

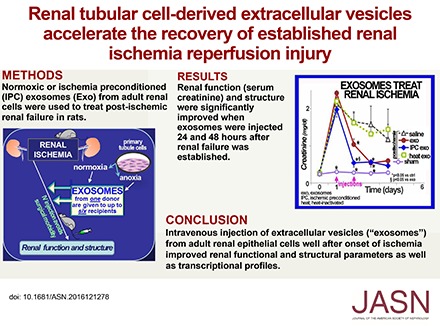
Abstract
Ischemic renal injury is a complex syndrome; multiple cellular abnormalities cause accelerating cycles of inflammation, cellular damage, and sustained local ischemia. There is no single therapy that effectively resolves the renal damage after ischemia. However, infusions of normal adult rat renal cells have been a successful therapy in several rat renal failure models. The sustained broad renal benefit achieved by relatively few donor cells led to the hypothesis that extracellular vesicles (EV, largely exosomes) derived from these cells are the therapeutic effector in situ. We now show that EV from adult rat renal tubular cells significantly improved renal function when administered intravenously 24 and 48 hours after renal ischemia in rats. Additionally, EV treatment significantly improved renal tubular damage, 4-hydroxynanoneal adduct formation, neutrophil infiltration, fibrosis, and microvascular pruning. EV therapy also markedly reduced the large renal transcriptome drift observed after ischemia. These data show the potential utility of EV to limit severe renal ischemic injury after the occurrence.
Keywords: hypoxia, acute renal failure, cell survival, mRNA, exosomes
Renal ischemia causes and frequently worsens existing AKI1 and CKD.2 AKI is morbid, potentially lethal,3–9 and an emergent medical problem.10 Unfortunately, there is no effective therapy for AKI.11 The medical failure to affect ischemic AKI is not surprising given its unpredictability and inherent complexity.12,13 Therefore, it is unlikely that this broad organ malfunction can be corrected by targeting single dysfunctional processes.13 The realization that renal ischemia also aggravates progressive CKD, regardless of its initial cause, is an additional impetus in the search for solutions.2,14,15 In this report, we describe the use of renal cell extracellular vesicles (EV) to treat ischemic renal injury in rats. We derived this work from prior experimentation that showed improved renal function and structure by relatively small numbers of infused renal cells.16–20 We suggest that the beneficial effect of those donor cells was due to their EV, and tested the hypothesis that renal EV can protect ischemic kidneys.
Ischemic preconditioning (IPC) is a highly conserved adaptation in which transient, sublethal ischemia protects from subsequent ischemic injury.21–25 However, translating the powerful effects of IPC to treat unpredictable renal ischemia is not feasible.26 It is not known how IPC confers protection, although an important clue is that IPC can be transferred by the blood of IPC donors,27,28 i.e., remote IPC (RIPC). One possible mechanism of RIPC transfer is by EV, released membrane-bound nanovesicles that mediate cell-to-cell communication and convey the distinctive state of originating cells.29–31 In this work, we show that EV from adult rat renal tubular cells can be delivered by peripheral vein to successfully treat rats with established severe ischemia/reperfusion AKI. Furthermore, EV from hypoxia preconditioned (IPC) cells are more effective, providing greater improvements in renal phenotype and genotype than EV from normoxic cells.
Results
EV
We employed two sets of EV (exosomes) derived from the same primary rat renal tubular cells: one set maintained in normoxic conditions (EXO) and the other exposed to 1% O2 hypoxia for 4 hours before collection of EV (IPC EXO). Cell number and EV protein content, size, and vesicle number were not different between EXO and IPC EXO (Table 1). Nano tracker analysis (Supplemental Figure 1) and electron microscopy showed similar size distribution between EV groups. The EV contained mRNAs encoding the male determinant SRY gene as well as the transfected serum amyloidA1 transcript. These were used to track EV in recipient kidneys. In addition, EV tracking was achieved by labeling a 10% fraction of EV with red Exo-Glow. This maneuver showed the exosomes broadly distributed within the kidney (Figure 1). Cell culture studies demonstrated that EV were rapidly taken up by proximal tubular epithelia; IPC EXO more than EXO (Supplemental Figure 2). This result is consistent with higher levels of adherent intercellular cell adhesion molecule1 (ICAM1),32 which reportedly promotes EV cell fusion33 (Figure 1).
Table 1.
Exosome parameters
| Parameter | EXO | IPC EXO | P Value |
|---|---|---|---|
| Cells/flask | 12.9×106±7×105 | 13.4×106±5×105 | NS |
| Protein/flask, μg | 70±7 | 91±29 | NS |
| Vesicle number/flask | 1.2±0.1×107 | 1.1±0.07×107 | NS |
| Mean EV size, nm | 100±3.94 | 106±3.78 | NS |
Data are mean±SEM. EV derived from normoxic (EXO) and IPC renal cells were comparable in size and number.
Figure 1.

Donor EV in recipient kidneys. (A and B) ICAM1 and CD63 protein levels in normoxic EV (EXO) and IPC EV (IPC EXO), measured on western blot, n=5. ICAM1 was higher (P=0.001) and CD63 lower (P<0.01) in IPC EXO. (C) Electron microscopy showed IPC EXO and EXO (not shown) averaged <200 nM diameter. (D) RT-PCR was used to identify the SRY male determining gene in EV (EXO) from male donor cells (positive control), and also in kidneys of female EV recipients (IPC EXO and EXO), but not in female kidneys of rats injected with saline (saline). Additional SRY-positive controls were amplified from normal male rat kidneys. (E) Representative image of three rat kidneys used to localize IPC EV labeled with red EXO-Glow. A 10% fraction of IPC EV was labeled, remixed with the unlabeled 90%, injected, and visualized by fluorescent microscopy in 50-μM vibratome sections. (F) RT-PCR was used to identify the murine SAA1 gene amplified in donor EV derived from cells transfected with SAA1 (EXO), and also in recipient kidneys of rats injected with EXO and IPC EXO (EXO and IPC EXO), but not in kidneys of rats injected with saline (saline). The negative and positive controls were amplifications of pCDNA1 and SAA1-pCDNA1 plasmids used for transfection. Ctrl, control; MW, molecular weight; pos, positive.
Renal Function
Four groups of five Sprague Dawley (SD) rats were studied: sham surgery (controls), bilateral renal ischemia injected with saline vehicle (untreated ischemia), or with EV from renal cells cultured under normoxic conditions (EXO), or with EV from posthypoxic kidney cells (IPC EXO). The sample size was determined by power analysis.34 The goal was to treat after the injury and EV or saline vehicle were given intravenously 24 and 48 hours postischemia. There was a rapid and similar increase in serum creatinine in all ischemic groups after 24 hours, and it decreased slowly in the ischemic rats given saline (Figure 2). In contrast, creatinine improved rapidly in ischemic rats injected with EXO, and the recovery was even faster in rats treated with IPC EXO (P<0.02 versus EXO). Creatinine levels in sham-operated rats remained within normal limits (Figure 2). Similarly, serum urea nitrogen demonstrated accelerated recovery in the postischemic rats treated with EXO or IPC EXO (not shown). Untreated ischemic rats (saline) also had abnormally large kidneys, and EV, particularly IPC EXO (P<0.01), significantly limited organomegaly, likely a reflection of improved inflammation and fibrosis. No mortality was observed.
Figure 2.

Improved renal function and kidney weight after treatment with EV. Left: Serum creatinine levels in the four groups of rats, n=5: Sham (open squares), untreated ischemic (filled triangles, saline), ischemic treated with normoxic EV (filled circles, EXO), and ischemic treated with IPC EV (filled diamonds, IPC EXO). Time zero designates day of ischemia/reperfusion; EV or saline were injected 1 and 2 days postischemia. (*) Next to sham symbols indicate significantly different than all others (P<0.05). (*) Next to IPC EXO symbols indicate significant difference from saline (P<0.05), and (§) indicates significantly different from EXO (P<0.05). (*) Next to EXO symbols designate significant difference from saline (P<0.05). Right: Kidney weights corrected for body weight. (*) On saline (untreated ischemia) bar indicates significantly different than all others. (*) On EXO and IPC EXO indicate significantly different than sham (P<0.05).
Renal Phenotype and Genotype
Untreated renal ischemia caused severe disruption of renal architecture, including tubular congestion and simplification, intratubular obstruction with casts, and inflammation (as compared with sham-operated controls, Figure 3A). In contrast, injection of EXO, and more so IPC EXO, limited each of these histologic abnormalities (Supplemental Figure 3). Heat-inactivated EV were ineffective in limiting postischemic injury (Supplemental Table 1), consistent with a role of EV RNA or specific proteins in the protection observed. Thus, comprehensive genetic profiling was performed and revealed significant activation of both pro- and antiapoptotic transcripts in untreated postischemic kidneys, when compared with sham and IPC EXO groups (Figure 3). Renal injury in untreated ischemia also included enhanced cellular proliferation, as indicated by higher protein levels of proliferative cell nuclear antigen (PCNA, a cell proliferation marker), as reported.35 Treatment with EXO or IPC EXO limited the proliferative postischemic response (Supplemental Figure 4). Although tubular cell proliferation contributes to recovery, proliferation of leukocytes and fibroblasts is detrimental,36 thus lower proliferation in the treated groups is likely a manifestation of decreased tubular injury.
Figure 3.

Improved renal structure after treatment with EV. (A) Representative images of renal sections stained with PAS. Left to right: sham rats, ischemic untreated rats (saline), ischemic rats treated with normoxic EV (EXO), and ischemic rats treated with IPC EV (IPC EXO). Tubular damage was widespread in untreated ischemia, including cystic-like tubular dilation. These changes were partly corrected by EXO, and even more by IPC EXO. In the bar diagram is shown the mean fraction of damaged tubules per field in each group: sham (n=20) and saline groups (n=20) were significantly different (P=2.03×10−16); sham and EXO (n=20; P=1.27−12) and sham and IPC EXO (n=20; 8.92−10) were also significantly different; EXO was significantly higher than IPC EXO (P=1.37−08). (*) Represents significantly different from sham, and (§) indicates significant difference between IPC EXO and EXO, P<0.05 for both. Specific renal proapoptotic transcripts were significantly activated by untreated ischemia (NS) when compared with sham or IPC EV–treated ischemic rats (IPC) (P<0.05). (B) Representative images of renal HNE. Heavy formation of HNE adducts was seen in untreated ischemic (saline) rats, whereas it was nearly undetectable in sham rats. Treatment with normoxic EV (exosome) or IPC EV (IPC EXO) limited the formation of HNE adducts. The bar diagram shows that HNE formation in the saline group (n=13) was significantly higher than in sham (n=5; P=2.74×10−6); sham and EXO (n=5) groups were also significantly different (P=0.02), but sham and IPC EXO (n=6) groups were not (P=0.05). EXO (P<0.01) and EXO IPC groups (P<0.001) were lower than the untreated ischemic group, whereas the IPC EXO group was lower than the EXO group (P<0.05). (*) Significantly different than sham group, P<0.05. Specific redox-related gene transcripts were reduced in the untreated ischemia group with respect to sham and also IPC EXO groups, indicating partial restoration of redox gene expression by IPC EXO treatment. In contrast, the pro-oxidant gene Olr (or Lox1) was activated in ischemia with respect to sham, and it was reduced by IPC EXO treatment. (C) Representative images of renal sections labeled with anti-neutrophil antibody. Large congregations of neutrophils were detected in the interstitial and intraluminal spaces of kidneys from untreated ischemic rats (saline), whereas were barely seen in kidneys of sham rats. In contrast, ischemic rats treated with EXO or IPC EXO had much lower numbers of neutrophils. The bar diagram shows mean neutrophil number per 400× field (PMN/hpf) in sham rats (sham, n=27 fields), untreated ischemic (saline, n=47), ischemic rats treated with normoxic EV (EXO, n=31), and ischemic rats treated with IPC EV (IPC EXO, n=32). Sham and saline groups were significantly different (P=1.94×10−5), sham and EXO groups were also significantly different (P=1.05×10−5), but sham and IPC EXO groups were not (P=0.65). In addition, EXO (P=8.74×10−5) and IPC EXO (P=2.72×10−5) were significantly lower than saline, and IPC EXO was significantly lower than EXO (P<0.001). Proinflammatory renal transcripts, activated in untreated ischemia and compared with sham rats or IPC-treated ischemic rats by fold change, include cytokines, chemokines, complement components, and adhesion molecules. In most cases, IPC EXO partly corrected the activation induced by ischemia (NS). (D) Glomerular complement C3 expression in sham rats was nearly undetectable, but it was heavily expressed in untreated ischemic rat kidneys. Treatment with EXO and IPC EXO limited C3 expression in ischemic rats. The bar diagram shows average pixel density of C3 immunofluorescence per 400× power field in sham rats (sham, n=24 fields), untreated ischemic (saline, n=16), ischemic rats treated with normoxic EV (EXO, n=8), and ischemic rats treated with IPC EV (IPC EXO, n=24). Saline was higher than sham group (P=5.97×10−26); sham and EXO were also significantly different (P=1.13×10−14), as well as sham and IPC EXO (P=1.78×10−8). In addition, EXO (P<0.001) and IPC EXO (9.43×10−10) were significantly lower than saline, and IPC EXO was significantly lower than EXO (P<0.01). (*) Significantly different than sham group, P<0.05; (§) indicates significant difference between IPC EXO and EXO, P<0.05. (E) Representative images of renal sections stained with Masson’s trichrome. In untreated ischemia (saline), interstitial renal fibrosis (blue) was extensive and replaced normal epithelia (red) when compared with controls (sham). Treatment of ischemia with normoxic EV (exosome) or IPC EV (IPC EXO) limited fibrosis. The bar diagram shows average fractional fibrosis in control (sham, n=16), untreated ischemia (saline, n=20), ischemia treated with normoxic EV (EXO, n=20), and ischemia treated with IPC EV (IPC EXO, n=20). Sham and saline were significantly different (P=2.94×10−6), sham and EXO were also significantly different (P<0.01), but sham and IPC EXO were not (P=0.08). In addition, EXO (P<0.001) and IPC EXO (P=3.31×10−6) were significantly lower than saline, and IPC EXO was significantly lower than EXO (P<0.02). (*) Significantly different than sham, P<0.05; (§) indicates significant difference between IPC EXO and EXO, P<0.05. Untreated ischemia (NS) activated a suite of profibrotic transcripts and the fold change among NS and sham and IPC EV (IPC EXO) is shown. These activated transcripts encoded extracellular matrix proteins, three members of the TGFb family, metalloproteinases, inhibitors of metalloproteinases, transcription factors, and other genes linked to renal fibrosis. (F) Representative images of renal peritubular microvasculature labeled with DyLight 594 conjugated Lycopersicon Esculentum (tomato lectin, LEL, red). The microvasculature was far less dense in untreated ischemia (saline, n=92) than in sham (n=62). However, treatment with normoxic EV (EXO, n=53) or IPC EV (IPC EXO, n=81) preserved microvascular networks in ischemic kidneys. The bar diagram shows the average fractional (fluorescence) positivity; sham and saline were significantly different (P=2.03−16), EXO (P=1.27−12) and IPC EXO (P=8.92−10) were also significantly lower than sham. However, EXO (P=8.81−11) and IPC EXO (P=7.80−17) were also significantly higher than saline, whereas IPC EXO was significantly higher than EXO (P=1.37−08). (*) Significantly different than sham, P<0.05; (§) indicates significant difference between IPC EXO and EXO, P<0.05. Untreated ischemia (NS) inhibited key proangiogenic transcripts, but also activated others—Ang, Adra, Cyr61, as well as antiangiogenic Thbs1. The significant (P<0.05) fold change among NS and sham and IPC EV (IPC EXO) is shown. Hpf, high power field; PAS, periodic acid Schiff; PMN, polymorphonuclear; V, versus.
The role of EV on oxidant stress induced by ischemia was examined in sections stained for 4-hydroxynonenal adducts (HNE).37 Whereas renal HNE formation was nearly undetectable in sham rats, strong HNE formation existed in untreated ischemic rats (Figure 3B); EXO and IPC EXO significantly limited the HNE labeling. Analysis of specific redox transcripts indicates that ischemia alters redox balance as compared with either sham or IPC EXO groups. Thus, treatment with EV from preconditioned cells (IPC EXO) restores renal genetic profiles to near that of nonischemic (sham) animals.
Renal inflammation complicates renal ischemia38 and, among the aggregates of renal inflammatory cells, short-lived neutrophils32 point to ongoing inflammation.38 Neutrophils in sham kidneys were rare, whereas kidneys from untreated ischemic rats contained large numbers of neutrophils (Figure 3C). In contrast, EXO and IPC EXO curtailed renal neutrophil accumulation postischemia. Another sensitive indicator of inflammation postischemia is upregulation of renal complement components.39 In Figure 3D, we show that glomerular C3 immuno-reactivity, near background in sham operated rats, was markedly increased in untreated ischemia. In contrast, EXO, and particularly IPC EXO, attenuated C3 protein expression. Moreover, the renal transcriptome in ischemia revealed broader activation, including C2, C3, and C4, with attenuation of the membrane attack complex inhibitor CD55. We also found activation of mRNAs encoding proinflammatory chemokines, cytokines, and adhesion molecules. These responses were suppressed by EXO and IPC EXO treatment, with significantly lower levels of proinflammatory transcripts in the IPC EXO group.
Substantial peritubular fibrosis followed renal ischemia within 6 days (Figure 3E). Treatment with EXO significantly limited the broad activation of profibrotic genes and decreased renal fibrosis. Infusions of IPC EXO were even more effective at limiting fibrosis in postischemic kidneys. It is noteworthy that ischemia upregulated genes that break down extracellular matrix, including MMP2, 7, 11, and 14, but also the MMP inhibitor TIMP1. IPC EXO limited the amplitude of these ischemic responses.
Renal ischemia severely compromised the renal microvasculature, consistent with prior observations.40 Renal sections, labeled with Lycopersicon Esculentum lectin (LEL), showed severe loss of capillaries, or of their lectin epitopes, in kidneys from untreated postischemic rats (Figure 3F). This vascular rarefication was prevented, in part, by EXO, and, for the most part, by IPC EXO. The renal genotype revealed that ischemia suppressed angiogenesis, in that angiostatic genes were activated whereas proangiogenic genes were inhibited. These antiangiogenic effects were antagonized by IPC EXO. Although renal ischemia severely decreased glomerular number (2.49±0.2/hpf versus 5.58±0.2 in sham), glomeruli were relatively preserved after EV treatment (3.46±0.2 in EXO; 4.12±0.2 in IPC EXO; P<0.001 versus untreated ischemia).
Western Blots
Three groups of proteins were measured on immunoblots; catalase (CAT) and SOD1 are constituents of the antioxidant system. Components of the hypoxia inducible factor (HIF) pathway, vascular endothelial growth factor (VEGF), glucose transporter1 (GLUT1 or Slc2a1), and erythropoietin (EPO), were included. Heat shock protein 27 (Hsp27) was included as a presumptive reno-protective protein. At 6 days postischemia, levels of CAT, SOD1, VEGF, EPO, and GLUT1 were reduced in untreated ischemic rats. However, treatment with IPC EXO largely prevented loss of these proteins. On the other hand, protein levels of renal HIF were not different 6 days postischemia (not shown). In contrast, Hsp27 protein levels were elevated postischemia, and IPC EXO treatment prevented Hsp27 upregulation (Supplemental Figure 5). Consistent with histologic and RNAseq data, immunoblots also showed increases in TGFb1, fas, and BIRC3 postischemia; these were all prevented with EV treatment (Supplemental Figure 6).
mRNA Pathways
Sequencing by RNAseq identified 12,159 renal transcripts. There were 3141 renal genes significantly altered in untreated ischemia as compared with the normal (sham) group. The number of altered genes was reduced to 1599 in the EXO-treated postischemic group, and IPC EXO further reduced the number to 71 altered transcripts, or only 2% of those altered in the untreated ischemic group. Hence, renal ischemia markedly altered the normal renal transcriptome. However, EXO treatment partly corrected and IPC EXO nearly fully corrected the large drift caused by ischemia. These effects can be visualized on a multidimensional scaling plot which reports the distances, or the inverse of similarities, among the groups (Figure 4). Distances on the multidimensional scaling plot correspond to leading log–fold changes between each pair of samples. The leading log–fold change is the average (root mean square) of the largest absolute log–fold changes between each pair of samples. The dimensions on the plot are leading log–fold changes, and demonstrate the genetic drift postischemia and its almost complete correction by IPC EXO. It is also noted that significant differences in gene expression among the four groups were determined by the false discovery rate (FDR<0.05), and only those genes that met this cutoff were included.
Figure 4.

Correction of transcriptome alterations after treatment with EV. Top, significantly altered genes with respect to control (sham) in untreated ischemia (saline = 3141 altered genes), treatment with normoxic EV (EXO = 1599 altered genes), and treatment with IPC EV (IPC EXO = 71 altered genes). A total of 12,159 transcripts were sequenced and identified per group. Bottom, multidimensional scaling plot, where distances correspond to leading log–fold changes between each pair of samples. The leading log–fold change is the average (root mean square) of the largest absolute log–fold changes between each pair of samples. The larger shapes represent the mean for the transcriptomes in each group and the smaller shapes represent individual rat transcriptomes. Filled diamonds = sham; gray triangles = untreated ischemic; striped circles = ischemic treated with normoxic, or neutral, EV; and speckled squares = treated with IPC EV. FC, fold change; Vs, versus.
All transcriptomes were then used for pathway analysis with WebGestalt (Vanderbilt University, Nashville, TN), and the generated data sets were incorporated into well defined KEGG pathways (http://www.genome.jp/kegg/pathway.html; Kyoto University, Japan). The pathways altered postischemia included, in descending order of statistical significance, metabolic, cell proliferation, focal adhesion, solute absorption, cell cycle, extracellular matrix receptor interaction, oxidative phosphorylation, glutathione metabolism, phagosome, apoptosis, cytokine-cytokine receptor interaction, and DNA replication (Supplemental Table 2). Compared with sham, renal ischemia caused the largest changes in metabolic pathways. Specifically, the largest number of altered transcripts were involved in metabolism and achieved the highest statistical significance. Treatment with IPC EXO effectively corrected the perturbations in metabolic gene expression such that expression was similar to sham (P>0.05). Postischemia, renal transcripts were partly normalized by IPC EXO treatment; 1526 were significantly higher and 1593 lower than in untreated rats. A subset of these transcripts was also corrected by EXO treatment. In analyzing the transcripts protected by both IPC EXO and EXO treatment we found 39 antiapoptotic and 29 anti-inflammatory transcripts. The most significant KEGG pathway was metabolic and by GO analysis it was transporter activity (Supplemental Figure 7).
RNAseq was used to measure 312 EV transcripts in RNA extracted from EXO and IPC EXO. There were 24 differentially expressed transcripts between EXO and IPC EXO (Supplemental Table 3). Ribosomal RNAs were abundant in renal EV, as shown by others.41 mRNAs encoding cytoplasmic ribosomal proteins Rps6 (2.3-fold) and Rps13 (1.3-fold) were also upregulated in IPC EXO. In contrast, mRNAs for structural Rn28S (0.28) and Rn45S (0.30) appeared suppressed (Supplemental Table 3). IPC EXO also carried higher mRNA levels for Drd4 (78-fold increased), a gene that protects from ischemic injury,42 as well as Lrrc56 (16-fold increased), and Ptdss2 (eight-fold increased). Interestingly, Ptdss enhances recovery from ischemic injury.43 IPC EXO also contained higher levels of Eps8l2 (4.5-fold increased), a transcript involved in actin remodeling.44 Phrf1, a transcript that participates in RNA processing,45 was increased 3.9-fold. Ugt2b1 and Ugt2b10, genes that encode detoxification enzymes reportedly suppressed by hypoxia,46 were each increased 3.2-fold. Rassf7, a transcript activated by hypoxia that regulates microtubule dynamics,47 was increased 2.9-fold. Although our EV mRNA compendium is not complete and its contents likely reflect only a portion of the entire renal EV library,41 we detected significant differences between IPC EXO and EXO among the 312 EV transcripts measured. The most significantly altered transcripts detected by Kegg pathway analysis were associated with the ribosome whereas GO analysis detected alterations in RNA processing (Supplemental Figure 8).
Discussion
Ischemic injury to the kidney and other organs can be deadly. Even with subsequent recovery, AKI is still linked to short- and long-term mortality.7,48 Chronic/recurrent renal ischemia also promotes the progression of CKD to ESRD.2,14,15 Moreover, ischemic AKI is common in transplanted kidneys, and is the main cause of delayed graft function.49,50
Unfortunately, there is no specific therapy for ischemic AKI, and supportive care has not improved outcomes.51 Thus, new approaches to AKI are needed. Animal and human data are consistent, showing that no single pathogenic event can be targeted to completely halt ischemic renal injury. Instead, data suggest that multiple abnormalities are intertwined in an expanding cycle of ischemia and inflammation.52
To provide a multifaceted therapy, we have transplanted adult renal tubular cells in experimental AKI and CKD.16,17,19,20 The advantages of organ-specific cells for repair have been documented.53–55 These studies led to the hypothesis that the relatively small number of donor cells infused17,20 acted at a distance on endogenous cells. Accordingly, we described the presence of growth factors and SAA1 protein in conditioned medium of donor cells.20 This report is consistent with data of others using conditioned media.56–65 We further hypothesized that exosomes or EV released from transplanted cells, and in conditioned medium, serve as a therapeutic agent that improves kidney function. Furthermore, we postulated that EV communicate an “IPC state.” Unfortunately, translating the powerful effect of IPC to the clinical arena is not feasible, because most ischemia is unpredictable, and inducing ischemia in a vital organ to prevent possible future injury is unethical.66 There are dozens of conflicting RIPC human trials, using diverse strategies (ClinicalTrials.gov). However, not knowing the mechanism of RIPC hinders design and implementation.24 Here, we tested the hypothesis that EV transfer is one mechanism of RIPC.
There are several points to be emphasized: Ischemic damage was marked by oxidative stress footprints as well as renal neutrophil infiltration and complement activation, likely in situ. Within 6 days, extensive fibrosis and loss of peritubular capillaries40 and glomeruli were seen. These postischemic changes were largely abrogated by EXO treatment, and greater improvements were seen with IPC EXO in virtually all parameters evaluated. Additionally, when the two groups, EXO and IPC EXO, are taken together (ten rats), the comparison strongly indicates that exosomes are superior to either vehicle (saline) or heat-inactivated exosomes postischemia. We acknowledge similarities in EXO and IPC EXO EV protection, suggesting that EXO EV were derived from cells that might have had some degree of hypoxic stress in normal culture. Multilayered ischemic damage was also manifested by suppressed antioxidant and HIF pathway proteins which were improved with IPC EXO treatment. It is also noteworthy that EV limited ischemia-induced activation of renal HSP27, suggesting that some stress responses were not required after EV treatment. It is remarkable that EV administration was successful well after renal ischemia, indicating that EV infusions are potentially therapeutic. This latent period also suggests that endogenous renal mechanisms allow survival for a time before treatment.
The renal transcriptome analysis revealed a major drift in transcriptomes postischemia away from normal (sham), whereas IPC EV largely contained the drift. Gene pathway analysis revealed drastic modifications by ischemia whereas IPC EXO prevented most alterations. These findings are consistent with a state of mixed renal dysfunction and dormancy/“stunning” for up to 24 hours after severe ischemia, which is partly resolved by IPC EXO. We presume that structural and cargo differences between EXO and IPC EXO might account for what may be differences in activity, i.e., due to greater membrane fusion potential, via ICAM1, or to transcripts that stabilized dormant ischemic cells in the IPC EXO group. We hypothesize that abundant ribosomal transcripts in IPC EXO and EXO might facilitate the transition from dormancy to restoration of function by reversing hypoxia-dependent inhibition of protein synthesis.67 In summary, treatment with EV from adult renal cells applied well after renal ischemia improved multiple parameters of structure, function, and transcriptome profile.
Concise Methods
Primary Renal Tubular Cells and EV
Primary renal tubular cells were obtained from male SD rats (Charles River, Wilmington, MA). All experiments were conducted in conformity with the “National Institutes of Health guide for the care and use of laboratory animals.” The rats were euthanized by removing both kidneys under general anesthesia. The kidney cortices were minced in S1 medium (below), and digested with type 4 collagenase (Worthington, Lakewood, NJ), 6 mg/dl, at 37°C in 38% O2 and 5% CO2 for 50 minutes. The renal tubules were then separated by percoll gradient,17 cultured for 3 days, and the tubular cells, now in monolayers, were transfected with pcDNA3.1-SAA1 (2 μg/ml) plasmid. pcDNA3.1-SAA1 was manufactured and sequenced in our laboratory, and its construction was previously reported.16,17 This plasmid is used because tubulogenic SAA1 can be used to track transfer to host kidney cells19 from donor EV (Figure 1). In addition, male determinant gene SRY was used to track donor male EV in recipient female kidneys. Transfection was performed with lipofectamine 2000 (Life Technologies, Grand Island, NY); efficiencies were >70%.19 The transfected cells were cultured in S1 medium: Each 2 L contains F-12 HAM, 10.7 g; DMEM, 8.32 g; L-glutamine, 0.29 g; HEPES, 4.78 g; sodium selenite, 1.7 μg; sodium pyruvate, 0.110 g; 3.2 ml phenol red and pH was adjusted to 7.4 with sodium bicarbonate (Sigma). S1 medium was supplemented with hepatocyte growth factor, 200 ng/ml, and EGF, 400 ng/ml (R&D Systems, Minneapolis, MN). The medium also contained hydrocortisone, 100 μg/ml; insulin, 35 μg/ml; transferrin, 32 μg/ml; sodium selenite, 42 ng/ml (Sigma, St. Louis, MO), with 10% FCS. The following day, renal tubular cells were divided into two groups: one group was placed in a hypoxic chamber (SCI-tive Dual; Baker Ruskinn, Sanford MA), 1% O2/5% CO2, and the other continued in 38% O2/5% CO2 for 4 more hours. The culture medium of both groups was then changed to oxygenated S1 medium with 10% FCS previously depleted of EV by 100,000 × g centrifugation twice for 90 minutes. The cells were cultured for 3 more days, and cell medium collected for EV isolation. The time periods of hypoxia and exosome collection were chosen on the basis of pilot studies in which survival of hypoxic cultured cells and exosome number were maximized. The medium was subjected to sequential centrifugation, 300 × g for 10 minutes, recovering supernatant; 2000 × g for 10 minutes, recovering supernatant; 10,000 × g for 30 minutes, recovering supernatant; and then 100,000 × g for 70 minutes twice, recovering the pellet. The EV pellet was suspended in 500 μl of PBS, and filtered through a 1.0 μM filter and then a 0.2 μM filter. The EV protein was measured with the Pierce Coomassie Plus (Bradford) Assay Kit (23236; Thermo Fisher Scientific). The EV were characterized by electron microscopy and by detecting EV CD63 on immunoblots (Figure 1).30 Nanotrack analysis was performed using ZetaView (Particle Metrix, Mebane, NC) according to the supplier’s instructions. A third group of EV consisted of IPC EV heated at 60°C for 45 minutes to inactivate mRNA.
Animal Protocols
At 8 weeks of age, female SD rats were anesthetized with inhaled isoflurane (0.5%–1%) and placed on a homeothermic table to maintain a body core temperature at approximately 37°C. In three groups of rats (below) both renal pedicles were occluded for 50 minutes with microaneurysm clamps, as described.16–18 In a fourth rat group, sham rats had an identical surgical procedure except that the renal pedicles were not clamped. The rats received EV 24 and 48 hours (equivalent quantities [100 μg] protein in 0.5 ml of sterile physiologic saline) after surgery via the tail vein. Blood samples were obtained daily from the saphenous or tail veins. Two doses were given to increase efficacy because we hypothesize that exosome release from engrafted cell transplants occurred over time on the basis of prior studies.16–20 Euthanasia occurred 6 days after ischemia: after ensuring adequate anesthesia with isoflurane, organs were removed and fixed in 3.8% paraformaldehyde. A fifth group was subjected to ischemia (as above) and received heat-inactivated EV at 24 and 48 hours after surgery.
Histology and Immunohistochemistry
Kidneys were fixed in 3.8% paraformaldehyde, paraffin embedded, and 5-μM sections obtained for Masson’s trichrome to stain connective tissue, and periodic acid Schiff (PAS) to image cellular morphology. The percentage of tubules in the outer medulla that showed epithelial necrosis, luminal debris, and/or tubular dilation was estimated on coded sections without the knowledge of the experimental group to which the animals belonged.68 Further quantification of congestion, intratubular obstruction (casts), inflammation, and tubule simplification was graded in cortex and medulla on the following scale: 0 = none, 1+ = <10%, 2+ = 10%–25%, 3+ = 26%–75%, 4+ = >75%.68 Renal neutrophils were visualized with primary anti-rat neutrophil rabbit antibody (ABIN2586050; antibodies-online.com) and secondary antibody with HRP tag (SK-4805; Vector, Burlingame, CA). The neutrophils were counted in blinded renal sections. The areas of glomerular and peritubular fibrosis were quantified using Metamorph imaging image processing software (Sunnyvale, CA) and expressed as fractional areas per 200× microscope field, covering all available surfaces in all coded kidneys. Atrophic tubules were counted in blinded PAS-stained sections. Renal sections were immunostained for HNE to estimate tissue peroxidation. The primary HNE rabbit antibody was applied at 1:300 dilution (ABIN873270; antibodies-online.com). Horse anti-rabbit antibody was used to develop: ImmPRESS HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit (MP-7401; Vector). Additional kidney sections made to visualize the microvasculature by immunofluorescence: kidneys were immersion-fixed in 3.8% paraformaldehyde, followed by cryoprotection in 20% sucrose/PBS, and mounting in optimal cutting temperature compound. Serial cross-sections (10 μm thick) were cut on a cryostat and mounted, and fixed to slides in 100% methanol at −20°C for 10 minutes and PBS-washed twice, followed by treatment with 0.3% Triton X-100 for 5 minutes and then PBS-washed twice. Sections were incubated with Lycopersicon Esculentum (Tomato) Lectin, conjugated with Alexa Fluor 594 (DL-1177; Vector) at 1:300, overnight at 4°C. DAPI (Molecular Probes, Eugene, OR) was applied at 300 nM for 5 minutes at room temperature. Images were taken through an inverted microscope (Zeiss Axio Imager.D2, Oberkochen, Germany). Positive fluorescence was quantified from >10 sections per animal by Aperio’s Positive Pixel Count algorithm (Aperio Technologies, Inc., Vista, CA). Negative controls for all targets were analyzed to ensure that the algorithm detected minimal false-positives and autofluorescence. Microvascular density was expressed as fractional positivity (positive pixels/total pixels on each of 400× fields per rat kidney).
Immuno (Western) Blotting
Kidney cortices were homogenized in 25 mM Tris, pH 7.6, 150 mM NaCl, 1% deoxycholate, 1% P-40, 0.1% SDS, and 2× Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL) and adjusted to a protein concentration of 2 mg/ml. The homogenates (20 µg) were fractionated by electrophoresis through 16.5% polyacrylamide Tris-Tricine gels. After electrophoresis, proteins were transferred to a nitrocellulose membrane. Blocking was carried out in 1% casein, 1× PBS for 1 hour. Incubation with primary antibody diluted in 1× PBS was for 1 hour. The following rabbit primary antibodies were from Abcam, Cambridge, MA: anti-SOD (1:1000, ab52950); anti-catalase (1:2000, ab16731); anti-erythropoietin (1:500, ab135390); anti-GLUT1 (1:250, ab652); anti-HSP27 (1:1000, ab5579); anti-actin (1:1000, ab8227), and mouse anti-actin (1:50,000, Ab6276). Anti-VEGF (1:500, sc507) and anti-CD63 (1:250, sc15363) were from Santa Cruz Biotechnology, Santa Cruz, CA. Goat anti-ICAM1 (1:200, af583) was from R&D Systems. Mouse anti-PCNA (1:3000, p8825) was from Sigma. Secondary antibodies were from Li-COR Biosciences, Lincoln, NE: IRDYE800CW donkey anti-rabbit (green, 1:15,000, 926–32213), IRDYE 680RD donkey anti-rabbit (red, 1:20,000, 926–68073), IRDYE800CW donkey anti-mouse (green, 1:15,000, 926–32212), IRDYE 680RD donkey anti-mouse (red, 1:20,000, 926–68072), and IRDYE 680RD donkey anti-goat (red, 1:20,000, 926–68074). The membrane was then washed in 1× PBS and incubated with secondary antibodies diluted in 1× PBS for 1 hour. Secondary antibodies were IRDye 680RD goat anti-rabbit IgG (1:15,000) (Li-COR Biosciences) and IRDye 800 CW goat anti-mouse IgG (1:20,000) (Li-COR Biosciences). After washing in 1× PBS, the filter was scanned using an Odyssey Infrared Imaging System (Li-COR Biosciences) for visualization of immunoreactive proteins. All steps were carried out at room temperature.
Electron Microscopy
EV were fixed in 2% paraformaldehyde/2% glutaraldehyde/0.1 M phosphate and then adsorbed to a 200–400 mesh carbon/formvar coated grid and the negative stain (NANOVAN; Nanoprobes, Yaphank, NY) added. The EV were visualized by electron microscopy18 (Tecnai G2 12 Bio TWIN Microscope [FEI, Hillsboro, OR] equipped with an AMT CCD camera [Advanced Microscopy Techniques, Danvers, MA]).
Renal and EV SAA1 and SYR mRNA
RT-PCR was also used to amplify SAA1 mRNA in recipient kidneys and EV. RNA extracted from homogenized renal cortices in lysis buffer was isolated with a purification kit as recommended by the vendor (CAT # 12183–555; Invitrogen), and cleaned with an RNeasy Mini kit as recommended by the vendor (CAT # 74104; Qiagen, Valencia, CA). For RT-PCR, 2 µg of total RNA were used to synthesize cDNA with an AffinityScript QPCR cDNA Synthesis Kit as recommended by the vendor (CAT # 600559; Agilent Technologies, Santa Clara, CA). The murine SAA1 mRNA was amplified as previously described19 using the following primers:
F: 5′-GA GTC TGG GCT GAG AA-3′
R: 5′-TG TCT GTT GGC TTC CTG GT-3′
The PCR products were separated in a 2.5% agarose gel.
The specific SRY DNA was amplified from extracted kidney and EV using the following primers19:
F:5′-AAGCGCCCCATGAATGC-3′
R: 5′-AGCCAACTTGCGCCTCTCT-3′
The PCR products were separated in a 2.5% agarose gel.
RNAseq and Differential Gene Expression
Whole renal and EV transcriptomes by RNAseq analysis39,69 were obtained by the Center for Medical Genomics, Indiana University School of Medicine sequencing core facility. Kidney total RNA and EV RNA were first evaluated for quantity and quality, using an Agilent Bioanalyzer. For kidney RNA quality, an RNA integrity number of seven or higher was required. The starting amount of RNA for library preparation was 500 ng for EV RNA and 1000 ng for kidney total RNA. PolyA mRNA capture of total RNA was performed using a Dynabeads mRNA DIRECT Micro Kit (61021; Ambion) as the first step. cDNA library preparation followed, inclusive of enzymatic fragmentation, hybridization and ligation of adaptors, reverse transcription, size selection, and amplification with barcode primers, following the Ion Total RNA-Seq Kit v2 User Guide, Pub. No. 4476286 Rev. E (Life Technologies). Each resulting barcoded library was quantified and its quality accessed by an Agilent Bioanalyzer, and multiple libraries pooled in equal molarity. Eight microliters of 100 pM pooled libraries were then applied to the Ion Sphere Particles (ISP) template preparation and amplification using Ion OneTouch 2 (Life Technologies), followed by ISP loading onto a PI chip and sequencing on an Ion Proton semiconductor (Life Technologies). Each PI chip allows loading of about 140 million ISP templates, generating approximately 80–100 million usable reads, up to 10–15 Gb. The reads were then mapped to the rat genome rn5 using TopHat2.70 Gene-based expression levels were quantified with NGSUtils.71 Read counts were normalized to the total number of sequencing reads falling into annotated gene regions in each sample, and further scaled on the basis of a trimmed mean of log-transformed counts per million (CPM) to correct for the variability of RNA composition in each sample.72 The genes with less than one read per million mappable reads in no less than half of samples in at least one condition were removed. The scaled CPM was used as gene-level quantification in each sample. The package “edgeR” was utilized to identify the genes that were differentially expressed between different biologic conditions.73 Benjamini and Hochberg’s algorithm was used to control the FDR. We determined FDR≤0.05 as the statistical cutoff for differential expression among different conditions. WEB-based GEne SeT AnaLysis Toolkit WEBGestalt (http://bioinfo.vanderbilt.edu/webgestalt/)74 was used to integrate the differentially expressed transcripts to the centrally curated database, KEGG (Kyoto Encyclopedia of Genes and Genomes), and to create functional categories with those differentially expressed transcripts. We performed several comparisons of renal transcripts: sham (control) and untreated ischemic; sham and normoxic EV (EXO); or sham and IPC EV. The IPC EV transcripts were separately compared with the normoxic EV (EXO).
Statistical Analyses
Sample size of five animals in each group gave 90% power to detect differences of 0.3 mg/dl in serum creatinine (with α set at 0.05).75 Data are expressed as mean±1 SEM. ANOVA was used to determine if differences among mean values reached statistical significance. Paired t test (two tailed, two sample, unequal variance) was used for comparisons between groups (GraphPad Prism, LaJolla, CA). Tukey’s test was used to correct for multiple comparisons. The null hypothesis was rejected at P<0.05.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Caroline Miller of the Indiana University (IU) Electron Microscopy Center and Jennifer Nelson and Dr. Sarah Tersey of the Islet and Physiology Core of the Center for Diabetes and Metabolic Diseases at IU for nanotrack analysis.
This work was supported by a National Institutes of Health grant R01 DK 082739, a research grant from Dialysis Clinics Inc. Paul Teschan Research and Development Fund, and a biomedical research grant from the IU School of Medicine to K.J.K. and Veterans Affairs Merit Review funds to J.H.D.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016121278/-/DCSupplemental.
References
- 1.Bonventre JV, Yang L: Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heyman SN, Khamaisi M, Rosen S, Rosenberger C: Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am J Nephrol 28: 998–1006, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Brivet FG, Kleinknecht DJ, Loirat P, Landais PJ; French Study Group on Acute Renal Failure : Acute renal failure in intensive care units--causes, outcome, and prognostic factors of hospital mortality; a prospective, multicenter study. Crit Care Med 24: 192–198, 1996 [DOI] [PubMed] [Google Scholar]
- 4.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW: Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR: Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am J Kidney Dis 53: 961–973, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horkan CM, Purtle SW, Mendu ML, Moromizato T, Gibbons FK, Christopher KB: The association of acute kidney injury in the critically ill and postdischarge outcomes: A cohort study*. Crit Care Med 43: 354–364, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Hoste EA, Bagshaw SM, Bellomo R, Cely CM, Colman R, Cruz DN, Edipidis K, Forni LG, Gomersall CD, Govil D, Honoré PM, Joannes-Boyau O, Joannidis M, Korhonen AM, Lavrentieva A, Mehta RL, Palevsky P, Roessler E, Ronco C, Uchino S, Vazquez JA, Vidal Andrade E, Webb S, Kellum JA: Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med 41: 1411–1423, 2015 [DOI] [PubMed] [Google Scholar]
- 8.Lassnigg A, Schmidlin D, Mouhieddine M, Bachmann LM, Druml W, Bauer P, Hiesmayr M: Minimal changes of serum creatinine predict prognosis in patients after cardiothoracic surgery: A prospective cohort study. J Am Soc Nephrol 15: 1597–1605, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Liaño F, Junco E, Pascual J, Madero R, Verde E: The Madrid Acute Renal Failure Study Group : The spectrum of acute renal failure in the intensive care unit compared with that seen in other settings. Kidney Int Suppl 66: S16–S24, 1998 [PubMed] [Google Scholar]
- 10.Siew ED, Davenport A: The growth of acute kidney injury: A rising tide or just closer attention to detail? Kidney Int 87: 46–61, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faubel S, Chawla LS, Chertow GM, Goldstein SL, Jaber BL, Liu KD: Acute Kidney Injury Advisory Group of the American Society of Nephrology : Ongoing clinical trials in AKI. Clin J Am Soc Nephrol 7: 861–873, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Agarwal A, Dong Z, Harris R, Murray P, Parikh SM, Rosner MH, Kellum JA, Ronco C; Acute Dialysis Quality Initiative XIII Working Group : Cellular and molecular mechanisms of AKI. J Am Soc Nephrol 27: 1288–1299, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eltzschig HK, Eckle T: Ischemia and reperfusion--from mechanism to translation. Nat Med 17: 1391–1401, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fine LG, Orphanides C, Norman JT: Progressive renal disease: The chronic hypoxia hypothesis. Kidney Int Suppl 65: S74–S78, 1998 [PubMed] [Google Scholar]
- 15.Tanaka S, Tanaka T, Nangaku M: Hypoxia as a key player in the AKI-to-CKD transition. Am J Physiol Renal Physiol 307: F1187–F1195, 2014 [DOI] [PubMed] [Google Scholar]
- 16.Kelly KJ, Kluve-Beckerman B, Dominguez JH: Acute-phase response protein serum amyloid A stimulates renal tubule formation: Studies in vitro and in vivo. Am J Physiol Renal Physiol 296: F1355–F1363, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Kelly KJ, Kluve-Beckerman B, Zhang J, Dominguez JH: Intravenous cell therapy for acute renal failure with serum amyloid A protein-reprogrammed cells. Am J Physiol Renal Physiol 299: F453–F464, 2010 [DOI] [PubMed] [Google Scholar]
- 18.Kelly KJ, Zhang J, Han L, Kamocka M, Miller C, Gattone VH 2nd, Dominguez JH: Improved structure and function in autosomal recessive polycystic rat kidneys with renal tubular cell therapy. PLoS One 10: e0131677, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly KJ, Zhang J, Han L, Wang M, Zhang S, Dominguez JH: Intravenous renal cell transplantation with SAA1-positive cells prevents the progression of chronic renal failure in rats with ischemic-diabetic nephropathy. Am J Physiol Renal Physiol 305: F1804–F1812, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly KJ, Zhang J, Wang M, Zhang S, Dominguez JH: Intravenous renal cell transplantation for rats with acute and chronic renal failure. Am J Physiol Renal Physiol 303: F357–F365, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonventre JV: Kidney ischemic preconditioning. Curr Opin Nephrol Hypertens 11: 43–48, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Islam CF, Mathie RT, Dinneen MD, Kiely EA, Peters AM, Grace PA: Ischaemia-reperfusion injury in the rat kidney: The effect of preconditioning. Br J Urol 79: 842–847, 1997 [DOI] [PubMed] [Google Scholar]
- 23.Jonker SJ, Menting TP, Warlé MC, Ritskes-Hoitinga M, Wever KE: Preclinical evidence for the efficacy of ischemic postconditioning against renal ischemia-reperfusion injury, a systematic review and meta-analysis. PLoS One 11: e0150863, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Gong R: Remote ischemic preconditioning for kidney protection: GSK3β-centric insights into the mechanism of action. Am J Kidney Dis 66: 846–856, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wever KE, Menting TP, Rovers M, van der Vliet JA, Rongen GA, Masereeuw R, Ritskes-Hoitinga M, Hooijmans CR, Warlé M: Ischemic preconditioning in the animal kidney, a systematic review and meta-analysis. PLoS One 7: e32296, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kierulf-Lassen C, Nieuwenhuijs-Moeke GJ, Krogstrup NV, Oltean M, Jespersen B, Dor FJ: Molecular mechanisms of renal ischemic conditioning strategies. Eur Surg Res 55: 151–183, 2015 [DOI] [PubMed] [Google Scholar]
- 27.Dickson EW, Lorbar M, Porcaro WA, Fenton RA, Reinhardt CP, Gysembergh A, Przyklenk K: Rabbit heart can be “preconditioned” via transfer of coronary effluent. Am J Physiol 277: H2451–H2457, 1999 [DOI] [PubMed] [Google Scholar]
- 28.Dickson EW, Reinhardt CP, Renzi FP, Becker RC, Porcaro WA, Heard SO: Ischemic preconditioning may be transferable via whole blood transfusion: Preliminary evidence. J Thromb Thrombolysis 8: 123–129, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Burger D, Viñas JL, Akbari S, Dehak H, Knoll W, Gutsol A, Carter A, Touyz RM, Allan DS, Burns KD: Human endothelial colony-forming cells protect against acute kidney injury: Role of exosomes. Am J Pathol 185: 2309–2323, 2015 [DOI] [PubMed] [Google Scholar]
- 30.Kourembanas S: Exosomes: Vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol 77: 13–27, 2015 [DOI] [PubMed] [Google Scholar]
- 31.Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, Ratajczak MZ: Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 20: 847–856, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Tak T, Tesselaar K, Pillay J, Borghans JA, Koenderman L: What’s your age again? Determination of human neutrophil half-lives revisited. J Leukoc Biol 94: 595–601, 2013 [DOI] [PubMed] [Google Scholar]
- 33.Lee HM, Choi EJ, Kim JH, Kim TD, Kim YK, Kang C, Gho YS: A membranous form of ICAM-1 on exosomes efficiently blocks leukocyte adhesion to activated endothelial cells. Biochem Biophys Res Commun 397: 251–256, 2010 [DOI] [PubMed] [Google Scholar]
- 34.Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, Crystal RG, Darnell RB, Ferrante RJ, Fillit H, Finkelstein R, Fisher M, Gendelman HE, Golub RM, Goudreau JL, Gross RA, Gubitz AK, Hesterlee SE, Howells DW, Huguenard J, Kelner K, Koroshetz W, Krainc D, Lazic SE, Levine MS, Macleod MR, McCall JM, Moxley RT 3rd, Narasimhan K, Noble LJ, Perrin S, Porter JD, Steward O, Unger E, Utz U, Silberberg SD: A call for transparent reporting to optimize the predictive value of preclinical research. Nature 490: 187–191, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witzgall R, Brown D, Schwarz C, Bonventre JV: Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest 93: 2175–2188, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leung KC, Tonelli M, James MT: Chronic kidney disease following acute kidney injury-risk and outcomes. Nat Rev Nephrol 9: 77–85, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Gao G, Wang W, Tadagavadi RK, Briley NE, Love MI, Miller BA, Reeves WB: TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J Clin Invest 124: 4989–5001, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly KJ, Burford JL, Dominguez JH: Postischemic inflammatory syndrome: A critical mechanism of progression in diabetic nephropathy. Am J Physiol Renal Physiol 297: F923–F931, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Kelly KJ, Liu Y, Zhang J, Dominguez JH: Renal C3 complement component: Feed forward to diabetic kidney disease. Am J Nephrol 41: 48–56, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, Bacallao RL, Molitoris BA, Sutton TA: Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol 300: F721–F733, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miranda KC, Bond DT, Levin JZ, Adiconis X, Sivachenko A, Russ C, Brown D, Nusbaum C, Russo LM: Massively parallel sequencing of human urinary exosome/microvesicle RNA reveals a predominance of non-coding RNA. PLoS One 9: e96094, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimada S, Hirabayashi M, Ishige K, Kosuge Y, Kihara T, Ito Y: Activation of dopamine D4 receptors is protective against hypoxia/reoxygenation-induced cell death in HT22 cells. J Pharmacol Sci 114: 217–224, 2010 [DOI] [PubMed] [Google Scholar]
- 43.Zhang W, Liu J, Hu X, Li P, Leak RK, Gao Y, Chen J: n-3 polyunsaturated fatty acids reduce neonatal hypoxic/ischemic brain injury by promoting phosphatidylserine formation and Akt signaling. Stroke 46: 2943–2950, 2015 [DOI] [PubMed] [Google Scholar]
- 44.Vaggi F, Disanza A, Milanesi F, Di Fiore PP, Menna E, Matteoli M, Gov NS, Scita G, Ciliberto A: The Eps8/IRSp53/VASP network differentially controls actin capping and bundling in filopodia formation. PLOS Comput Biol 7: e1002088, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuryev A, Patturajan M, Litingtung Y, Joshi RV, Gentile C, Gebara M, Corden JL: The C-terminal domain of the largest subunit of RNA polymerase II interacts with a novel set of serine/arginine-rich proteins. Proc Natl Acad Sci U S A 93: 6975–6980, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schults MA, Sanen K, Godschalk RW, Theys J, van Schooten FJ, Chiu RK: Hypoxia diminishes the detoxification of the environmental mutagen benzo[a]pyrene. Mutagenesis 29: 481–487, 2014 [DOI] [PubMed] [Google Scholar]
- 47.Recino A, Sherwood V, Flaxman A, Cooper WN, Latif F, Ward A, Chalmers AD: Human RASSF7 regulates the microtubule cytoskeleton and is required for spindle formation, Aurora B activation and chromosomal congression during mitosis. Biochem J 430: 207–213, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.U.S. Renal Data System : USRDS 2015 Annual Data Report: Atlas of End-Stage Renal Disease in the United States, Bethesda, Md, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2015 [Google Scholar]
- 49.Ojo AO, Wolfe RA, Held PJ, Port FK, Schmouder RL: Delayed graft function: Risk factors and implications for renal allograft survival. Transplantation 63: 968–974, 1997 [DOI] [PubMed] [Google Scholar]
- 50.Weber M, Dindo D, Demartines N, Ambühl PM, Clavien PA: Kidney transplantation from donors without a heartbeat. N Engl J Med 347: 248–255, 2002 [DOI] [PubMed] [Google Scholar]
- 51.Brown JR, Rezaee ME, Marshall EJ, Matheny ME: Hospital mortality in the United States following acute kidney injury. BioMed Res Int 2016: 4278579, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schnaper HW, Hubchak SC, Runyan CE, Browne JA, Finer G, Liu X, Hayashida T: A conceptual framework for the molecular pathogenesis of progressive kidney disease. Pediatr Nephrol 25: 2223–2230, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang GT, Gronthos S, Shi S: Mesenchymal stem cells derived from dental tissues vs. those from other sources: Their biology and role in regenerative medicine. J Dent Res 88: 792–806, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li TS, Cheng K, Malliaras K, Smith RR, Zhang Y, Sun B, Matsushita N, Blusztajn A, Terrovitis J, Kusuoka H, Marbán L, Marbán E: Direct comparison of different stem cell types and subpopulations reveals superior paracrine potency and myocardial repair efficacy with cardiosphere-derived cells. J Am Coll Cardiol 59: 942–953, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tedesco FS, Dellavalle A, Diaz-Manera J, Messina G, Cossu G: Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J Clin Invest 120: 11–19, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng K, Rai P, Plagov A, Lan X, Kumar D, Salhan D, Rehman S, Malhotra A, Bhargava K, Palestro CJ, Gupta S, Singhal PC: Transplantation of bone marrow-derived MSCs improves cisplatinum-induced renal injury through paracrine mechanisms. Exp Mol Pathol 94: 466–473, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Du T, Ju G, Wu S, Cheng Z, Cheng J, Zou X, Zhang G, Miao S, Liu G, Zhu Y: Microvesicles derived from human Wharton’s jelly mesenchymal stem cells promote human renal cancer cell growth and aggressiveness through induction of hepatocyte growth factor. PLoS One 9: e96836, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Ariunbold U, Suhaimi N, Sunn N, Guo J, McMahon JA, McMahon AP, Little M: Collecting duct-derived cells display mesenchymal stem cell properties and retain selective in vitro and in vivo epithelial capacity. J Am Soc Nephrol 26: 81–94, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Machiguchi T, Nakamura T: Cellular interactions via conditioned media induce in vivo nephron generation from tubular epithelial cells or mesenchymal stem cells. Biochem Biophys Res Commun 435: 327–333, 2013 [DOI] [PubMed] [Google Scholar]
- 60.Tögel F, Zhang P, Hu Z, Westenfelder C: VEGF is a mediator of the renoprotective effects of multipotent marrow stromal cells in acute kidney injury. J Cell Mol Med 13[8B]: 2109–2114, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Koppen A, Joles JA, van Balkom BW, Lim SK, de Kleijn D, Giles RH, Verhaar MC: Human embryonic mesenchymal stem cell-derived conditioned medium rescues kidney function in rats with established chronic kidney disease. PLoS One 7: e38746, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang DH, Li FR, Zhang Y, Wang YQ, Yuan FH: Conditioned medium from renal tubular epithelial cells stimulated by hypoxia influences rat bone marrow-derived endothelial progenitor cells. Ren Fail 32: 863–870, 2010 [DOI] [PubMed] [Google Scholar]
- 63.Yuen DA, Connelly KA, Advani A, Liao C, Kuliszewski MA, Trogadis J, Thai K, Advani SL, Zhang Y, Kelly DJ, Leong-Poi H, Keating A, Marsden PA, Stewart DJ, Gilbert RE: Culture-modified bone marrow cells attenuate cardiac and renal injury in a chronic kidney disease rat model via a novel antifibrotic mechanism. PLoS One 5: e9543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zarjou A, Kim J, Traylor AM, Sanders PW, Balla J, Agarwal A, Curtis LM: Paracrine effects of mesenchymal stem cells in cisplatin-induced renal injury require heme oxygenase-1. Am J Physiol Renal Physiol 300: F254–F262, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou Y, Xiong M, Fang L, Jiang L, Wen P, Dai C, Zhang CY, Yang J: miR-21-containing microvesicles from injured tubular epithelial cells promote tubular phenotype transition by targeting PTEN protein. Am J Pathol 183: 1183–1196, 2013 [DOI] [PubMed] [Google Scholar]
- 66.Tapuria N, Kumar Y, Habib MM, Abu Amara M, Seifalian AM, Davidson BR: Remote ischemic preconditioning: A novel protective method from ischemia reperfusion injury--a review. J Surg Res 150: 304–330, 2008 [DOI] [PubMed] [Google Scholar]
- 67.Staudacher JJ, Naarmann-de Vries IS, Ujvari SJ, Klinger B, Kasim M, Benko E, Ostareck-Lederer A, Ostareck DH, Bondke Persson A, Lorenzen S, Meier JC, Blüthgen N, Persson PB, Henrion-Caude A, Mrowka R, Fähling M: Hypoxia-induced gene expression results from selective mRNA partitioning to the endoplasmic reticulum. Nucleic Acids Res 43: 3219–3236, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kelly KJ, Meehan SM, Colvin RB, Williams WW, Bonventre JV: Protection from toxicant-mediated renal injury in the rat with anti-CD54 antibody. Kidney Int 56: 922–931, 1999 [DOI] [PubMed] [Google Scholar]
- 69.Kelly KJ, Liu Y, Zhang J, Goswami C, Lin H, Dominguez JH: Comprehensive genomic profiling in diabetic nephropathy reveals the predominance of proinflammatory pathways. Physiol Genomics 45: 710–719, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL: TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Breese MR, Liu Y: NGSUtils: A software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 29: 494–496, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson MD, Oshlack A: A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson MD, McCarthy DJ, Smyth GK: edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang J, Duncan D, Shi Z, Zhang B: WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res 41: W77–W83, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kadam P, Bhalerao S: Sample size calculation. Int J Ayurveda Res 1: 55–57, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.