1236) (Sigma-Aldrich) for an additional 30 minutes. Endogenous peroxidase was blocked for 20 minutes, after which sections were incubated with each primary antibody at 4°C overnight, then washed and incubated for 45 minutes with Envison labeled polymer (Dako, Glostrup, Denmark) or Histofine simple stain MAX-PO (M) (Nichrei, Tokyo, Japan). Diaminobenzidine was used as a chromogen. Counter staining was with hematoxylin.
For dual immunohistochemical staining, formalin-fixed, paraffin-embedded sections were dewaxed, rehydrated, subjected to microwave heating (and protease digestion for immunoglobulin), and incubated with primary antibody (IgM and AQP2) at 4°C overnight. The sections were then washed and incubated for 45 minutes with Histofine simple stain AP (R) (Nichrei, Tokyo, Japan). A Histofine new fukushin substrate kit (Nichrei, Tokyo, Japan) with Levamisole solution (Dako, Glostrup, Denmark) served as a chromogen. These sections were then again subjected to microwave heating and endogenous peroxidase was blocked, after which they were incubated with primary antibody (CD3, CD20, and CD138) at 4°C overnight, washed, and incubated for 45 minutes with Envison labeled polymer or Histofine simple stain MAX-PO (M) (Nichrei, Tokyo, Japan). Diaminobenzidine was used as a chromogen. Counter staining was with hematoxylin.
Histologic Analyses
For patients with IgMPC-TIN and with TIN, IgM-CD138 dual-positive cells were counted per hpf (original magnification, ×400) and graded on the basis of the average of the three most densely populated areas.44,45 The IgM-positive plasma cell fraction among total plasma cells was calculated as the percentage of dual-positive IgM-CD138 cells among the total CD138-positive cells in the renal interstitium of patients with IgMPC-TIN and in the portal tract of patients with PBC. The IgM-positive plasma cell fraction among total IgM-positive cells was calculated as the percentage of dual-positive IgM-CD138 cells among the total IgM-positive cells in the renal interstitium of patients with IgMPC-TIN. Proton pump and anion exchanger levels in the CCDs were quantified on the basis of immunohistochemical staining using cellSens Dimension imaging software (Olympus, Tokyo, Japan).
Statistical Analyses
Statistical calculations were made using SigmaPlot 13 (Systat Software, Inc., San Jose, CA). To compare pre- and post-treatment values, we used paired t test for normally distributed data and Wilcoxon signed rank test for skewed distributions. In addition, to compare the averaged numbers of infiltrating IgM-positive plasma cells in IgMPC-TIN and control TIN, and IgM-positive plasma cell fractions in IgMPC-TIN and PBC, we used the Mann–Whitney rank sum test. To compare the averaged numbers of infiltrating IgM-positive plasma cells in IgMPC-TIN and IgG4-related kidney disease and TIN with Sjögren syndrome and other control TIN, we used the Kruskal–Wallis on ranks test and Dunn’s method for multiple comparisons. To compare proton pump and anion exchanger levels in IgMPC-TIN and control TIN without RTA, we used the t test for normally distributed data and the Mann–Whitney rank sum test for skewed distributions. A ROC curve was calculated to derive the most suitable cutoff numbers of infiltrating IgM-positive plasma cells per hpf to differentiate between IgMPC-TIN and non–IgMPC-TIN using Youden index.46 Values of P<0.05 were considered significant.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Nobuo Takimoto, Hiromi Takahashi, and Hideki Maegawa for their valuable technical assistance, and Itaru Yamaguchi for his helpful discussion.
This work was supported by JSPS KAKENHI Grant Numbers JP15H04836, JP26460645, JP17K09692 from the Japan Society for the Promotion of Science, and by a Grant-in-Aid for Progressive Renal Disease Research from the Ministry of Health Labor and Welfare of Japan.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016101074/-/DCSupplemental.
References
- 1.Churg J, Cotran R, Sinniah R, Sakaguchi H, Sobin L: Renal Disease: Classification and Atlas of Tubulo-Interstitial Diseases, Tokyo, Igaku-Shoin, 1985 [Google Scholar]
- 2.Colvin R, Fang L: Interstitial nephritis. In: Renal Pathology with Clinical and Functional Correlations, edited by Tisher C, Brenner B, Philadelphia, JB Lippincott, 1994, pp 723–768 [Google Scholar]
- 3.Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, Kawano M, Yamamoto M, Takahashi H, Matsui S, Nakada S, Origuchi T, Hirabayashi A, Homma N, Tsubata Y, Takata T, Wada Y, Saito A, Fukase S, Ishioka K, Miyazaki K, Masaki Y, Umehara H, Sugai S, Narita I: Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int 78: 1016–1023, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Saeki T, Kawano M: IgG4-related kidney disease. Kidney Int 85: 251–257, 2014 [DOI] [PubMed] [Google Scholar]
- 5.Takahashi N, Kimura H, Kawajiri Y, Mikami D, Yamamoto C, Kasuno K, Imai N, Kuroda T, Nishi S, Yamamoto M, Yoshida H: Tubulointerstitial nephritis with IgM-positive plasmacytoid large lymphocyte infiltration in a patient with primary biliary cirrhosis and Sjögren’s syndrome. Clin Nephrol 74: 74–80, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Sanderson RD, Lalor P, Bernfield M: B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul 1: 27–35, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawano M, Mizushima I, Yamaguchi Y, Imai N, Nakashima H, Nishi S, Hisano S, Yamanaka N, Yamamoto M, Takahashi H, Umehara H, Saito T, Saeki T: Immunohistochemical characteristics of IgG4-related tubulointerstitial nephritis: Detailed analysis of 20 Japanese Cases. Int J Rheumatol 2012: 609795, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bohannon C, Powers R, Satyabhama L, Cui A, Tipton C, Michaeli M, Skountzou I, Mittler RS, Kleinstein SH, Mehr R, Lee FE, Sanz I, Jacob J: Long-lived antigen-induced IgM plasma cells demonstrate somatic mutations and contribute to long-term protection. Nat Commun 7: 11826, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talwalkar JA, Lindor KD: Primary biliary cirrhosis. Lancet 362: 53–61, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Parés A, Rimola A, Bruguera M, Mas E, Rodés J: Renal tubular acidosis in primary biliary cirrhosis. Gastroenterology 80: 681–686, 1981 [PubMed] [Google Scholar]
- 11.Lino M, Binaut R, Noël LH, Patey N, Rustin P, Daniel L, Serpaggi J, Varaut A, Vanhille P, Knebelmann B, Grünfeld JP, Fakhouri F: Tubulointerstitial nephritis and Fanconi syndrome in primary biliary cirrhosis. Am J Kidney Dis 46: e41–e46, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Bando H, Hashimoto N, Hirota Y, Sakaguchi K, Hisa I, Inoue Y, Imanishi Y, Seino S, Kaji H: Severe hypophosphatemic osteomalacia with Fanconi syndrome, renal tubular acidosis, vitamin D deficiency and primary biliary cirrhosis. Intern Med 48: 353–358, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Komatsuda A, Wakui H, Ohtani H, Masai R, Okuyama S, Nimura T, Suzuki N, Sawada K: Tubulointerstitial nephritis and renal tubular acidosis of different types are rare but important complications of primary biliary cirrhosis. Nephrol Dial Transplant 25: 3575–3579, 2010 [DOI] [PubMed] [Google Scholar]
- 14.Yamaguchi S, Maruyama T, Wakino S, Tokuyama H, Hashiguchi A, Tada S, Homma K, Monkawa T, Thomas J, Miyashita K, Kurihara I, Yoshida T, Konishi K, Hayashi K, Hayashi M, Itoh H: A case of severe osteomalacia caused by Tubulointerstitial nephritis with Fanconi syndrome in asymptomotic primary biliary cirrhosis. BMC Nephrol 16: 187, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saeki T, Nakajima A, Ito T, Takata T, Imai N, Yoshita K, Kabasawa H, Yamazaki H, Narita I: Tubulointerstitial nephritis and Fanconi syndrome in a patient with primary Sjögren’s syndrome accompanied by antimitochondrial antibodies: A case report and review of the literature. Mod Rheumatol 4: 1–4, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Macdougall IC, Isles CG, Whitworth JA, More IA, MacSween RN: Interstitial nephritis and primary biliary cirrhosis: A new association? Clin Nephrol 27: 36–40, 1987 [PubMed] [Google Scholar]
- 17.Kamouchi M, Tsuji H, Hirakata H, Okamura K, Ishitsuka T, Murai K, Onoyama K, Fujishima M: Tubulointerstitial disorders in the kidney associated with primary biliary cirrhosis (PBC). Clin Nephrol 35: 134–135, 1991 [PubMed] [Google Scholar]
- 18.Kodama T, Imai H, Wakui H, Ohtani H, Komatsuda A, Miura AB: Tubulointerstitial nephritis with renal tubular acidosis and asymptomatic primary biliary cirrhosis accompanied by antibody to a 52-kDa mitochondrial protein alone. Clin Nephrol 45: 401–405, 1996 [PubMed] [Google Scholar]
- 19.Terrier B, Fakhouri F, Berezne A, Bouldouyre MA, Guilpain P, Sogni P, Terris B, Noël LH, Guillevin L, Mouthon L: Osteomalacia revealing celiac disease and primary biliary cirrhosis-related Fanconi syndrome in a patient with systemic sclerosis. Clin Exp Rheumatol 26: 467–470, 2008 [PubMed] [Google Scholar]
- 20.Bansal T, Takou A, Khwaja A: Progressive chronic kidney disease secondary to tubulointerstitial nephritis in primary biliary cirrhosis. Clin Kidney J 5: 442–444, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwakura T, Fujigaki Y, Matsuyama T, Fujikura T, Ohashi N, Yasuda H, Kato A, Baba S: Tubulointerstitial nephritis and primary biliary cirrhosis with a T cell-dominant profile of infiltrating cells and granulomas in both organs. Intern Med 52: 467–471, 2013 [DOI] [PubMed] [Google Scholar]
- 22.Oertelt S, Rieger R, Selmi C, Invernizzi P, Ansari AA, Coppel RL, Podda M, Leung PS, Gershwin ME: A sensitive bead assay for antimitochondrial antibodies: Chipping away at AMA-negative primary biliary cirrhosis. Hepatology 45: 659–665, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Yamagiwa S, Kamimura H, Takamura M, Aoyagi Y: Autoantibodies in primary biliary cirrhosis: Recent progress in research on the pathogenetic and clinical significance. World J Gastroenterol 20: 2606–2612, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim WR, Lindor KD, Locke GR 3rd, Therneau TM, Homburger HA, Batts KP, Yawn BP, Petz JL, Melton LJ 3rd, Dickson ER: Epidemiology and natural history of primary biliary cirrhosis in a US community. Gastroenterology 119: 1631–1636, 2000 [DOI] [PubMed] [Google Scholar]
- 25.Sood S, Gow PJ, Christie JM, Angus PW: Epidemiology of primary biliary cirrhosis in Victoria, Australia: High prevalence in migrant populations. Gastroenterology 127: 470–475, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Whaley K, Webb J, McAvoy BA, Hughes GR, Lee P, MacSween RN, Buchanan WW: Sjogren’s syndrome. 2. Clinical associations and immunological phenomena. Q J Med 42: 513–548, 1973 [PubMed] [Google Scholar]
- 27.Cohen EP, Bastani B, Cohen MR, Kolner S, Hemken P, Gluck SL: Absence of H(+)-ATPase in cortical collecting tubules of a patient with Sjogren’s syndrome and distal renal tubular acidosis. J Am Soc Nephrol 3: 264–271, 1992 [DOI] [PubMed] [Google Scholar]
- 28.DeFranco PE, Haragsim L, Schmitz PG, Bastani B: Absence of vacuolar H(+)-ATPase pump in the collecting duct of a patient with hypokalemic distal renal tubular acidosis and Sjögren’s syndrome. J Am Soc Nephrol 6: 295–301, 1995 [DOI] [PubMed] [Google Scholar]
- 29.Medina JF, Martínez-Ansó, Vazquez JJ, Prieto J: Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis. Hepatology 25: 12–17, 1997 [DOI] [PubMed] [Google Scholar]
- 30.Melero S, Spirlì C, Zsembery A, Medina JF, Joplin RE, Duner E, Zuin M, Neuberger JM, Prieto J, Strazzabosco M: Defective regulation of cholangiocyte Cl-/HCO3(-) and Na+/H+ exchanger activities in primary biliary cirrhosis. Hepatology 35: 1513–1521, 2002 [DOI] [PubMed] [Google Scholar]
- 31.Vázquez JJ, Vázquez M, Idoate MA, Montuenga L, Martínez-Ansó E, Castillo JE, García N, Medina JF, Prieto J: Anion exchanger immunoreactivity in human salivary glands in health and Sjögren’s syndrome. Am J Pathol 146: 1422–1432, 1995 [PMC free article] [PubMed] [Google Scholar]
- 32.François H, Mariette X: Renal involvement in primary Sjögren syndrome. Nat Rev Nephrol 12: 82–93, 2016 [DOI] [PubMed] [Google Scholar]
- 33.Solís Herruzo JA, Solís Muñoz P, Muñoz Yagüe T: The pathogenesis of primary biliary cirrhosis. Rev Esp Enferm Dig 101: 413–423, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM: The generation of antibody-secreting plasma cells. Nat Rev Immunol 15: 160–171, 2015 [DOI] [PubMed] [Google Scholar]
- 35.Baba Y, Matsumoto M, Kurosaki T: Signals controlling the development and activity of regulatory B-lineage cells. Int Immunol 27: 487–493, 2015 [DOI] [PubMed] [Google Scholar]
- 36.Paronetto F, Schaffner F, Popper H: Immunocytochemical and serologic observations in primary biliary cirrhosis. N Engl J Med 271: 1123–1128, 1964 [DOI] [PubMed] [Google Scholar]
- 37.van den Oord JJ, Fevery J, de Groote J, Desmet VJ: Immunohistochemical characterization of inflammatory infiltrates in primary biliary cirrhosis. Liver 4: 264–274, 1984 [DOI] [PubMed] [Google Scholar]
- 38.Daniels JA, Torbenson M, Anders RA, Boitnott JK: Immunostaining of plasma cells in primary biliary cirrhosis. Am J Clin Pathol 131: 243–249, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Takahashi T, Miura T, Nakamura J, Yamada S, Miura T, Yanagi M, Matsuda Y, Usuda H, Emura I, Tsuneyama K, He XS, Gershwin ME: Plasma cells and the chronic nonsuppurative destructive cholangitis of primary biliary cirrhosis. Hepatology 55: 846–855, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menéndez-Caro JL, Alvarez-Mon M, Girón JA, Manzano L, Garrido A, Abreu L, Albillos A, Durántez A: Increased IgM B cell differentiation lymphokine production by T lymphocytes from patients with primary biliary cirrhosis. J Hepatol 20: 446–453, 1994 [DOI] [PubMed] [Google Scholar]
- 41.Clarkson MR, Giblin L, O’Connell FP, O’Kelly P, Walshe JJ, Conlon P, O’Meara Y, Dormon A, Campbell E, Donohoe J: Acute interstitial nephritis: clinical features and response to corticosteroid therapy. Nephrol Dial Transplant 19: 2778–2783, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Rodríguez Soriano J: Renal tubular acidosis: The clinical entity. J Am Soc Nephrol 13: 2160–2170, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox RI, Kassan SS, Pillemer SR, Talal N, Weisman MH; European Study Group on Classification Criteria for Sjögren’s Syndrome : Classification criteria for Sjögren’s syndrome: A revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 61: 554–558, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raissian Y, Nasr SH, Larsen CP, Colvin RB, Smyrk TC, Takahashi N, Bhalodia A, Sohani AR, Zhang L, Chari S, Sethi S, Fidler ME, Cornell LD: Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol 22: 1343–1352, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, Klöppel G, Heathcote JG, Khosroshahi A, Ferry JA, Aalberse RC, Bloch DB, Brugge WR, Bateman AC, Carruthers MN, Chari ST, Cheuk W, Cornell LD, Fernandez-Del Castillo C, Forcione DG, Hamilos DL, Kamisawa T, Kasashima S, Kawa S, Kawano M, Lauwers GY, Masaki Y, Nakanuma Y, Notohara K, Okazaki K, Ryu JK, Saeki T, Sahani DV, Smyrk TC, Stone JR, Takahira M, Webster GJ, Yamamoto M, Zamboni G, Umehara H, Stone JH: Consensus statement on the pathology of IgG4-related disease. Mod Pathol 25: 1181–1192, 2012 [DOI] [PubMed] [Google Scholar]
- 46.Youden WJ: Index for rating diagnostic tests. Cancer 3: 32–35, 1950 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.