

Abstract
Phospholipase D4 (PLD4), a single-pass transmembrane glycoprotein, is among the most highly upregulated genes in murine kidneys subjected to chronic progressive fibrosis, but the function of PLD4 in this process is unknown. Here, we found PLD4 to be overexpressed in the proximal and distal tubular epithelial cells of murine and human kidneys after fibrosis. Genetic silencing of PLD4, either globally or conditionally in proximal tubular epithelial cells, protected mice from the development of fibrosis. Mechanistically, global knockout of PLD4 modulated innate and adaptive immune responses and attenuated the upregulation of the TGF-β signaling pathway and α1-antitrypsin protein (a serine protease inhibitor) expression and downregulation of neutrophil elastase (NE) expression induced by obstructive injury. In vitro, treatment with NE attenuated TGF-β–induced accumulation of fibrotic markers. Furthermore, therapeutic targeting of PLD4 using specific siRNA protected mice from folic acid–induced kidney fibrosis and inhibited the increase in TGF-β signaling, decrease in NE expression, and upregulation of mitogen-activated protein kinase signaling. Immunoprecipitation/mass spectrometry and coimmunoprecipitation experiments confirmed that PLD4 binds three proteins that interact with neurotrophic receptor tyrosine kinase 1, a receptor also known as TrkA that upregulates mitogen-activated protein kinase. PLD4 inhibition also prevented the folic acid–induced upregulation of this receptor in mouse kidneys. These results suggest inhibition of PLD4 as a novel therapeutic strategy to activate protease-mediated degradation of extracellular matrix and reverse fibrosis.
Keywords: chronic kidney disease, fibrosis, immunology, TGF-beta
Fibrosis is the underlying pathology of CKD, a complex debilitating condition affecting >70 million people worldwide.1 Of several known mechanisms, it is often a balance between the immune components as well as the synthesis and degradation of extracellular matrix (ECM) components that determines whether the kidney injury resolves or progresses to fibrosis.2,3 Understanding the mechanistic basis of such multivariate interactions within the scar microenvironment, including interplay between the immune system and the proteases, may enable identification of novel target-based precision therapeutics for kidney fibrosis.
We previously reported phospholipase D4 (PLD4) to be among the most upregulated genes in a mouse model of folic acid (FA)–induced kidney fibrosis.4 PLD4 belongs to phospholipase D family, which comprises six members, i.e., PLD1–PLD6.5 Of these, PLD1 and PLD2 are well characterized and are known as classical PLDs. Unlike PLD1 and PLD2 proteins, which contain Phox homology (PX) and pleckstrin homology (PH) domains in the N-terminal region and two conserved His-x-Lys-x-x-x-x-Asp sequences (where x is any amino acid, HKD) motifs in the C-terminal region, PLD4, a nonclassical PLD, has a putative transmembrane domain instead of PX and PH domains.5–7 PLD1 and PLD2 are well known to play a vital role in the pathogenesis of various diseases such as cancer (breast, ovarian, lung, colon, renal, pancreatic, prostate, and brain cancer), infectious disorders (influenza viruses), and neurodegenerative disorders (encephalomyelitis, stroke, and Alzheimer disease)8 by modulating the mammalian target of rapamycin (mTOR) pathway,9 hypoxia inducible factor 1 α (HIF1α) signaling pathway,10 angiogenesis,11 and phagocyte cell migration.12 Although much is known about the classic PLDs (PLD1 and PLD2), the functional role of PLD4 has not been investigated beyond its implication in systemic sclerosis,13 rheumatoid arthritis,14 and the phagocytosis of microglia.15 Our study was aimed at investigating the mechanistic role of PLD4 in kidney fibrosis and examining if modulating PLD4 by genetic and pharmacologic approaches alters the course of fibrosis progression.
Results
PLD4 Expression Is Upregulated in Mice and Human Kidneys after Fibrosis
We found that among the PLD family members, PLD2 and PLD4 expression significantly (P<0.05) increased at mRNA level when treated with FA (Figure 1, A and B) or subjected to unilateral ureteral obstruction (UUO) (Figure 1, D and E) two mechanistically distinct mouse models of kidney fibrosis.4,16–18 There was also a significant increase in the protein expression of PLD4 in both FA (Figure 1C) and UUO (Figure 1F) models. In order to examine the pan-fibrotic expression of PLD4, its levels were measured in a carbon tetrachloride (CCl4)–induced liver fibrosis model. PLD4 expression significantly increased in mice with liver fibrosis (Supplemental Figure 1), extending the relevance of PLD4 in fibrosis beyond kidneys. Translatability of PLD4 expression in human disease was confirmed by observing a significant increase in PLD4 expression in patients with biopsy-proven tubulointerstitial fibrosis (Figure 1G).
Figure 1.

Expression of fibrotic markers as well as PLD4 increases in FA- and UUO-induced kidney fibrosis. Protein expression of the fibrotic markers α-SMA, fibronectin (FN), and collagen 1α1 (Col 1α1) in the kidneys of mice (A) injected with FA and (D) subjected to UUO (n=5/group). (B and E) mRNA levels of PLD1, PLD2, PLD3, and PLD4 and (C and F) protein level of PLD4 in the kidneys of mice injected with FA and subjected to UUO, respectively. Data were normalized to GAPDH and are presented as mean±SEM (n=5/group) of the fold change over normal. *P<0.05. (G) Immunostaining and the relative quantitation of α-SMA and PLD4 in the kidney sections from patients with biopsy-proven tubulointerstitial fibrosis versus patients with no evidence of fibrosis (n=5/group). Scale bars, 50 µm. Data are presented as mean±SEM of the fold change over normal. *P<0.05. C, contralateral; CoK, contralateral kidney; N, normal; U, UUO.
PLD4 Global Knockout Mice Are Protected against Kidney Fibrosis
To decipher the functional role of PLD4 in kidney fibrosis, we conducted loss-of-function experiments using PLD4 global knockout (PLD4−/−) mice. Knockdown of PLD4 was confirmed by the absence of PLD4 at baseline (Supplemental Figure 2, A and B) as well as lack of increase in PLD4 expression in the PLD4−/− mice after fibrosis (Supplemental Figure 2, C and D). The PLD4−/− mice are viable and did not show any histologic abnormality (Supplemental Figure 2E). There was a significant attenuation (P<0.05) in the expression of fibrotic markers α-smooth muscle actin (α-SMA), fibronectin, and collagen 1α1 (Figure 2, A–C, Supplemental Figure 3, A–D) in the PLD4−/− mice compared with the wild-type (PLD4+/+) mice subjected to either FA administration or surgical UUO. The protection from fibrosis was further confirmed pathologically by observing a significant decrease in collagen accumulation and scar tissue formation as evidenced by Picrosirius Red and Masson’s trichrome staining in both the models (Figure 2C).
Figure 2.

PLD4−/− mice are protected from FA- and UUO-induced kidney fibrosis. Protein expression of the fibrotic markers α-SMA, fibronectin (FN), and collagen 1α1 (Col 1α1) in the kidneys of PLD4+/+ and PLD4−/− mice injected with (A) FA and (B) subjected to UUO (n=5–7/group). (C) Representative photomicrographs depicting the expression of fibrotic markers (α-SMA, FN, and Col 1α1) in mouse kidneys (n=5/group), and Picrosirius Red and Masson’s trichrome staining depicting collagen deposition in mouse kidneys at baseline and after fibrosis (n=3/group). Scale bars, 50 µm. Relative quantitation of immunostaining and Picrosirius Red and Masson’s trichrome staining. Data are presented as mean±SEM of the fold change over PLD4+/+ normal. *P<0.05. (D) Schematic representation of the timeline of tamoxifen and FA administration in mice. Mice were injected with FA (250 mg/kg, ip) followed by treatment with tamoxifen (2 mg, diluted in corn oil and 3% ethanol, ip) at 1, 3, and 5 days after FA injection. Mice were euthanized on day 7 after FA injection (n=5/group). (E) Specificity of PLD4 knockdown in kidney proximal tubular epithelial cells was confirmed by coimmunostaining PLD4 with LTL, a proximal tubular marker (thick arrows, LTL-positive tubules; thin arrows, LTL-negative tubules), using the kidneys obtained from SLC34a1GCE/+ PLD4fl/fl (PLD4fl/fl) and wild-type mice SLC34a1GCE/+ PLD4wt/wt (PLD4wt/wt) (n=5/group). Scale bars, 50 μm. (F) Protein levels of α-SMA, FN, and Col 1α1 in the kidneys of PLD4wt/wt and PLD4fl/fl mice injected with FA (n=5/group). C, contralateral; ip, intraperitoneally; N, normal; U, UUO; +/+, PLD4 wild-type; −/−, PLD4 knockout.
Proximal Tubule Epithelial Cell–Specific PLD4 Knockdown Confers Protection against Kidney Fibrosis
Because PLD4 is also expressed in leukocytes, the global knockout mouse model could not distinguish whether PLD4 expressed in leukocytes or epithelial cells mediates fibrosis. Therefore, we generated conditional mice lacking PLD4 in the kidney proximal tubular epithelial cells. We generated these mice by crossing PLD4fl/fl mice with mice expressing Cre recombinase under the control of the tamoxifen-inducible SLC34a1 promoter, a transport protein expressed only in the proximal tubule cells.19 We compared SLC34a1GCE/+ PLD4fl/fl (GCE stands for eGFPCreERT2) with SLC34a1GCE/+ PLD4wt/wt mice; Cre was present in both the experimental groups, and all mice were injected with tamoxifen. In SLC34a1GCE/+ PLD4wt/wt mouse kidneys (baseline), PLD4 was broadly expressed in proximal tubule cells as evident from the costaining of PLD4 and Lotus tetragonolobus lectin (LTL, proximal tubule marker). In SLC34a1GCE/+ PLD4fl/fl mouse kidneys (baseline), PLD4 was specifically deleted only in LTL-positive proximal tubule cells (thick arrows) but not in distal tubular segments (thin arrows) (Figure 2E). Tamoxifen was injected into SLC34a1GCE/+ PLD4wt/wt and SLC34a1GCE/+ PLD4fl/fl mice on days 1, 3, and 5 after FA injection and mice were euthanized 7 days after FA injection (Figure 2D). SLC34a1GCE/+ PLD4fl/fl mice showed significantly decreased expression of α-SMA, fibronectin, and collagen 1α1 compared with the SLC34a1GCE/+ PLD4wt/wt mice (Figure 2F, Supplemental Figure 3E). Although leukocyte-associated PLD4 might still play a role in fibrogenesis, our data demonstrates that PLD4 expressed in proximal tubular epithelial cells play a predominant role in propagating fibrosis in the kidneys.
PLD4 Modulates the Immune Responses and Upregulates α1-Antitrypsin (a Serine Protease Inhibitor) in Kidney Fibrosis
To investigate the mechanisms of protection from fibrosis, we performed RNA sequencing (Supplemental Figure 4) in the kidneys of global PLD4−/− and PLD4+/+ mice at baseline and after UUO. We observed differential expression of genes involved in the innate and adaptive immune responses between the PLD4−/− and PLD4+/+ mice at baseline and after UUO (Figure 3, A and B, Supplemental Figure 4F). Experimentally, we confirmed a significant reduction (P<0.05) of neutrophils (CD11b+Ly6G+) and monocytes (CD11b+Ly6C+) in the PLD4−/− mice at day 5 after UUO (Figure 3C, Supplemental Figure 5A). On the other hand, CD8+ and MHC-II+ cells were significantly higher (P<0.05) at baseline and after UUO, whereas CD4+ cells were significantly higher (P<0.05) only after UUO in the PLD4−/− mice (Figure 3C, Supplemental Figure 5A). There were no differences in the levels of natural killer cells (NK1.1+ cells) or B cells (CD19+ cells) between the PLD4−/− and PLD4+/+ mice (Supplemental Figure 5B). We also measured cytokines in the kidney (Figure 3D, Supplemental Figure 5C) and found that mRNA levels of INF-γ, TNF-α, IL-1β, IL-6, IL-17, and IL-10 were significantly higher (P<0.05) in the PLD4−/− mice after UUO (Figure 3D). Also, there was a significant decrease in Arg1 (a M2 macrophage marker), but no difference in iNOS (a M1 macrophage marker), between the PLD4−/− and PLD4+/+ mice after UUO (Figure 3D, Supplemental Figure 5C). INF-γ and TNF-α are known to be antifibrotic cytokines whereas M2 macrophages are known to be profibrotic because of their ability to inhibit and induce TGF-β secretion, respectively.20–22 INF-γ and TNF-α also exert their antifibrotic effect by suppressing the synthesis of TGF-β–induced collagen and connective tissue growth factor, respectively.23 We observed downregulation of TGF-β signaling in the PLD4−/− mice after UUO as evidenced by a significant (P<0.05) decrease in the expression of pSmad 2, Smad2, pSmad3, and Smad3 (Figure 3E, Supplemental Figure 5D).
Figure 3.

PLD4−/− mouse kidneys have differentially expressed innate and adaptive immune genes and decreased level of a protease inhibitor, AAT, compared with the PLD4+/+ mice. (A) Heat map depicting differentially expressed genes between PLD4+/+ and PLD4−/− mouse kidneys at baseline and after UUO (days 5 and 10) (n=4/group). (B) Pathway analysis of differentially expressed genes between PLD4+/+ and PLD4−/− mouse kidneys at baseline. (C) Flow cytometric analysis of innate and adaptive immune system components in the kidneys of mice at baseline and after UUO (day 5) (n=3/group). (D) mRNA levels of cytokines in the kidneys of mice at baseline and after UUO (days 5 and 10). Protein expression of (E) TGF-β signaling molecules, pSmad2, Smad2, pSmad3, and Smad3; and (F) AAT and NE in mouse kidneys. Data were normalized to GAPDH and are presented as mean±SEM (n=5–7/group) of the fold change over PLD4+/+ normal. *P<0.05. C, contralateral; N, normal; U, UUO; +/+, PLD4 wild-type; −/−, PLD4 knockout.
RNA sequencing (Supplemental Figure 4) also revealed downregulation of serpina1 family genes in the PLD4−/− mice (Figure 3A, Supplemental Figure 4F). Among serpina1 family genes, seprina1d was the most downregulated gene, and serpina1d translates to α1-antitrypsin (AAT), which is a serine protease inhibitor. Downregulation of AAT was confirmed by observing a significantly decreased protein expression of AAT in the PLD4−/− mice after fibrosis (Figure 3F, Supplemental Figure 5E). The association between PLD4 and AAT was further established by observing a significant increase in the AAT expression (P<0.05) in PLD4-overexpressed HEK293T cells (Supplemental Figure 6). AAT specifically inhibits neutrophil elastase (NE), a serine protease known to degrade the ECM proteins such as fibronectin and collagen.24 Significantly increased (P<0.05) expression of NE was observed in the PLD4−/− mice (Figure 3F, Supplemental Figure 5E), suggesting that degradation of the ECM proteins by NE was partly playing a role in protecting these mice from fibrosis.
The role of NE in decreasing the expression of fibrotic markers was further confirmed in vitro using a coculture system of primary human kidney fibroblasts and HEK293T cells (transfected with pCMV or pCMV-PLD4). NE treatment (50 nM) for 48 hours significantly decreased the levels of α-SMA and fibronectin compared with the untreated cells. TGF-β treatment (10 ng/ml) for 48 hours led to overexpression of the fibrotic markers in the cocultured cells. TGF-β treatment in the PLD4-overexpressed HEK293T cells cocultured with the primary human kidney fibroblasts led to further increase in the expression of α-SMA and fibronectin. Cotreatment of cells with NE (50 nM) and TGF-β for 48 hours significantly decreased the level of fibrotic markers compared with TGF-β–only treatment. The NE-mediated decrease in TGF-β–induced fibrotic markers was significantly lower in the coculture system with pCMV-PLD4–transfected HEK293T cells compared with the pCMV-transfected cells, which might be because of PLD4-induced AAT expression. This observation strengthens our in vivo findings reporting the role of PLD4 in inducing fibrogenesis, at least in part, by downregulating NE expression (Supplemental Figure 7). Expression of the fibrotic markers in cells treated with TGF-β (10 ng/ml) for 48 hours followed by NE treatment (50 nM) for 6 hours was not significantly different compared with the TGF-β alone–treated cells (Supplemental Figure 7). This indicates that NE treatment alone might not be effective in reversing fibrosis at a later time point.
Targeting PLD4 Using Small Interfering RNA Protects against Kidney Fibrosis
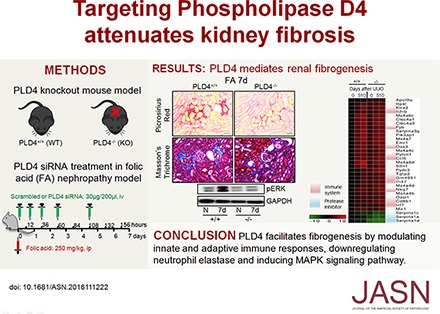
We next investigated whether silencing PLD4 in mice using a small interfering RNA (siRNA) strategy protects from kidney fibrosis (Figure 4A). PLD4 siRNA (validated in vitro using mouse inner medullary collecting duct [mIMCD3] cells, Supplemental Figure 8A) was delivered to the kidney (Figure 4B, Supplemental Figure 8, B and C) and protected mice from kidney fibrosis as evident from the decreased expression of fibrotic markers (Figure 4, B and C, Supplemental Figure 8C). The therapeutic effect of PLD4 siRNA was mediated via the same mechanisms, i.e., downregulation of TGF-β signaling (Figure 4D, Supplemental Figure 8D), decreased expression of AAT, and increased expression of NE (Figure 4E, Supplemental Figure 8E). PLD4 silencing decreased kidney fibrosis without significantly reducing the initiation of kidney injury, as evident from the levels of plasma creatinine, BUN, and kidney injury molecule 1 (KIM1) expression in the kidney on day 2 after FA injection (Figure 4, F and G).
Figure 4.

Therapeutic targeting of PLD4 by specific siRNA protects mice from kidney fibrosis. (A) Schematic representation of the treatment protocol with FA and siRNA (scrambled or PLD4). Mice were injected with FA (250 mg/kg, ip) followed by treatment with scrambled or PLD4 siRNA (30 µg/200 µl, iv) at 2, 20, 38, 62, and 110 hours after FA injection. Mice were euthanized on day 7 after FA injection (n=5/group). (B) Protein levels of PLD4 and the fibrotic markers α-SMA, fibronectin (FN), and collagen 1α1 (Col 1α1) in mouse kidneys (n=5/group). (C) Picrosirius Red and Masson’s trichrome staining depicting collagen deposition in mouse kidneys (n=3/group). Scale bars, 50 µm. Data are presented as mean±SEM (n=5/group) of the fold change over vehicle+scrambled siRNA. *P<0.05. Protein expression of (D) TGF-β signaling molecules, pSmad2, Smad2, pSmad3, and Smad3; and (E) AAT and NE in mouse kidneys (n=5/group). (F) Level of plasma creatinine and BUN in mice and (G) protein level of KIM1 in mouse kidneys (n=4/group). Data are presented as mean±SEM (n=4–5/group) of the fold change over vehicle+scrambled siRNA. *P<0.05. (H) Schematic representation depicting how PLD4 drives fibrosis. PLD4 leads to a decrease in anti-fibrotic cytokines, upregulation of TGFβ signaling and downregulation of neutrophil elastase expression by preventing a decrease in α1-antitrypsin expression. All these result in increased ECM accumulation that progresses to fibrosis. ip, intraperitoneally; iv, intravenously; Scr, scrambled siRNA; V+scr, vehicle+scrambled siRNA.
PLD4 Interacts with Calmegin, Lectin, Mannose Binding 2, and Suppressor of Lin-12–Like Protein 1 and Upregulates MAPK Signaling via Tropomyosin-Related Kinase A Pathway to Mediate Fibrogenesis
To further delineate PLD4-mediated signaling pathways in promoting fibrogenesis, we next took an immunoprecipitation/mass spectrometry–based interactome proteomics approach25 (Supplemental Figure 9) and found several proteins to be the binding partners of PLD4 (Figure 5A), most of which are endoplasmic reticulum proteins, although some are localized in the Golgi apparatus, mitochondria, and plasma membrane (Figure 5B). The subcellular localization of PLD4 was assessed using HeLa cells (Figure 5C) as well as HEK293 cells (Figure 5D), and we found PLD4 to be localized in the endoplasmic reticulum, Golgi apparatus, and mitochondria. We experimentally confirmed the interaction between PLD4 and each of the top three proteins—calmegin (CLGN), lectin, mannose binding 2 (LMAN2), and suppressor of lin-12–like protein 1 (SEL1L)—(on the basis of the weighted and normalized D score [WDN score]) using coimmunoprecipitation/western blot (Co-IP/WB) (Figure 5E). Interestingly, CLGN, LMAN2, and SEL1L are known to have a common interactor, neurotrophic receptor tyrosine kinase 1 (NTRK1), also known as tropomyosin-related kinase A (TrkA) (Supplemental Figure 10, https://thebiogrid.org), which in turn upregulates MAPK via fibroblast growth factor receptor substrate 2 (FRS2α). PLD4 inhibition (genetic knockdown as well as PLD4 siRNA treatment) significantly decreased the protein expression of CLGN and SEL1L in mouse kidneys; however, LMAN2 expression was not affected. There was a significant decrease in the expression of TrkA, and the downstream signaling proteins, FRS2α, pERK, and ERK, with PLD4 inhibition, suggesting that PD4 mediates fibrogenesis by upregulating MAPK signaling via TrkA pathway (Figure 5, F and G, Supplemental Figure 11).
Figure 5.

PLD4 upregulates MAPK signaling via TrkA pathway to mediate fibrogenesis. (A) Overview of the PLD4 interaction network on the basis of the immunoprecipitation/mass spectrometry (IP/MS) analysis (respective WDN scores indicated in parentheses). (B) Diagrammatic representation of the localization of interacting partners of PLD4. Orange circles represent PLD4 and gray circles represent the binding partners of PLD4. Immunofluorescence analysis of the subcellular localization of PLD4 by costaining PLD4 with ER (Calnexin)-, Golgi (Golga1)-, and mitochondria (Tom20)-specific proteins in (C) HeLa cells and (D) HEK293 cells. Scale bars, 20 µm. (E) Coimmunoprecipitation of the top three interacting partners of PLD4 (CLGN, LMAN2, and SEL1L, selected on the basis of the WDN score) using PLD4-overexpressed HEK293T cells. Protein expression of CLGN, LMAN2, SEL1L, TrkA, FRS2α, pERK, and ERK in the kidneys of (F) PLD4+/+ and PLD4−/− mice and (G) siRNA (scr and PLD4)-treated mice injected with FA (n=5/group). ER, endoplasmic reticulum; IP, immunoprecipitation; Mito, mitochondria; scr, scrambled; +/+, PLD4 wild-type; −/−, PLD4 knockout.
Discussion
Collectively, our results demonstrate that PLD4 expressed on the kidney proximal tubular epithelial cells is a novel target against kidney fibrosis. We show that PLD4 facilitates fibrogenesis by modulating innate and adaptive immune responses and promoting the TGF-β signaling pathway. Moreover, PLD4 induces the expression of AAT and the subsequent downregulation of NE expression, thereby, at least in part, leading to the accumulation of ECM proteins (Figure 4H).
Although inflammation is well known to drive fibrogenesis,26 here we observed that PLD4−/− mice were protected from fibrosis in spite of elevated proinflammatory cytokines and T cells. Consistent with our findings, proinflammatory cytokines, IFN-γ, and TNF-α, have been shown to be antifibrotic using in vitro and in vivo models of kidney, liver, and lung fibrosis.27–29 The proposed antifibrotic mechanisms of IFN-γ are antagonism toward TGF-β–induced signaling and collagen deposition by signal transducer and activator of transcription (STAT)–1 activation in lung, as well as inhibition of hepatic stellate cell activation.20,30 Similarly, TNF-α exerts an antifibrotic effect by inhibiting TGF-β/Smad signaling through the activation of AP-1 components, activating the inhibitory Smad7 by NF-κB/RelA and suppressing TGF-β1–induced myofibroblast activation.22,31,32 Furthermore, treatment with recombinant IL-2 and TNF-α significantly reduced the appearance of fibrocytes and the severity of fibrosis in the UUO model.33 Increased proinflammatory cytokines in PLD4−/− mice could, at least in part, be because of a decreased level of AAT, an endogenous inhibitor of proinflammatory cytokine production in whole blood.34 Renal AAT gene induction was observed in experimental and clinical kidney damage,35 which corroborates our finding.
Another finding from our study is that significantly less fibrosis in global PLD4−/− mice was associated with an increased level of NE, indicating a protective role of NE in kidney fibrosis. Although NE is known to damage the lungs,36 we observed that, in kidneys, increased expression of NE mirrors the decreased extent of fibrosis. Interestingly, the histologic examination of the lungs of PLD4−/− mice did not show any abnormality. However, given the destructive effect of NE in lungs, the role of NE in kidney fibrosis needs to be definitively confirmed by performing additional studies such as reversing the protective effect of PLD4-siRNA against kidney fibrosis with either (1) reconstitution of AAT level in PLD4-siRNA–treated mice similar to that in scrambled siRNA-treated mice and/or (2) using blocking antibodies to NE. We speculate that increased NE might be exerting an antifibrotic effect in conjunction with the proinflammatory cytokines. NE degrades collagen in three-dimensional culture by acting synergistically with proinflammatory cytokines.37 Decreased level of elastase due to increased expression of monocyte/neutrophil elastase inhibitor was associated with accumulation of elastin, an ECM protein, in kidneys of mice with diabetic nephropathy.38 Interestingly, although the neutrophils were significantly less in the PLD4−/− mice after UUO, these mice had higher expression of NE, which potentially could be because of a significantly lower expression of AAT in the kidneys.
PLD4 interaction network and Co-IP/WB confirm that PLD4 binds with CLGN, LMAN2, and SEL1L and induces fibrosis via TrkA-mediated MAPK signaling pathway. TrkA, a common interactor of the binding partners of PLD4 (CLGN, LMAN2, and SEL1L), activates members of the MAPK pathway via FRS2α, a signaling adapter that directly binds to the phosphotyrosine residue of TrkA.39,40 Moreover, TrkA expression is elevated in several nephropathies, including diabetic nephropathy.41 The MAPK pathway plays a vital role in ECM synthesis in vitro42 and in progressing bleomycin-induced pulmonary fibrosis,43 and UUO-induced kidney fibrosis in vivo.44,45 Downregulation of TrkA-mediated MAPK signaling with PLD4 inhibition depicts the role of PLD4 in inducing this pathway. However, which among CLGN, LMAN2, and SEL1L plays a critical role in inducing TrkA needs to be further explored. PLD4-induced AAT expression also might, at least in part, be playing a role in upregulating MAPK signaling, because AAT is known to stimulate fibroblast proliferation and procollagen production via MAPK signaling pathway.46 Together, our findings provide several novel insights to conclude that PLD4 regulates fibrogenesis in kidney. Moreover, overexpression of α-SMA, FN, Col 1α1, and PLD4 in CCl4-induced fibrotic liver suggests that PLD4 could be playing a causal role in liver fibrosis as well making PLD4 a potential pan-fibrotic target.
In summary, these findings implicate that abrogation of PLD4 led to (1) increased antifibrotic cytokines, (2) downregulated TGF-β signaling, (3) increased NE due to decreased AAT level, and (4) downregulated MAPK signaling, and these effects rescued the mice from scar tissue formation. Our work not only establishes the functional significance of PLD4 overexpression in kidney fibrosis but also highlights the therapeutic potential of blocking PLD4 as a new approach to treat kidney fibrosis, an unmet medical need.
Concise Methods
Animals
All animal maintenance and treatment protocols were approved by the Harvard Medical School Animal Care and Use Committees (Institutional Animal Care and Use Committees), and complied with the National Institutes of Health Guide for Care and Use of Laboratory Animals. Experiments were performed on male C57BL/6J mice (8–10 weeks old) purchased from Charles River Laboratories. Global PLD4−/− mice and PLD4fl/fl with C57BL/6J background were obtained from Dr. David Nemazee at the Scripps Research Institute, La Jolla, California, and were then bred and maintained in our animal facility. SLC34a1-GFPCreERT2 mice were obtained from Dr. Benjamin D. Humphreys, Washington University School of Medicine, St. Louis, Missouri. Detailed methods are described in the Supplemental Methods.
Western Blot Analysis, Quantitative Real-Time PCR, and Histology
All of the techniques were performed using standard protocols established in the laboratory.47–49 Primary antibodies used are indicated in the Supplemental Material, and primer pairs used are listed in Supplemental Table 1.
Immunoprecipitation and Proteomic Analysis
Immunoprecipitation and proteomic analysis were performed as described previously25,50,51 (Supplemental Figure 9). Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was used for the plasmid transfection of human embryonic kidney 293T (HEK293T) cells according to manufacturer’s specifications. The cells were harvested 48 hours post-transfection for further analysis. Viral particles were generated in HEK293T cells through transfection with a pHAGE lentiviral vector, containing a C-terminal HA-Flag–tagged gene of interest and four helper vectors (VSVG, Tat1b, Mgpm2, and CMV-Rev). Virus-containing supernatants were used to infect HEK293T cells. Cells stably expressing the tagged proteins were selected with Puromycin (Invitrogen, Carlsbad, CA) for a week. For IP, cells (approximately 107) were lysed in lysis buffer and rotated for 30 minutes at 4°C to obtain whole-cell extracts. Lysates were cleared by centrifugation at 4°C for 15 minutes at maximum speed. Lysates were incubated with anti-Flag beads, and proteins were eluted with a Flag peptide. Proteins were then subjected to TCA precipitation and trypsinized before passage through StageTips. Samples were run in technical duplicate on a Thermo LTQ mass spectrometer, and spectra search was conducted with Sequest before target-decoy peptide filtering and linear discriminant analysis. Protein Assembler was used to convert spectral counts to average peptide spectral matches, which were uploaded into the CompPASS algorithm housed within the Core facility. For CompPASS analysis of HEK293T cells, a database of 48 baits was used. The CompPASS system identifies high-confidence candidate interacting proteins (HCIPs) on the basis of the WDN score, and proteins with WDN score >1.0 are considered HCIPs.
Library Preparation and RNA Sequencing
RNA samples (n=4 mice/group) were checked for quantity and quality (RIN value >8.0) using Agilent 2200 TapeStation instrument and by SYBR qPCR assay. Libraries were prepared using Illumina TruSeq Stranded mRNA sample preparation kits (San Diego, CA) from 1 μg of purified total RNA according to the manufacturer’s protocol. The finished dsDNA libraries were quantified by Qubit fluorometer, Agilent TapeStation 2200, and RT-qPCR was performed using the Kapa Biosystems library quantification kit (Wilmington, MA) according to the manufacturer’s protocols. Uniquely indexed libraries were pooled in equimolar ratios and sequenced on a single Illumina NextSeq500 run with single-end 75-bp reads by the Dana–Farber Cancer Institute Molecular Biology Core Facilities. STAR aligner was used to map sequenced reads to the mm9 genome assembly and to quantify gene level expression. Testing for differential gene expression was performed using DESeq2. The full dataset is available in the National Center for Biotechnology Information Gene Expression Omnibus database with the accession number GSE87212.
Flow Cytometry
Mouse kidneys were digested as described previously,52 washed with FACS buffer, FC blocked, and stained for surface expression of CD11b (BV711; BD Biosciences, CA), Ly6C (FITC; Biolegend, CA), Ly6G (PE; Biolegend), CD3 (eFluor 450; eBioscience, CA), CD8 (APC-eFluor 780; eBioscience), CD4 (FITC; eBioscience), NK1.1 (PerCP-Cy 5.5; BD Biosciences), CD19 (PerCP-Cy 5.5; BD Biosciences), and MHC-II (I-A/I-E PE; Biolegend). Antibody concentrations used for staining were according to manufacturers’ specifications. Gating for live cells was on the basis of staining the cells with DAPI. Data were acquired using a BD LSRII flow cytometer at the Harvard Medical School Systems Biology FACS Core Facility and were analyzed with FlowJo software (TreeStar).
Statistical Analyses
Data are expressed as mean±SEM. Statistical significance was calculated by nonparametric t test. For multiple comparisons, statistical difference was calculated by one-way ANOVA. Post hoc analysis was performed with Tukey’s test when ANOVA showed significant differences. P<0.05 was considered statistically significant. Statistical analysis was performed using GraphPad Prism (GraphPad, Inc., La Jolla, CA).
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Dr. Virginia Guarani and Dr. J. Wade Harper, Department of Cell Biology, Harvard Medical School for designing and performing the Immunoprecipitation/mass spectrometry experiments. We thank Dr. Jennifer Waters and Dr. Talley Lambert of the Harvard Medical School’s Nikon Imaging Center for their help in quantitative microscopy. We also thank Dr. Vanesa Bijol, Department of Pathology, Brigham and Women’s Hospital for providing de-identified human kidney tissue samples from patients with severe kidney fibrosis. We are thankful to Dr. David Nemazee at the Scripps Research Institute, La Jolla, California, for generously providing us with global PLD4−/− mice as well as PLD4fl/fl mice.
Work in the Vaidya laboratory was supported by the Outstanding New Environmental Sciences award from the National Institutes of Health (NIH)/National Institute of Environmental Health Sciences (NIEHS) (ES017543) and the Innovation in Regulatory Science Award from Burroughs Wellcome Fund (BWF-1012518). S.B. is supported by the Harvard Medical School Center of Excellence in Systems Pharmacology NIH grant P50 GM107618 and the Giovanni Armenise-Harvard Foundation. Liver fibrosis studies from J.P.L. and N.J. were supported by R01 ES017537 from NIH/NIEHS.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016111222/-/DCSupplemental.
References
- 1.Braun L, Sood V, Hogue S, Lieberman B, Copley-Merriman C: High burden and unmet patient needs in chronic kidney disease. Int J Nephrol Renovasc Dis 5: 151–163, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurts C, Panzer U, Anders HJ, Rees AJ: The immune system and kidney disease: Basic concepts and clinical implications. Nat Rev Immunol 13: 738–753, 2013 [DOI] [PubMed] [Google Scholar]
- 3.Eddy AA: Serine proteases, inhibitors and receptors in renal fibrosis. Thromb Haemost 101: 656–664, 2009 [PMC free article] [PubMed] [Google Scholar]
- 4.Craciun FL, Bijol V, Ajay AK, Rao P, Kumar RK, Hutchinson J, Hofmann O, Joshi N, Luyendyk JP, Kusebauch U, Moss CL, Srivastava A, Himmelfarb J, Waikar SS, Moritz RL, Vaidya VS: RNA sequencing identifies novel translational biomarkers of kidney fibrosis. J Am Soc Nephrol 27: 1702–1713, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshikawa F, Banno Y, Otani Y, Yamaguchi Y, Nagakura-Takagi Y, Morita N, Sato Y, Saruta C, Nishibe H, Sadakata T, Shinoda Y, Hayashi K, Mishima Y, Baba H, Furuichi T: Phospholipase D family member 4, a transmembrane glycoprotein with no phospholipase D activity, expression in spleen and early postnatal microglia. PLoS One 5: e13932, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stahelin RV, Ananthanarayanan B, Blatner NR, Singh S, Bruzik KS, Murray D, Cho W: Mechanism of membrane binding of the phospholipase D1 PX domain. J Biol Chem 279: 54918–54926, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Sung TC, Roper RL, Zhang Y, Rudge SA, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht J, Frohman MA: Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. EMBO J 16: 4519–4530, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown HA, Thomas PG, Lindsley CW: Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat Rev Drug Discov 16: 351–367, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Käch A, Ziegler U, Ong AC, Wallace DP, Arcaro A, Serra AL: The role of phospholipase D in modulating the MTOR signaling pathway in polycystic kidney disease. PLoS One 8: e73173, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghim J, Moon JS, Lee CS, Lee J, Song P, Lee A, Jang JH, Kim D, Yoon JH, Koh YJ, Chelakkot C, Kang BJ, Kim JM, Kim KL, Yang YR, Kim Y, Kim SH, Hwang D, Suh PG, Koh GY, Kong YY, Ryu SH: Endothelial deletion of phospholipase D2 reduces hypoxic response and pathological angiogenesis. Arterioscler Thromb Vasc Biol 34: 1697–1703, 2014 [DOI] [PubMed] [Google Scholar]
- 11.Chen Q, Hongu T, Sato T, Zhang Y, Ali W, Cavallo JA, van der Velden A, Tian H, Di Paolo G, Nieswandt B, Kanaho Y, Frohman MA: Key roles for the lipid signaling enzyme phospholipase d1 in the tumor microenvironment during tumor angiogenesis and metastasis. Sci Signal 5: ra79, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehman N, Di Fulvio M, McCray N, Campos I, Tabatabaian F, Gomez-Cambronero J: Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. Blood 108: 3564–3572, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin J, Chou C, Lima M, Zhou D, Zhou X: Systemic sclerosis is a complex disease associated mainly with immune regulatory and inflammatory genes. Open Rheumatol J 8: 29–42, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okada Y, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Kawaguchi T, Stahl EA, Kurreeman FA, Nishida N, Ohmiya H, Myouzen K, Takahashi M, Sawada T, Nishioka Y, Yukioka M, Matsubara T, Wakitani S, Teshima R, Tohma S, Takasugi K, Shimada K, Murasawa A, Honjo S, Matsuo K, Tanaka H, Tajima K, Suzuki T, Iwamoto T, Kawamura Y, Tanii H, Okazaki Y, Sasaki T, Gregersen PK, Padyukov L, Worthington J, Siminovitch KA, Lathrop M, Taniguchi A, Takahashi A, Tokunaga K, Kubo M, Nakamura Y, Kamatani N, Mimori T, Plenge RM, Yamanaka H, Momohara S, Yamada R, Matsuda F, Yamamoto K: Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat Genet 44: 511–516, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Otani Y, Yamaguchi Y, Sato Y, Furuichi T, Ikenaka K, Kitani H, Baba H: PLD$ is involved in phagocytosis of microglia: Expression and localization changes of PLD4 are correlated with activation state of microglia. PLoS One 6: e27544, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K: Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long DA, Woolf AS, Suda T, Yuan HT: Increased renal angiopoietin-1 expression in folic acid-induced nephrotoxicity in mice. J Am Soc Nephrol 12: 2721–2731, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Gerarduzzi C, Kumar RK, Trivedi P, Ajay AK, Iyer A, Boswell S, Hutchinson JN, Waikar SS, Vaidya VS: Silencing SMOC2 ameliorates kidney fibrosis by inhibiting fibroblast to myofibroblast transformation. JCI Insight 2, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kusaba T, Lalli M, Kramann R, Kobayashi A, Humphreys BD: Differentiated kidney epithelial cells repair injured proximal tubule. Proc Natl Acad Sci U S A 111: 1527–1532, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eickelberg O, Pansky A, Koehler E, Bihl M, Tamm M, Hildebrand P, Perruchoud AP, Kashgarian M, Roth M: Molecular mechanisms of TGF-(beta) antagonism by interferon (gamma) and cyclosporine A in lung fibroblasts. FASEB J 15: 797–806, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Su S, Zhao Q, He C, Huang D, Liu J, Chen F, Chen J, Liao JY, Cui X, Zeng Y, Yao H, Su F, Liu Q, Jiang S, Song E: miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. Nat Commun 6: 8523, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verrecchia F, Pessah M, Atfi A, Mauviel A: Tumor necrosis factor-alpha inhibits transforming growth factor-beta /Smad signaling in human dermal fibroblasts via AP-1 activation. J Biol Chem 275: 30226–30231, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Leask A, Abraham DJ: TGF-beta signaling and the fibrotic response. FASEB J 18: 816–827, 2004 [DOI] [PubMed] [Google Scholar]
- 24.Watanabe H, Hattori S, Katsuda S, Nakanishi I, Nagai Y: Human neutrophil elastase: Degradation of basement membrane components and immunolocalization in the tissue. J Biochem 108: 753–759, 1990 [DOI] [PubMed] [Google Scholar]
- 25.Sowa ME, Bennett EJ, Gygi SP, Harper JW: Defining the human deubiquitinating enzyme interaction landscape. Cell 138: 389–403, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ueha S, Shand FH, Matsushima K: Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Front Immunol 3: 71, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao Y, Zhang J, Tan DQ, Chen XY, Ye DF, Peng JP, Li JT, Zheng YQ, Fang L, Li YK, Fan MX: Interferon-gamma improves renal interstitial fibrosis and decreases intrarenal vascular resistance of hydronephrosis in an animal model. Urology 77: 761.e8–761.e13, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Emmez H, Kardes O, Dogulu F, Kurt G, Memis L, Baykaner MK: Role of antifibrotic cytokine interferon-gamma in the prevention of postlaminectomy peridural fibrosis in rats. Neurosurgery 62: 1351–1357, discussion 1357–1358, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Redente EF, Keith RC, Janssen W, Henson PM, Ortiz LA, Downey GP, Bratton DL, Riches DW: Tumor necrosis factor-α accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol 50: 825–837, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baroni GS, D’Ambrosio L, Curto P, Casini A, Mancini R, Jezequel AM, Benedetti A: Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology 23: 1189–1199, 1996 [DOI] [PubMed] [Google Scholar]
- 31.Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg AA, Rojkind M, Böttinger EP: A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev 14: 187–197, 2000 [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg MT, Han YP, Yan C, Shaw MC, Garner WL: TNF-alpha suppresses alpha-smooth muscle actin expression in human dermal fibroblasts: An implication for abnormal wound healing. J Invest Dermatol 127: 2645–2655, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Göbel N, Talke Y, Schweda F, Mack M: CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A 106: 17892–17897, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pott GB, Chan ED, Dinarello CA, Shapiro L: Alpha-1-antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J Leukoc Biol 85: 886–895, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zager RA, Johnson AC, Frostad KB: Rapid renal alpha-1 antitrypsin gene induction in experimental and clinical acute kidney injury. PLoS One 9: e98380, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai YF, Yu HP, Chang WY, Liu FC, Huang ZC, Hwang TL: Sirtinol inhibits neutrophil elastase activity and attenuates lipopolysaccharide-mediated acute lung injury in mice. Sci Rep 5: 8347, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu YK, Liu XD, Sköld CM, Umino T, Wang HJ, Spurzem JR, Kohyama T, Ertl RF, Rennard SI: Synergistic neutrophil elastase-cytokine interaction degrades collagen in three-dimensional culture. Am J Physiol Lung Cell Mol Physiol 281: L868–L878, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Thongboonkerd V, Barati MT, McLeish KR, Benarafa C, Remold-O’Donnell E, Zheng S, Rovin BH, Pierce WM, Epstein PN, Klein JB: Alterations in the renal elastin-elastase system in type 1 diabetic nephropathy identified by proteomic analysis. J Am Soc Nephrol 15: 650–662, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Brodeur GM: Neuroblastoma: Biological insights into a clinical enigma. Nat Rev Cancer 3: 203–216, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Meakin SO, MacDonald JI, Gryz EA, Kubu CJ, Verdi JM: The signaling adapter FRS-2 competes with Shc for binding to the nerve growth factor receptor TrkA. A model for discriminating proliferation and differentiation. J Biol Chem 274: 9861–9870, 1999 [DOI] [PubMed] [Google Scholar]
- 41.Fragiadaki M, Hill N, Hewitt R, Bou-Gharios G, Cook T, Tam FW, Domin J, Mason RM: Hyperglycemia causes renal cell damage via CCN2-induced activation of the TrkA receptor: Implications for diabetic nephropathy. Diabetes 61: 2280–2288, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song YW, Zhang T, Wang WB: Gluococorticoid could influence extracellular matrix synthesis through Sox9 via p38 MAPK pathway. Rheumatol Int 32: 3669–3673, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD: MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol 46: 380–388, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pat B, Yang T, Kong C, Watters D, Johnson DW, Gobe G: Activation of ERK in renal fibrosis after unilateral ureteral obstruction: Modulation by antioxidants. Kidney Int 67: 931–943, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Stambe C, Atkins RC, Tesch GH, Masaki T, Schreiner GF, Nikolic-Paterson DJ: The role of p38alpha mitogen-activated protein kinase activation in renal fibrosis. J Am Soc Nephrol 15: 370–379, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Dabbagh K, Laurent GJ, Shock A, Leoni P, Papakrivopoulou J, Chambers RC: Alpha-1-antitrypsin stimulates fibroblast proliferation and procollagen production and activates classical MAP kinase signalling pathways. J Cell Physiol 186: 73–81, 2001 [DOI] [PubMed] [Google Scholar]
- 47.Ajay AK, Kim TM, Ramirez-Gonzalez V, Park PJ, Frank DA, Vaidya VS: A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J Am Soc Nephrol 25: 105–118, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cárdenas-González M, Osorio-Yáñez C, Gaspar-Ramírez O, Pavković M, Ochoa-Martínez A, López-Ventura D, Medeiros M, Barbier OC, Pérez-Maldonado IN, Sabbisetti VS, Bonventre JV, Vaidya VS: Environmental exposure to arsenic and chromium in children is associated with kidney injury molecule-1. Environ Res 150: 653–662, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jadhav S, Ajay AK, Trivedi P, Seematti J, Pellegrini K, Craciun F, Vaidya VS: RNA-binding protein musashi homologue 1 regulates kidney fibrosis by translational inhibition of p21 and numb mRNA. J Biol Chem 291: 14085–14094, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Behrends C, Sowa ME, Gygi SP, Harper JW: Network organization of the human autophagy system. Nature 466: 68–76, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guarani V, McNeill EM, Paulo JA, Huttlin EL, Fröhlich F, Gygi SP, Van Vactor D, Harper JW: QIL1 is a novel mitochondrial protein required for MICOS complex stability and cristae morphology. eLife 4: e06265, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martina MN, Bandapalle S, Rabb H, Hamad AR: Isolation of double negative αβ T cells from the kidney. J Vis Exp 87: e51192, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.