

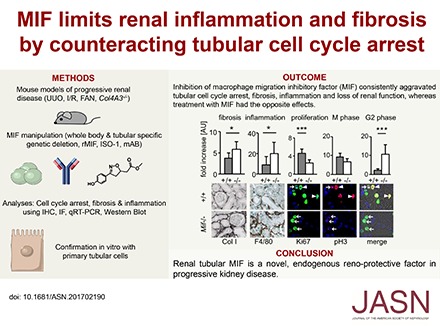
Abstract
Renal fibrosis is a common underlying process of progressive kidney diseases. We investigated the role of macrophage migration inhibitory factor (MIF), a pleiotropic proinflammatory cytokine, in this process. In mice subjected to unilateral ureteral obstruction, genetic deletion or pharmacologic inhibition of MIF aggravated fibrosis and inflammation, whereas treatment with recombinant MIF was beneficial, even in established fibrosis. In two other models of progressive kidney disease, global Mif deletion or MIF inhibition also worsened fibrosis and inflammation and associated with worse kidney function. Renal MIF expression was reduced in tubular cells in fibrotic compared with healthy murine and human kidneys. Bone marrow chimeras showed that Mif expression in bone marrow-derived cells did not affect fibrosis and inflammation after UUO. However, Mif gene deletion restricted to renal tubular epithelial cells aggravated these effects. In LPS-stimulated tubular cell cultures, Mif deletion led to enhanced G2/M cell-cycle arrest and increased expression of the CDK inhibitor 1B (p27Kip1) and of proinflammatory and profibrotic mediators. Furthermore, MIF inhibition reduced tubular cell proliferation in vitro. In all three in vivo models, global Mif deletion or MIF inhibition caused similar effects and attenuated the expression of cyclin B1 in tubular cells. Mif deletion also resulted in reduced tubular cell apoptosis after UUO. Recombinant MIF exerted opposing effects on tubular cells in vitro and in vivo. Our data identify renal tubular MIF as an endogenous renoprotective factor in progressive kidney diseases, raising the possibility of pharmacologic intervention with MIF pathway agonists, which are in advanced preclinical development.
Keywords: fibrosis, tubular epithelium, cell cycle arrest
CKD is a common end point of a multitude of renal diseases and an increasingly prevalent global health problem.1 The underlying pathologic process of CKD is renal fibrosis associated with unremitting renal inflammation or inadequate homeostatic repair responses. Although a large number of studies have identified molecules and pathways driving renal fibrosis and inflammation in experimental models, our understanding of endogenous factors that may limit fibrosis and renal inflammation remains incomplete.2–7
Macrophage migration inhibitory factor (MIF) is a pleiotropic chemokine-like cytokine with proinflammatory and growth-promoting functions. MIF drives inflammation in various diseases including sepsis, autoimmune arthritis, atherosclerosis, and GN.8–13 In addition to clinical trials of MIF inhibition in SLE, ongoing phase 2 trials are testing the inhibition of MIF in patients with different malignancies.14,15 We have recently shown that MIF also has noninflammatory local effects on renal glomerular cells to promote their pathologic proliferation and drive GN.16 Here we show that in progressive CKDs with fibrosis, local tubular MIF is renoprotective by promoting proliferation of tubular cells. This improves regeneration and abrogates cell cycle arrest, thereby keeping the proinflammatory and profibrotic phenotype of tubular cells in check.
Results
MIF in Obstructive Nephropathy
Compared with WT mice, 5 days after unilateral ureteral obstruction (UUO), Mif−/− mice had significantly increased fibrosis and more myofibroblasts as shown by quantitative immunohistochemical analyses of collagen type I and type IV, fibronectin, and α–smooth muscle actin (α-SMA); a biochemical collagen assay in renal cortical tissue lysates; and by quantitative real-time PCR (qRT-PCR) for Col1, Acta2, and Fibronectin (Figure 1A, Supplemental Figure 1A). Mif−/− mice showed significantly increased infiltration of inflammatory cells as shown by quantification of F4/80, ErHr3, and CD3 staining (Figure 1A). We found similarly aggravated renal interstitial fibrosis and inflammation in Mif−/− mice at later time points of UUO, i.e., on day 10 (Supplemental Figure 1A).
Figure 1.

MIF was antifibrotic in obstructive nephropathy. (A) In a model of renal fibrosis, the UUO on day 5, compared with wild-type littermates (+/+), Mif deficient (−/−) mice had significantly increased renal fibrosis and inflammation as shown by computer-assisted morphometry of sections stained for fibrosis markers, i.e., collagen type I (Col I), collagen IV (Col IV), fibronectin, and α-SMA, and markers of inflammatory infiltrates, i.e., macrophages and monocytes (ErHr3 and F4/80) and T-cells (CD3). Biochemical assay for collagen content (Sircol) confirmed these results. (B) Compared with vehicle-treated mice, MIF neutralization with a specific small-molecule inhibitor, ISO-1 (40 mg/kg body wt daily i.p.), initiated in established disease on day 3 of UUO also significantly increased accumulation of myofibroblasts, collagen deposition, and immune cell infiltration compared with vehicle-treated (Veh.) mice. (C) Vice versa, compared with vehicle-treated mice, treatment with murine rMIF (2 mg/kg body wt daily i.p.) in established fibrosis in UUO decreased renal fibrosis and inflammation. Data are mean±SD; n=8 per group in all experiments. Values of healthy contralateral kidneys of WT were set as 1, represented by the dashed line. *P<0.05; **P<0.01. AU, arbitrary unit.
We next treated mice with UUO with either a neutralizing anti-MIF mAb or an IgG1 isotype control (both 5 mg/kg body wt daily i.p. starting 2 days before UUO) and found a significant increase in fibrosis and inflammation in mice in which MIF was neutralized (Supplemental Figures 1B and 2D). We additionally studied the effects of small-molecule MIF antagonist ISO-117 application initiated in already established disease. Compared with vehicle, ISO-1–treated mice (40 mg/kg body wt daily i.p. starting 3 days after UUO) showed significantly increased fibrosis and inflammation (Figure 1B, Supplemental Figure 2D).
In an inverse setting, we treated mice with biologically active murine recombinant MIF (rMIF) or vehicle in a therapeutic approach from day 3 to 7 or from day 5 to 10 of UUO, and in both situations found a significant reduction of fibrosis and inflammation (Figure 1C, Supplemental Figures 1C and 2D, respectively).
Collectively, these data show that in murine obstructive nephropathy, MIF has consistent and potent antifibrotic and anti-inflammatory effects across the full time scale of the disease process.
MIF in Ischemia/Reperfusion and Folic Acid Nephropathies
To analyze whether the renoprotective effect of MIF is a more general finding, we used two additional models of renal fibrosis, the unilateral ischemia/reperfusion–induced injury model (I/R) and folic acid nephropathy (FAN), a model of toxic tubular injury.
Compared with WT with I/R, Mif−/− mice had significantly increased renal fibrosis and inflammatory infiltrates (Figure 2A, Supplemental Figure 2D). Similarly, compared with vehicle-treated mice, ISO-1 treatment initiated on day 3 after I/R aggravated fibrosis and inflammation on day 21 (Figure 2B, Supplemental Figure 2D). We next asked whether the observed effects of MIF translated into changes in renal function. For this, we analyzed WT and Mif−/− mice with I/R on day 16, which had received a nephrectomy of the noninjured kidney on day 14. Both fibrosis and inflammation were significantly increased in Mif−/− mice compared with WT (Figure 2C, Supplemental Figure 2D), and this was mirrored by increased serum creatinine, BUN, and proteinuria, and reduced creatinine clearance, documenting significant loss of renal function (Figure 2C).
Figure 2.

MIF was antifibrotic in various models of renal fibrosis. (A) In unilateral I/R-induced renal fibrosis (I/R; day 21), genetic deletion of Mif (−/−; n=12) aggravated renal fibrosis and inflammation compared with wild-type littermates (+/+; n=10), as shown by computer-assisted morphometric quantification of immunohistochemistry staining for collagen I (Col I), α-SMA, ErHr3+, and F4/80+ monocytes/macrophages. (B) In unilateral I/R model, compared with vehicle, MIF neutralization with ISO-1 initiated in early disease (40 mg/kg body wt daily i.p., start day 3 after operation) increased also fibrosis and inflammation. (C) I/R injury on day 14 combined with uninephrectomy (UniX) of healthy contralateral kidney allowed the measurement of renal function. Compared with wild-type littermates (+/+; n=9), genetic deletion of Mif (−/−; n=7) significantly aggravated renal fibrosis (Col I and α-SMA) and inflammation (F4/80 and ErHr3), and this translated into significant deterioration of renal function, i.e., showing increased proteinuria and BUN and decreased creatinine clearance. (D) On day 56 of toxin-induced fibrosis, i.e., FAN, compared with wild-type littermates (+/+; n=6), Mif deficiency (−/−; n=6) led to increased fibrosis and inflammation and thereby to decreased renal function. Data are mean±SD. Values of healthy contralateral kidneys of WT were set as 1, represented by the dashed line. *P<0.05; **P<0.01; **P<0.001. AU, arbitrary unit; Veh., vehicle.
Similarly, compared with WT, Mif−/− mice had significantly worse fibrosis and inflammation on day 56 of FAN, which was also mirrored by significantly reduced renal function (Figure 2D, Supplemental Figure 2D). Taken together, MIF inhibition leads to aggravated renal damage and loss of renal function in different models of renal interstitial injury.
MIF in Fibrosis Secondary to Glomerular Injury and in Healthy Kidneys
Previous studies showed that MIF neutralization improved the disease course and reduced inflammation in primary inflammatory and autoimmune glomerular disease models, i.e., in models of crescentic and lupus GN.16–18 To address the question of whether this is true for nonautoimmune glomerular diseases, we analyzed Col4a3−/− mice, a model of Alport syndrome with noninflammatory, progressive glomerular injury and secondary interstitial fibrosis and death due to kidney failure. We first treated Col4a3−/− mice with either control IgG or anti-MIF antibody for 3 weeks, starting at the age of 6 weeks, when the mice already exhibit manifest renal disease. When compared with IgG-treated mice, anti-MIF antibody–treated mice showed a significantly increased mortality during the treatment period (Supplemental Figure 3A), which prevented us from analyzing the end points of renal fibrosis and inflammation. Therefore, we next treated Col4a3−/− mice with either vehicle or ISO-1 for a shorter period of 2 weeks starting at the age of 6 weeks with mortality of only one single mouse from the ISO-1–treated group. Compared with vehicle group, ISO-1–treated Col4a3−/− mice showed significantly more interstitial fibrosis and inflammation, but no significant difference in glomerular injury (Supplemental Figure 3, B and C). Previously, we have shown that in mice with a model of immune-mediated crescentic GN, i.e., nephrotoxic serum nephritis, Mif−/− mice had significantly reduced glomerular damage and interstitial inflammatory infiltrates 2 weeks after nephrotoxic serum nephritis induction 16. Here, we have reanalyzed tubulointerstitial damage and fibrosis in this model, showing that compared with WT mice, Mif−/− also had reduced tubulointerstitial injury and fibrosis (Supplemental Figure 3D). These data suggested that, depending on the underlying disease mechanism, MIF neutralization has renoprotective effects in (auto) immune models, whereas it is harmful in chronic models, in which inflammation is secondary to epithelial injury.
These data show that MIF is also renoprotective in secondary tubulointerstitial fibrosis due to noninflammatory glomerular disease. We have found no effects on fibrosis or inflammation in healthy, nonfibrotic kidneys in any of the models analyzed (Supplemental Figure 4), suggesting that the effects of MIF are only relevant during pathologic conditions.
Cellular Source of MIF in Renal Fibrosis
We next analyzed the localization and regulation of MIF in renal fibrosis. Compared with healthy kidneys, MIF expression was significantly and progressively reduced during the course of murine UUO, as shown by immunofluorescence, Western blot, and qRT-PCR analyses (Figure 3, A and B). Specificity of the staining was confirmed using two different anti-MIF antibodies and a number of controls (Supplemental Figure 5). This loss of expression was particularly obvious in renal tubular cells (Figure 3A). A similar reduction of MIF was also observed in the other models studied: I/R day 21 (Figure 3C) and FAN day 56 (Figure 3D). At early stages of fibrosis, a shuttling of MIF to the nucleus was observed (Figure 3A, Supplemental Figure 5C), possibly being involved in gene regulation and in line with a recent report on nuclear shuttling of MIF.19
Figure 3.

MIF was localized mainly to tubular cells and was reduced in fibrosis. (A) MIF is expressed by tubular cells of distal and proximal tubules (asterisk) and even more strongly in collecting ducts (arrowhead) in healthy wild-type mice. Immunofluorescence staining showed a prominent nuclear MIF staining (arrow) after 2 days of UUO and a progressive decrease during the time course of fibrosis. (B) Immunoblot and mRNA expression confirmed the downregulation of MIF during UUO (n=5 each time point; a representative blot of n=2 is shown). (C) At day 21 after I/R injury and (D) at day 56 of FAN a decreased MIF protein expression was detected, using immunofluorescence and Western blot, compared with healthy or SHAM-operated mice, respectively (n=5 each group). (E) Compared with human kidneys without detectable renal disease and fibrosis, human fibrotic kidneys showed a significant downregulation of MIF (n=6 each group). Data are mean±SD. Values of SHAM-operated or healthy mice and humans were set as 1, represented by the orange, dashed line. *P<0.05; **P<0.01 versus healthy/SHAM. AU, arbitrary units.
Compared with human kidney tissue samples without kidney diseases and with unremarkable histopathologic findings, patients with renal fibrosis also showed reduced expression of MIF both by Western blot and immunofluorescence (Figure 3E, Supplemental Figure 6).
Previous studies have proposed that leukocytes and renal cells might be important sources of MIF in various glomerulonephritides.16,18,20 To address the cellular source of MIF responsible for its renoprotective effects in fibrosis, we performed UUO in WT mice that underwent bone marrow ablation followed by reconstitution with Mif−/− or WT bone marrow. As expected, fibrosis and inflammation significantly increased in Mif−/− mice with Mif−/− bone marrow compared with WT mice with WT bone marrow (Figure 4A). Importantly, WT mice receiving Mif−/− bone marrow had similar fibrosis and inflammation as WT mice with WT bone marrow, whereas Mif−/− mice with WT bone marrow had similar fibrosis as Mif−/− mice with Mif−/− bone marrow (Figure 4A, Supplemental Figure 2D). These data show that bone marrow–derived MIF is not involved in mediating the observed renoprotective effects in fibrosis.
Figure 4.

Local tubular cell–derived MIF was responsible for the antifibrotic effects. (A) First bone marrow transplantation was performed in UUO model of fibrosis. Compared with WT recipients with bone marrow from WT donors (WTBM/WT; n=9), Mif−/− recipients with bone marrow from Mif−/− donors (Mif−/−BM/Mif−/−; n=7) had increased matrix deposition, myofibroblast accumulation, and immune cell infiltration, as expected. Similar fibrosis and inflammation was found in WT recipients with bone marrow from Mif−/− donors (Mif−/−BM/WT; n=8) compared with WT mice with WT bone marrow (WTBM/WT), showing that bone marrow–derived MIF is not involved in mediating the antifibrotic effects. Mif−/− mice with WT bone marrow (WTBM/Mif−/−; n=10) had similar expression as Mif−/− mice with Mif−/− bone marrow (Mif−/−BM/Mif−/−), collectively suggesting that MIF expressed in nonmyeloid cells is involved in the observed antifibrotic effects. (B) To specifically address whether local renal tubular cell–derived MIF is involved in the antifibrotic effects, tubular-specific Mif–deficient mice were generated by crossbreeding of a tubular-specific Cre driver (Pax8Cre/+) with Mifflox/flox mice. The deletion was confirmed by Western blot and immunofluorescence staining, showing normal interstitial but undetectable tubular MIF staining. (C) Compared with control Pax8+/+::Mifflox/flox mice (+/+; n=6), Pax8Cre/+::Mifflox/flox mice (Cre/+; n=5) had significantly more severe fibrosis and inflammation in UUO day 5 analyzed by immunohistochemical evaluation of collagen I, α-SMA, F4/80, and ErHr3 staining. Data are mean±SD. Values of healthy contralateral kidneys of WTBM/WT or Pax8+/+::Mifflox/flox were set as 1, represented by the orange, dashed line. *P<0.05 versus WTBM/WT; §P<0.05 versus Mif−/−BM/WT; #P<0.05 versus WTBM/WT; ##P<0.01 versus WTBM/WT; $P<0.05 versus Mif−/−BM/WT; $$P<0.01 versus Mif−/−BM/WT. AU, arbitrary unit; BMT, bone marrow transplantation; WT, wild-type.
On the basis of the MIF expression pattern, we next asked whether local renal tubular–derived MIF might be involved. For this, we crossbred the tubular-specific Pax8Cre/+ mice with Mifflox/flox mice, resulting in renal tubular cell–specific deletion of MIF as confirmed by Western blot and immunofluorescence (Figure 4B). Compared with control Pax8+/+::Mifflox/flox mice, Pax8Cre/+::Mifflox/flox mice had significantly more severe fibrosis and inflammation on UUO day 5 (Figure 4C).
Taken together, these data indicate that renal tubular MIF is reduced in both animal models and humans with renal fibrosis. In contrast to bone marrow–derived MIF, local tubular–derived MIF mediates the observed renoprotective effects.
MIF Effects on Cell Cycle Arrest
To dissect the mechanisms behind the renoprotective effects of MIF, we compared WT and Mif−/− mice on day 2 of UUO. At this early time point, both fibrosis and inflammation were slightly, but already significantly, increased compared with nonobstructed kidneys. At this stage, only inflammation was significantly more pronounced in Mif−/− mice whereas fibrosis was comparable in both groups (Figure 5A). An array of inflammatory mediators showed that from among the ten detectable inflammatory mediators: C5a, sICAM, IFN-γ, IL-1α, IL-1rα, CXCL10, CXCL1, TNF-α, CCL2, and TIMP-1, only CCL2 and TIMP-1 were differentially regulated in Mif−/− mice when compared with WT mice on day 2 of UUO (Figure 5B, Supplemental Figure 7A). We focused on CCL2, because it is a well recognized chemokine for monocytes and macrophages and was shown to promote renal fibrosis.21,22 We confirmed this unexpected downregulation of CCL2 by MIF at both the protein and mRNA level in kidneys (Figure 5C) and at other time points of UUO as well as in I/R (Supplemental Figure 7, B and C). The increased renal expression of CCL2 also translated into increased serum levels of CCL2 in Mif−/− mice with UUO (Supplemental Figure 7B). We next confirmed the regulation of CCL2 by MIF in primary murine renal tubular cells (pmTECs) in vitro. In resting conditions in pmTECs, neither MIF stimulation nor MIF inhibition using ISO-1 altered Ccl2 expression (Figure 5, D–F). Upon activation of pmTECs using LPS or BSA, ISO-1 augmented and MIF abrogated Ccl2 production (Figure 5, D–F). The challenge of pmTECs with BSA and the effects of MIF and MIF inhibition provide a possible explanation of how glomerular injury in Alport mice, which is characterized by a prominent proteinuria and thereby albumin leakage into the urine, translates to tubular injury which MIF is able to modulate.
Figure 5.

MIF reduced expression of CCL2 in tubular cells. (A) In very early stages of fibrosis, i.e., UUO day 2, the earliest detectable changes in Mif−/− mice, compared with wild-type littermates (+/+), were increased inflammation as shown were by the number of infiltrating monocytes and macrophages (ErHr3 and F4/80), whereas renal fibrosis was similar between the two groups, as shown by collagen type I (Col I) and α-SMA (n=10 per group). (B) Densitometry of cytokine array with protein lysates from UUO day 2 kidneys from Mif−/− (−/−; n=2) and wild-type (+/+; n=2) mice demonstrated an upregulation of CC-chemokine ligand 2 (CCL2) and Tissue Inhibitor of Metalloproteinases 1 (TIMP1) in Mif−/−, whereas all other analyzed cytokines were not substantially altered between the groups. (C) Confirmation of increased renal CCL2 expression in Mif−/− compared with WT in UUO day 2 by qRT-PCR and ELISA showed significantly increased CCL-2 expression in Mif−/− mice compared with wild-type littermates. (D) In vitro, specific small-molecule inhibitor of MIF, ISO-1, induced Ccl2 mRNA expression in primary murine tubular cells (pmTEC) activated with LPS for 6 hours from wild-type. Vice versa, incubation with rMIF reduced Ccl2 mRNA expression in wild-type pmTEC after activation with (E) BSA and (F) LPS (both for 6 hours). All in vitro experiments were performed at least in triplicate. Data are mean±SD. Values of healthy contralateral kidneys of WT were set as 1, represented by the dashed line. *P<0.05; **P<0.01. AU, arbitrary unit.
We next analyzed the proliferation of renal tubular cells and interstitial fibroblasts. On day 2 of UUO, tubular cells in Mif−/− mice showed a significantly reduced proliferation rate compared with WT (Figure 6A). This finding was consistent at later time points, in different fibrosis models, and using different assays including counts of mitotic figures and incorporation of bromodeoxyuridine (BrdU)-labeling cells which went through the S phase (Figure 6B, Supplemental Figure 8, A, C, and D). In line, mice treated with rMIF showed a significantly increased proliferation of tubular cells (Supplemental Figure 8B). On day 2 of UUO, fibroblast proliferation was similar between the groups. Using different assays, at later time points of UUO and other models, fibroblast proliferation was significantly increased in Mif−/− mice and significantly reduced in MIF-treated mice (Figure 6, A and B versus Supplemental Figure 8, A–C).
Figure 6.

MIF abrogated cell cycle arrest in tubular cells and reduced activation of fibroblasts. (A) At day 2 of UUO, compared with wild-type littermates (+/+; n=10), Mif (−/−; n=10) mice showed a significantly decreased number of proliferating tubular cells as analyzed by Ki67 staining (arrowhead pointing to Ki67 and DAPI double-positive nuclei of tubular cells). Tubular damage analyzed by LCN2 staining and proliferation of fibroblasts, positive for both Ki67 and α-SMA (arrows), was similar in both groups. (B) At a later time point after UUO, on day 5, tubular cells also showed decreased proliferation whereas fibroblasts proliferated more in Mif−/− compared with wild-type littermates, as analyzed by BrdU incorporation (n=5 per group). (C) Further, compared with wild-type littermates, tubular cells in Mif−/− mice showed significantly more cells in G2 cell cycle arrest and reduced number of cells in mitosis, analyzed by chromatin pattern in immunofluorescence staining for phosphorylated serine 10 of histone H3 (pH3). Arrowheads point to cells in M stage, arrows to cells in G2 cell cycle arrest. Mif−/− mice showed a significantly decreased expression of cell cycle regulators cyclin B1 and cyclin E1 and an increased expression of cyclin inhibitor p27Kip1 (Cdkn1b), analyzed by (D) Western blot and (E) qRT-PCR. (F) Coimmunofluorescence staining of Ki67 and pH3 of primary murine tubular cells (pmTEC) isolated from Mif−/− and WT mice confirmed the in vivo finding of increased number of cells in G2 phase in Mif−/− compared with wild-type. (G) Compared with pmTEC isolated from wild-type littermates, cell proliferation was reduced in pmTEC isolated from Mif−/− mice and induced by incubation with murine rMIF after 48 hours. (H) In pmTEC challenged with LPS for 6 hours, incubation with rMIF decreased, whereas ISO-1, a specific small-molecule inhibitor of MIF, increased expression of p27 Kip1 and pH3, indicating increased cell cycle arrest. (I) Conditioned media of pmTEC incubated for 2 hours with ISO-1 or rMIF reduced or increased, respectively, the proliferation of primary renal fibroblasts after 48 hours compared with conditioned medium of vehicle (DMSO/PBS)-treated tubular cells. Data are mean±SD. Values of healthy contralateral kidneys of WT were set as 1, represented by the orange, dashed line. *P<0.05; **P<0.01; ***P<0.001. AU, arbitrary unit; DAPI, 4′6-Diamidin-2-phenylindol.
The significantly reduced proliferation of tubular cells, accompanied by a later increased proliferation of fibroblasts and fibrosis as well as the unexpected effects of MIF on CCL2 expression in Mif−/− mice, prompted us to further dissect the underlying mechanism. Cell cycle arrest of tubular cells is a major mechanism driving renal fibrosis and inflammation by inducing the expression of profibrotic and proinflammatory mediators like TGF-β, PDGF-B, or CCL2.23–27 Previous data suggested that MIF might abrogate cell cycle arrest in pancreatic and cervical adenocarcinoma cells.28,29 We therefore asked if MIF might influence cell cycle arrest of tubular cells. We quantified cell cycle arrest in vivo using Ki67/pH3 coimmunofluorescence and morphologic cell-based analyses (Supplemental Figure 9, A and B). Already on day 2 of UUO, Mif−/− mice had significantly more tubular cells in the G2 phase compared with WT (Figure 6C). Similar findings were obtained in UUO day 5, and FAN and I/R day 21 (Supplemental Figure 9, C and D). Previous in vitro data showed that MIF regulates SCF ubiquitin ligase activity, which controls the transition between G1/S and G2/M phase by its association with the Jab1/CSN5 subunit of the COP9 signalosome.11,30 It was shown that in human embryonic kidney cells (HEK293), MIF knockdown decreased the expression of positive regulators of cell cycle progression, cyclins and cyclin-dependent kinases (CDK). Conversely, the CDK-inhibitors, which antagonize cyclins and CDKs, and thereby block cell cycle progression, were significantly elevated after MIF knockdown.31 Our experiments showed that Mif−/− mice had significantly increased protein expression of the CDK inhibitor 1B (p27Kip1) and decreased expression of cyclin B1, which induces progression from the G2 to the M phase (Figure 6, D and E, Supplemental Figure 9E).
In vitro, MIF induced, and its genetic deletion reduced, proliferation of primary murine renal tubular cells (Figure 6G). Compared with WT cells, significantly more primary murine TECs (pmTECs) from Mif−/− mice were arrested in the G2 phase (Figure 6F). It was not possible to expand pmTECs from Mif−/− mice beyond the first passage due to lack of proliferation, possibly because of cell cycle arrest. Therefore, we used the specific MIF inhibitor ISO-1 for further scrutiny. In LPS-activated tubular cells, MIF reduced and ISO-1 increased the expression of both p27 and phosphorylated serine 10 of histone H3 (pH3) (Figure 6H). In addition to CCL2, we analyzed the profibrotic mediators TGF-β and PDGF-B. In vivo, both Tgfb and Pdgfb were significantly increased in animals upon MIF neutralization, whereas they were reduced in mice treated with rMIF (Supplemental Figure 2, A–C), closely resembling the data on cell cycle arrest. Such paracrine signaling from injured or cell cycle–arrested tubular cells is likely one major source of factors activating renal (myo) fibroblasts.2,4 To analyze the role of MIF in the crosstalk between tubular cells and fibroblasts, we generated conditioned media from primary tubular cells, which were stimulated with rMIF or treated with ISO-1. Compared with conditioned medium from unstimulated tubular cells, conditioned medium from MIF-stimulated tubular cells reduced proliferation rate of primary renal fibroblasts, whereas conditioned medium from ISO-1–treated tubular cells increased fibroblast proliferation (Figure 6I).
We next analyzed apoptosis. On day 2 of UUO, no significant difference in apoptosis of tubular cells between WT and Mif−/− was observed, as analyzed by quantification of TUNEL and cleaved caspase 3 staining (Supplemental Figure 10A). At later time points of UUO, i.e., on day 5 and on day 7, a decrease in tubular cells undergoing apoptosis was observed in both Mif−/− or ISO-1–treated mice compared with the controls, respectively (Supplemental Figure 10, B and C). Vice versa, treatment with rMIF in UUO significantly increased the number of tubular cells undergoing apoptosis (Supplemental Figure 10D).
In contralateral and healthy nonfibrotic kidneys, we found no differences in proliferation, cell cycle arrest, apoptosis, or the expression of Ccl2, Tgfb, and Pdgfb between the groups (data not shown).
We conclude that MIF abrogated cell cycle arrest in tubular cells, reducing the production of the chemokine CCL2 and the profibrotic mediators TGF-β and PDGF-B, while attenuating the proliferation of fibrogenic fibroblasts, together resulting in reduced renal inflammation and fibrosis.
Discussion
Here, we show that in the setting of chronic renal injury and fibrosis in the tubulointerstitial compartment of the kidney, MIF acts as a potent antifibrotic and anti-inflammatory factor. This is noteworthy because virtually all chronic and severe acute kidney diseases are characterized by prominent tubulointerstitial inflammation and fibrosis. Most previous studies showed that MIF exacerbates acute inflammatory and autoimmune diseases, including lupus nephritis and crescentic GN, and also renal cystic disease, mechanistically mainly by aggravating inflammation and pathologic cell proliferation.16,18,32,33 Only a few findings have suggested that MIF might have protective effects in specific organs and disease settings. We have shown previously that MIF promotes liver fibrosis in a nonalcoholic steatohepatitis model via its receptor CD74,34 but is antifibrotic in obstructive and toxic models of liver fibrosis.35 MIF was also found to have cardioprotective effects, in part due to activation of AMP-activated protein kinase (AMPK) signaling during myocardial I/R,36–39 and AMPK activation is also operative in MIF’s protective action in liver fibrosis.35 In contrast to the liver, we show that the renoprotective effects of MIF were similar in all models of fibrosis independent of the underlying disease induction mechanism, which included mechanical obstruction, I/R, toxin, or glomerular injury. The effects were observed at all time points analyzed, which represented early, established, and also advanced disease and fibrosis stages. In addition, similar effects were observed in male and female mice. Our data suggest that, compared with other organs, i.e., the liver, in the kidneys, MIF has similar effects on fibrosis development in a broad range of injurious stimuli, arguing for a common, central mechanism driving progressive kidney diseases and fibrosis.
Cell cycle arrest of tubular epithelial cells has emerged as a common and crucial process driving progressive renal diseases.23–26 The consequences of cell cycle arrest and the major mechanisms responsible for kidney disease progression are the inability of arrested tubular cells to proliferate and thus regenerate the kidney as well as their phenotypic switch driving both renal fibrosis and inflammation in a paracrine manner.6,23 Our data identified MIF as a potent factor inducing cell cycle entry and abrogating cell cycle arrest in tubular cells, thereby improving regeneration, reducing secretion of profibrotic and proinflammatory factors, and consequently leading to reduced fibrosis and inflammation and better preservation of renal function. We observed the effects on cell cycle entry and arrest already very early after renal injury, suggesting that MIF might be an important initiating molecular mechanism for tubular regeneration. Only a few in vitro studies in tumor cells have previously suggested a role of MIF in cell cycle arrest.29 We show herein that the effect of MIF on renal tubular cell cycle arrest is robust and occurs in nontransformed cells in vitro and in vivo, supporting a broader regulatory role for MIF beyond malignancies.
Our data showed renoprotective effects of MIF in models of tubulointerstitial kidney diseases. This is in contrast to previous reports describing a detrimental role of MIF in models of immune-mediated GN and polycystic kidney disease (PKD).17,33,40 The main reason for these divergent effects is most likely the different effect of MIF on inflammatory versus cytoprotective and regenerative pathways in disease pathogenesis.
The most extensively described functions of MIF are its proinflammatory and chemokine-like effects.9,10,41 In our animal models, we consistently found MIF’s dominant role to be tissue-protective and associated with less inflammation and reduced CCL2 expression. This suggests that in the tubulointerstitial compartment during kidney injury and fibrosis, beneficial early effects of MIF on cell cycle arrest resulting in reduced CCL2 expression and secretion are more prominent and overrule MIF’s direct proinflammatory and chemokine-like effects. Our data also support the importance of cell cycle arrest as an important mechanism relevant for the development and progression of renal diseases. The disease context is likely the main reason for the divergent effects of MIF on CCL2 expression and inflammation. MIF might increase chemokine expression and is proinflammatory in diseases with prominent involvement of the immune system, such as we have shown for autoimmune diseases.14,16,42 Contrarily, in chronic injury with fibrosis, where inflammatory component is much less important and local epithelium is mainly involved in the pathogenesis, as we show here in both primary tubulointerstitial models but also in a primary glomerular model with secondary tubular epithelial damage, i.e., in Alport mice, the noninflammatory effects predominate. In this regard and compared with other pathologic situations and other organs, the cellular source of MIF might also explain our findings. Our data show that the renoprotective effects can be fully ascribed to the local tubular cell–derived MIF but not bone marrow–derived MIF.
MIF promoted proliferation of tubular cells after tubular injury, thereby promoting regeneration of renal epithelium. MIF is known to promote the proliferation of different cell types in pathologic conditions, e.g., in malignancies, proliferative glomerulonephritides, or PKD.12,16,28,29,33,43,44 In PKD, for instance, tubular cell proliferation is detrimental and leads to cyst formation and secondary inflammation and fibrosis.33 In contrast to the above situations, in CKD, reflected by the models we have used, cell proliferation is an attempt of renal epithelium for compensatory regeneration, and is beneficial.45,46
Clearance of irreparably damaged cells is the main physiologic role of apoptosis. Failure to undergo apoptosis is implicated in various, in particular chronic atrophic, diseases.47,48 Cells in G2/M cell cycle arrest were suggested to be resistant to apoptosis. Thereby, cell cycle–arrested cells survive, accumulate, and further promote fibrosis.24,49 Our data showing significantly reduced apoptosis in fibrosis when MIF is neutralized, versus increased apoptosis when MIF is substituted, support this hypothesis. MIF was shown to induce apoptosis by its endonuclease activity and nuclear shuttling in neurons,19 suggesting that in the kidneys MIF might also regulate apoptosis directly, independently of the effects on cell cycle arrest.
MIF inhibition is undergoing first clinical trials (NCT01765790; NCT01541670).14,15 Several anticancer and anti-inflammatory drugs have significant renal side effects and our data suggest that this might also be the case for MIF inhibition. Such potential side effects may be limited only to patients with preexisting renal disease and fibrosis, because we observed no effects of MIF inhibition in nonfibrotic and healthy kidneys. Vice versa, therapeutic strategies making use of MIF pathway agonists that result in pharmacologic augmentation of MIF activity might be a potential novel treatment approach for patients with CKD and renal fibrosis.38,44,50 One example of such an agonist is MIF20, a triazole-based, small molecule that promotes CD74 signal transduction and is showing promising results in experimental ischemic cardiac damage.51,52 In particular, manipulation of local tubular MIF by upregulation or pharmacologic augmentation, e.g., using cell-specific targeting, could be envisaged as a possible future therapeutic approach for renal fibrosis.
In summary, we have identified a hitherto unexpected role for local renal MIF, which acts as an endogenous renoprotective molecule by abrogating cell cycle arrest of tubular cells and thereby limits inflammation and fibrosis.
Concise Methods
Animal Experiments
All animal experiments were approved by the local and governmental review boards. The animals were held in cages with constant temperature and humidity with ad libitum access to tap water and standard chow. First, 12-week-old male Mif−/− (n=26)44 and WT littermates all on C57BL6/N background (n=26) underwent UUO. Renal tissue (for histologic evaluation, and protein and RNA isolation) from both the obstructed and contralateral kidney was obtained when the mice were euthanized, on day 2 (n=10 per group), day 5 (n=8 per group), and day 10 (n=8 per group) after UUO. For MIF neutralization via inhibitory antibodies, 16 11-week-old male C57BL6/N mice (n=8) were injected 2 days before UUO with an anti-MIF neutralizing antibody (NIHIIID.9)17 or control isotype-matched IgG1 (n=8) (both at 5 mg/kg body wt i.p.) and afterward daily until day 5 of UUO. For therapeutic approach, 24 12-week-old mice underwent UUO and were daily i.p.-injected from day 3 after operation with vehicle (DMSO/PBS, n=8), rmMIF (synthesized as described previously9) (n=8), or ISO-1 (n=8) to day 7 of UUO. A further 16 12-week-old male C57BL6/N mice underwent UUO and received rmMIF (n=8) or vehicle (n=8) from day 5 daily i.p. until euthanasia 10 days after UUO. The endotoxin level of rMIF ranged between 5 and 10 pg endotoxin per microgram rMIF protein. For BrdU incorporation each mouse was injected i.p. 4 hours before euthanasia with 1 mg BrdU.
The bone marrow reconstitution experiments in Mif−/− and WT littermates were performed as described previously using a well established protocol for which a reconstitution rate of 96%±2% was reached.10,16,50,53,54 For bone marrow transplantation, 10-week-old female Mif−/− (n=17) and C57BL/6N (n=17) wild-type littermates were used as recipients and 10-week-old male Mif−/− (n=6) and C57BL/6N (n=7) wild-type littermates as donors. Recipients were irradiated with 6.8 Gy for 8 minutes two times with a pause of 4 hours. A total of 2×106 bone marrow cells obtained from femur and tibia of donor mice were injected via tail vein in recipient mice 6 hours after the first irradiation. All mice were treated with Sulfadoxinum and Trimethoprimum via drinking water for 2 weeks. After two additional weeks, i.e., 4 weeks after bone marrow transplantation, mice underwent UUO and were analyzed on day 5 after disease induction.
For tubular cell–specific Mif deletion, Mifflox/flox mice were crossbred with Pax8Cre/+. Pax8+/+::Mifflox/flox (n=6) or Pax8Cre/+::Mifflox/flox (n=5) and underwent UUO for 5 days. All mice were on a C57BL6/N background.
As second model, unilateral I/R injury was performed in 12-week-old female Mif−/− (n=12) and WT littermate (n=10) mice for 21 days. A further 16 C57BL6/N mice underwent I/R for 21 days and were treated daily i.p. with vehicle (n=8) or ISO-1 (n=8) from day 3.
Next, unilateral I/R injury was performed in 12-week-old female Mif−/− (n=7) and WT littermate (n=9) mice for 14 days. Afterward, the healthy, contralateral kidney was removed and animals were euthanized 2 days later. One day before uninephrectomy and 1 day before euthanasia, the mice were placed in metabolic cages for 24 hours for stool-free urine collection for biochemical analyses. Standard blood and urine parameters were measured.
As a third model, FAN was performed in 10-week-old female (Mif−/− n=6) and WT littermates (n=6) by weekly i.p. injection of folic acid (250 mg/kg of body wt and dissolved in NaHCO3). All mice were euthanized after 8 weeks. One day before folic acid injection and 1 day before euthanasia, the mice were placed in metabolic cages for 24-hour, stool-free urine collection for biochemical analyses. Standard blood and urine parameters were measured.
As a fourth model of renal injury, 18 male Col4a3−/− (Alport) mice, all on Sv129 background, were injected with anti-MIF mAb (n=9) or control isotype-matched IgG1 (n=9) (both 5 mg/kg body wt i.p.) daily for 3 weeks, and 16 male Col4a3−/− (Alport) mice were injected with ISO-1 (n=8) or control (n=8) daily for 2 weeks.
In all experiments, mice were euthanized by exsanguination via abdominal aorta in ketamine (100 mg/kg body wt) and xylazine (10 mg/kg body wt) i.p. anesthesia; kidneys were taken and immediately processed for further analyses. If not analyzed immediately, all samples apart from fixed tissues for histology and immunohistochemistry were stored at −80°C.
Renal Morphology and Immunohistochemistry
Tissue for light microscopy and immunohistochemistry was fixed in methyl Carnoy’s solution or formalin and embedded in paraffin. The sections (1-µm) were stained with periodic acid–Schiff reagent and counterstained with hematoxylin.
All immunohistochemical staining, except for MIF, was performed on methyl Carnoy’s–fixed renal tissues. The sections (1-µm) were processed as previously described.55,56 Primary and secondary antibodies are shown in Table 1. The evaluation of immunohistochemistry was performed by using the ImageJ v1.48 software (http://imagej.nih.gov/ij/). The percentage of positively stained, cortical area in each tissue was calculated separately in 20 interstitial fields representing almost the whole cortical area. All histomorphologic analyses were done in a blinded manner.
Table 1.
List of primary and secondary antibodies
| Target | Clone/Label | Host | Application | Supplier |
|---|---|---|---|---|
| α-SMA | 1A4 | Mouse | IHC; IF | Dako |
| β-Actin | A5441 | Mouse | WB | Sigma-Aldrich |
| Caspase-3, cleaved | Rabbit | IHC | Cell Signaling | |
| CD3 | Rat | IHC | Serotec AbD | |
| CD68 | PGM1 | Mouse | IHC | Dako |
| CD74 | Rat | IF | BD Pharmingen | |
| Collagen type I | Goat | IHC | Southern Biotech | |
| Collagen type III | Goat | IHC | Southern Biotech | |
| ErHr3 | Rat | IHC | BMA Biomedicals | |
| F4/80 | CI:A3–1 | Rat | IHC | Serotec AbD |
| Fibronectin | Rabbit | IHC | Millipore | |
| GAPDH | Mouse | WB | Novus Biologicals | |
| Histone H3 pS10 | Mouse | WB, IF | Abcam | |
| KI67 | SP6 | Rabbit | IF | Abcam |
| LCN2 | AF3508 | Goat | IHC | R&D Systems |
| MIF | Rabbit | IF | Life Technology | |
| MIF | Rabbit | WB | Sigma-Aldrich | |
| P27kip1 | Mouse | WB | BD Pharmingen | |
| PDGFRβ | Rabbit | IF | Abcam | |
| Rabbit IgG | biotin | Goat | IHC | Vector |
| Rat IgG | biotin | Goat | IHC | Vector |
| Mouse IgG | biotin | Horse | IHC | Vector |
| Goat IgG | biotin | Horse | IHC | Vector |
| Mouse IgG2A | biotin | Goat | IHC | Antikoerper-online |
| Rabbit IgG | Alexa-647 | Donkey | IF | Life Technology |
| Mouse IgG2A | Alexa-488 | Goat | IF | Life Technology |
| Mouse IgG | Alexa-488 | Donkey | IF | Life Technology |
| Mouse IgG1 | Alexa-555 | Goat | IF | Life Technology |
| Rabbit Ig | HRP | Goat | WB | Dako |
| Mouse Ig | HRP | Goat | WB | Vector |
IHC, immunohistochemistry; IF, immunofluorescence; WB, Western blot.
Immunofluorescence and Double Staining
MIF staining was performed on formalin-fixed tissue with heat-induced antigen retrieval in citrate buffer (pH 6.0). Costaining for α-SMA or pH3 with Ki67 was performed on methyl Carnoy’s–fixed tissue with heat-induced antigen retrieval in citrate buffer (pH 6.0). Primary and secondary antibodies are shown in Table 1. TUNEL staining of tissue sections was performed as recommended by the manufacturer (Roche).
RNA Extraction and qRT-PCR
Total RNA was collected from renal cortex using the RNAeasy Mini Kit (Qiagen, Hilden, Germany). RNA purity determination and cDNA synthesis were performed as described previously.57 For the normalization of the data, the standard ΔΔCT method was used with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a housekeeping gene. The expression of genes of interest was calculated as relative expression units in comparison to wild type or control group (arbitrarily set as 1). Sequences of primers used for qRT-PCR are shown in Table 2.
Table 2.
List of primers used for real-time RT-PCR (all genes were murine)
| Gene | Sequence (Sense) | Sequence (Antisense) | Probe |
|---|---|---|---|
| Mif | 5′-CGTGCCGCTAAAAGTCATGA-3′ | 5′-GCAAGCCCGCACAGTACAT-3′ | SYBR Green |
| Col 1A1 | 5′-TCGCTTCACCTACAGCACCC-3′ | 5′-TGACTGTCTTGCCCCAAGTTC-3′ | SYBR Green |
| Col 3A1 | 5′-TGAAGATGTCGTTGATGTGCAG-3′ | 5′-GCAGTGGTATGTAATGTTCTGGGAG-3′ | SYBR Green |
| Tgfb | 5′-GGACTCTCCACCTGCAAGACC-3′ | 5′-GGATGGCTTCGATGCGC-3′ | SYBR Green |
| CDKN1A | 5′-GTGGCCTTGTCGCTGTCTT-3′ | 5′-GCGCTTGGAGTGATAGAAATCTG-3′ | SYBR Green |
| P27 | 5′-GCAAAACAAAAGGGCCAACA-3′ | 5′-GGGCGTCTGCTCCACAGT-3′ | SYBR Green |
| CCND1 | 5′-ACAACTCTATCCGGCCCGA-3′ | 5′-GGCCAGGTTCCACTTGAGC-3′ | SYBR Green |
| CCNE1 | 5′-GTTGGCTGCTTAGAATTTCCTTATG-3′ | 5′-ACTGATAACCTGAGACCTTCTGCAT-3′ | SYBR Green |
| Gapdh | 5′-GGCAAATTCAACGGCACAGT-3′ | 5′-AGATGGTGATGGGCTTCCC-3′ | SYBR Green |
| Ccl2 | 5′-TGGCTCAGACAGATGCAGT-3′ | 5′-ATTGGGATCATCTTGCTGGTG-3′ | SYBR Green |
| Acta2 | 5′-CGTGGATGCCCGCTGA-3′ | 5′-TACGCTTCCGCTGCCC-3′ | SYBR Green |
| Fibronectin | 5′-GATGGAATCCGGGAGCTTTT-3′ | 5′-TGCAAGGCAACCACACTGAC-3′ | 5′-CCGGCCTGAGGCCCTGCAG-3′ |
| Pdgfb | 5′-CCATCCGCTCCTTTGATGAT-3′ | 5′-AAGTCCAGCTCAGCCCCAT-3′ | 5′-CGCCTGCTGCACAGAGACTCCGTA-3′ |
Isolation of Primary Tubular Cells
Mice were anesthetized using ketamine/xylazine, euthanized by exsanguination via abdominal aorta, and directly perfused via the left ventricle with Fe3O4 (Sigma-Aldrich) diluted in 20 ml 0.9% NaCl. The kidneys then were transferred into RPMI 1640 medium (Life Technologies) containing 1% penicillin/streptomycin and cut into small fragments. Afterward, fragments were treated for 30 minutes at 37°C with 1 mg/ml collagenase Type IV (Worthington). The small kidney fragments were gently sieved through a 100-µm cell strainer, centrifuged, and resuspended in PBS. Glomeruli containing Fe3O4 were deleted using a magnetic particle concentrator (DynaMag TM-2; Life Technologies). Remaining tubulointerstitial cells were seeded and selected by specific culture media. Primary tubular cells (pmTECs) were cultured in advanced DMEM/F12 supplemented with rmEGF (100 ng/ml), HEPES (14 mM), hydrocortisone (100 nmol/L), penicillin (100 U/ml), streptomycin (100 µg/ml), and L-Glutamine (2 mM). Cells were harvested for protein lysate or RNA isolation in passage 2.
Immunocytofluorescence
Primary murine renal tubular cells (pmTECs) were seeded directly after isolation on collagen-coated coverslips and fixed with 4% paraformaldehyde at 37°C for 30 minutes. After permeabilization with Triton X-100 in PBS and washing with PBS, cells were stained with Ki67 (Abcam), Histone H3 PS10 (Abcam), and DAPI (Roche). For evaluation of the cell cycle, all DAPI and Ki67 double-positive nuclei were counted. Afterward, Ki67-positive cells were divided into M and G2 phase by nuclear pattern of histone H3 phosphoS10 (H3p), which is specific for these two phases of cell cycle, and the percentage for each phase was calculated in relation to total Ki67+ cells.
In Vitro Stimulation Assays
pmTECs were isolated and cultured as described above. For stimulation, pmTECs were seeded into six-well plates and incubated with 30 ng/ml ISO-1 (Synthon-Lab) or 100 ng/ml rmMIF, and stimulated with 200 ng/ml LPS (Sigma-Aldrich), for 6 hours. Afterward, cells were harvested for protein lysates or RNA isolation. To obtain conditioned media 2 hours after incubation with rMIF or ISO-1, medium was removed and the tubular cells were rinsed twice with PBS and fresh medium was added. Twenty-four hours later, the cell-free conditioned media from tubular cells were transferred directly to the fibroblasts.
Cell Proliferation Assays
For the analysis of cell proliferation, murine tubular cells were seeded into 24-well plates and growth-synchronized by serum deprivation for 24 hours before stimulation with 100 ng/ml rmMIF or vehicle. Manual cell counting in Neubauer chambers was performed at daily intervals after stimulation.
Lysate Preparation and Western Blot Analysis
Cells and tissue were lysed in RIPA buffer (50 mM Tris-HCl, 150 mM Nonidet P-40, 1 mM sodium deoxycholate, 1 mM EDTA, and 1 mM sodium orthovanadate) containing complete protease inhibitor cocktail (Roche) at 4°C for 15 minutes, sonicated three times for 10 seconds, and centrifuged at 10,000 × g for 15 minutes at 4°C. Supernatants containing cellular proteins were collected and protein concentrations determined by BCA protein assay with BSA standard (Interchim).
Denatured protein samples were separated by electrophoresis in 10% SDS-PA gels, transferred to nitrocellulose membranes, blocked with 2% (w/v) BSA in PBS, washed with TTBS, and incubated with primary antibodies diluted in TTBS overnight at 4°C. The following antibodies were applied: mouse anti–human GAPDH (Novus Biologicals), rabbit anti–rat MIF (Sigma-Aldrich), anti-P27Kip1 (BD Pharmingen), and anti-actin (Sigma-Aldrich). Primary antibodies were detected using HRP-conjugated antibodies and visualized by ECL (Roche). GAPDH quantification was performed on the same nitrocellulose blots to normalize for loading incongruities. Band intensities were quantified by Scion Image software.
Quantification of Collagen Content
Collagen content in 60-µg kidney tissue lysates was quantified by Sircol soluble collagen assay (Biocolor). The assay was performed as recommended by the manufacturer.
Statistical Analyses
All values are expressed as mean±SD. After testing for Gaussian distribution with D’Agostino–Pearson omnibus and Shapiro–Wilk normality tests, we have used the appropriate test for multiple comparisons, using ANOVA with post hoc Tukey test or Kruskal–Wallis with post hoc Dunn’s test, and for comparison of two groups t test and Mann–Whitney test for normally and not-normally distributed parameters, respectively. All tests were unpaired and two-tailed. Statistical significance was defined as P≤0.05.
Disclosures
R.B. is listed as an inventor on a Yale University patent application describing the therapeutic utility of MIF agonism.
Supplementary Material
Acknowledgments
The technical assistance of Anna Maria Hübbers, Hongqi Lue, Marianna Tatarek-Nossol, Marie Cherelle Timm, and Simon Wilhelm Otten is gratefully acknowledged.
This work was supported by financial research grants of the Else-Kröner Fresenius Foundation (EKFS 2012_A216) to P.B. and J.B.; German Research Foundation Sonderforschungsbereich-Transregio 57 “Mechanisms of organ fibrosis” to P.B., J.F., and J.B.; BO 3755/3-1 to P.B.; and Munich Cluster for Systems Neurology (EXC 1010 SyNergy) to J.B.; German Ministry of Education and Research (Consortium sop focal segmental glomerulosclerosis number 01GM1518A) to P.B.; the Interdisciplinary Centre for Clinical Research within the Faculty of Medicine at the Rheinish-Westphalian Technical University, Aachen University to P.B. (K7-3); and the National Institutes of Health (R.B.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2017020190/-/DCSupplemental.
References
- 1.Global Burden of Disease Study 2013 Collaborators : Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: A systematic analysis for the Global Burden of Disease study 2013. Lancet 386: 743–800, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boor P, Sebeková K, Ostendorf T, Floege J: Treatment targets in renal fibrosis. Nephrol Dial Transplant 22: 3391–3407, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Boor P, Floege J: Chronic kidney disease growth factors in renal fibrosis. Clin Exp Pharmacol Physiol 38: 441–450, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Boor P, Floege J: The renal (myo-)fibroblast: A heterogeneous group of cells. Nephrol Dial Transplant 27: 3027–3036, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Boor P, Floege J: Renal allograft fibrosis: Biology and therapeutic targets. Am J Transplant 15: 863–886, 2015 [DOI] [PubMed] [Google Scholar]
- 6.Grande MT, Sánchez-Laorden B, López-Blau C, De Frutos CA, Boutet A, Arévalo M, Rowe RG, Weiss SJ, López-Novoa JM, Nieto MA: Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med 21: 989–997, 2015 [DOI] [PubMed] [Google Scholar]
- 7.Wynn TA, Ramalingam TR: Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bendrat K, Al-Abed Y, Callaway DJ, Peng T, Calandra T, Metz CN, Bucala R: Biochemical and mutational investigations of the enzymatic activity of macrophage migration inhibitory factor. Biochemistry 36: 15356–15362, 1997 [DOI] [PubMed] [Google Scholar]
- 9.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R: MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 365: 756–759, 1993 [DOI] [PubMed] [Google Scholar]
- 10.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C: MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med 13: 587–596, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, Grell M, Finkelmeier D, Brunner H, Bernhagen J: Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 408: 211–216, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Mitchell RA, Bucala R: Tumor growth-promoting properties of macrophage migration inhibitory factor (MIF). Semin Cancer Biol 10: 359–366, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Morand EF, Bucala R, Leech M: Macrophage migration inhibitory factor: An emerging therapeutic target in rheumatoid arthritis. Arthritis Rheum 48: 291–299, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Greven D, Leng L, Bucala R: Autoimmune diseases: MIF as a therapeutic target. Expert Opin Ther Targets 14: 253–264, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Hussain F, Freissmuth M, Völkel D, Thiele M, Douillard P, Antoine G, Thurner P, Ehrlich H, Schwarz HP, Scheiflinger F, Kerschbaumer RJ: Human anti-macrophage migration inhibitory factor antibodies inhibit growth of human prostate cancer cells in vitro and in vivo. Mol Cancer Ther 12: 1223–1234, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Djudjaj S, Lue H, Rong S, Papasotiriou M, Klinkhammer BM, Zok S, Klaener O, Braun GS, Lindenmeyer MT, Cohen CD, Bucala R, Tittel AP, Kurts C, Moeller MJ, Floege J, Ostendorf T, Bernhagen J, Boor P: Macrophage migration inhibitory factor mediates proliferative GN via CD74. J Am Soc Nephrol 27: 1650–1664, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leng L, Chen L, Fan J, Greven D, Arjona A, Du X, Austin D, Kashgarian M, Yin Z, Huang XR, Lan HY, Lolis E, Nikolic-Paterson D, Bucala R: A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. J Immunol 186: 527–538, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan HY, Bacher M, Yang N, Mu W, Nikolic-Paterson DJ, Metz C, Meinhardt A, Bucala R, Atkins RC: The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med 185: 1455–1465, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, Kim B, Bao L, Harraz MM, Chang C, Chen R, Wang JE, Kam TI, Jeong JS, Xie Z, Neifert S, Qian J, Andrabi SA, Blackshaw S, Zhu H, Song H, Ming GL, Dawson VL, Dawson TM: A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354(6308): aad6872, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lan HY, Yang N, Nikolic-Paterson DJ, Yu XQ, Mu W, Isbel NM, Metz CN, Bucala R, Atkins RC: Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int 57: 499–509, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Tesch GH: MCP-1/CCL2: A new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol 294: F697–F701, 2008 [DOI] [PubMed] [Google Scholar]
- 22.Wada T, Yokoyama H, Matsushima K, Kobayashi K: Chemokines in renal diseases. Int Immunopharmacol 1: 637–645, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Bonventre JV: Primary proximal tubule injury leads to epithelial cell cycle arrest, fibrosis, vascular rarefaction, and glomerulosclerosis. Kidney Int Suppl (2011) 4: 39–44, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonventre JV: Maladaptive proximal tubule repair: Cell cycle arrest. Nephron Clin Pract 127: 61–64, 2014 [DOI] [PubMed] [Google Scholar]
- 25.Canaud G, Bonventre JV: Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol Dial Transplant 30: 575–583, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV: Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M, Kalluri R: Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 21: 998–1009, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denz A, Pilarsky C, Muth D, Rückert F, Saeger HD, Grützmann R: Inhibition of MIF leads to cell cycle arrest and apoptosis in pancreatic cancer cells. J Surg Res 160: 29–34, 2010 [DOI] [PubMed] [Google Scholar]
- 29.Guo P, Wang J, Liu J, Xia M, Li W, He M: Macrophage immigration inhibitory factor promotes cell proliferation and inhibits apoptosis of cervical adenocarcinoma. Tumour Biol 36: 5095–5102, 2015 [DOI] [PubMed] [Google Scholar]
- 30.Nemajerova A, Mena P, Fingerle-Rowson G, Moll UM, Petrenko O: Impaired DNA damage checkpoint response in MIF-deficient mice. EMBO J 26: 987–997, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L, Ji C, Chen J, Li Y, Fu X, Xie Y, Gu S, Mao Y: A global genomic view of MIF knockdown-mediated cell cycle arrest. Cell Cycle 7: 1678–1692, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Lan HY, Mu W, Yang N, Meinhardt A, Nikolic-Paterson DJ, Ng YY, Bacher M, Atkins RC, Bucala R: De Novo renal expression of macrophage migration inhibitory factor during the development of rat crescentic glomerulonephritis. Am J Pathol 149: 1119–1127, 1996 [PMC free article] [PubMed] [Google Scholar]
- 33.Chen L, Zhou X, Fan LX, Yao Y, Swenson-Fields KI, Gadjeva M, Wallace DP, Peters DJ, Yu A, Grantham JJ, Li X: Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease. J Clin Invest 125: 2399–2412, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinrichs D, Berres ML, Coeuru M, Knauel M, Nellen A, Fischer P, Philippeit C, Bucala R, Trautwein C, Wasmuth HE, Bernhagen J: Protective role of macrophage migration inhibitory factor in nonalcoholic steatohepatitis. FASEB J 28: 5136–5147, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinrichs D, Knauel M, Offermanns C, Berres ML, Nellen A, Leng L, Schmitz P, Bucala R, Trautwein C, Weber C, Bernhagen J, Wasmuth HE: Macrophage migration inhibitory factor (MIF) exerts antifibrotic effects in experimental liver fibrosis via CD74. Proc Natl Acad Sci U S A 108: 17444–17449, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rassaf T, Weber C, Bernhagen J: Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc Res 102: 321–328, 2014 [DOI] [PubMed] [Google Scholar]
- 37.Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, Merk M, Zierow S, Bernhagen J, Ren J, Bucala R, Li J: Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation 122: 282–292, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi D, Atsina K, Qu L, Hu X, Wu X, Xu B, Piecychna M, Leng L, Fingerle-Rowson G, Zhang J, Bucala R, Young LH: The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J Clin Invest 124: 3540–3550, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH: Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 451: 578–582, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Lan HY, Nikolic-Paterson DJ, Mu W, Atkins RC: Local macrophage proliferation in the pathogenesis of glomerular crescent formation in rat anti-glomerular basement membrane (GBM) glomerulonephritis. Clin Exp Immunol 110: 233–240, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, McColl SR, Hickey MJ: Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol 177: 8072–8079, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Hoi AY, Hickey MJ, Hall P, Yamana J, O’Sullivan KM, Santos LL, James WG, Kitching AR, Morand EF: Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol 177: 5687–5696, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Ohta S, Misawa A, Fukaya R, Inoue S, Kanemura Y, Okano H, Kawakami Y, Toda M: Macrophage migration inhibitory factor (MIF) promotes cell survival and proliferation of neural stem/progenitor cells. J Cell Sci 125: 3210–3220, 2012 [DOI] [PubMed] [Google Scholar]
- 44.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Müller W, Bucala R: The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci U S A 100: 9354–9359, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu F, Liu W, Li T, Wan J, Tian J, Zhou Z, Li H, Liu Y, Hou FF, Nie J: Numb contributes to renal fibrosis by promoting tubular epithelial cell cycle arrest at G2/M. Oncotarget 7: 25604–25619, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vincent IS, Okusa MD: Biology of renal recovery: Molecules, mechanisms, and pathways. Nephron Clin Pract 127: 10–14, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanz AB, Santamaría B, Ruiz-Ortega M, Egido J, Ortiz A: Mechanisms of renal apoptosis in health and disease. J Am Soc Nephrol 19: 1634–1642, 2008 [DOI] [PubMed] [Google Scholar]
- 48.Shimizu A, Yamanaka N: Apoptosis and cell desquamation in repair process of ischemic tubular necrosis. Virchows Arch B Cell Pathol Incl Mol Pathol 64: 171–180, 1993 [DOI] [PubMed] [Google Scholar]
- 49.Pietenpol JA, Stewart ZA: Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. Toxicology 181-182: 475–481, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Liehn EA, Kanzler I, Konschalla S, Kroh A, Simsekyilmaz S, Sönmez TT, Bucala R, Bernhagen J, Weber C: Compartmentalized protective and detrimental effects of endogenous macrophage migration-inhibitory factor mediated by CXCR2 in a mouse model of myocardial ischemia/reperfusion. Arterioscler Thromb Vasc Biol 33: 2180–2186, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang J, Tong C, Yan X, Yeung E, Gandavadi S, Hare AA, Du X, Chen Y, Xiong H, Ma C, Leng L, Young LH, Jorgensen WL, Li J, Bucala R: Limiting cardiac ischemic injury by pharmacological augmentation of macrophage migration inhibitory factor-AMP-activated protein kinase signal transduction. Circulation 128: 225–236, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jorgensen WL, Gandavadi S, Du X, Hare AA, Trofimov A, Leng L, Bucala R: Receptor agonists of macrophage migration inhibitory factor. Bioorg Med Chem Lett 20: 7033–7036, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Chen T, Leng L, Fan J, Cao K, Duan Z, Zhang X, Shao C, Wu M, Tadmori I, Li T, Liang L, Sun D, Zheng S, Meinhardt A, Young W, Bucala R, Ren Y: MIF produced by bone marrow-derived macrophages contributes to teratoma progression after embryonic stem cell transplantation. Cancer Res 72: 2867–2878, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zernecke A, Schober A, Bot I, von Hundelshausen P, Liehn EA, Möpps B, Mericskay M, Gierschik P, Biessen EA, Weber C: SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ Res 96: 784–791, 2005 [DOI] [PubMed] [Google Scholar]
- 55.Djudjaj S, Chatziantoniou C, Raffetseder U, Guerrot D, Dussaule JC, Boor P, Kerroch M, Hanssen L, Brandt S, Dittrich A, Ostendorf T, Floege J, Zhu C, Lindenmeyer M, Cohen CD, Mertens PR: Notch-3 receptor activation drives inflammation and fibrosis following tubulointerstitial kidney injury. J Pathol 228: 286–299, 2012 [DOI] [PubMed] [Google Scholar]
- 56.Boor P, Konieczny A, Villa L, Kunter U, van Roeyen CR, LaRochelle WJ, Smithson G, Arrol S, Ostendorf T, Floege J: PDGF-D inhibition by CR002 ameliorates tubulointerstitial fibrosis following experimental glomerulonephritis. Nephrol Dial Transplant 22: 1323–1331, 2007 [DOI] [PubMed] [Google Scholar]
- 57.Ostendorf T, Rong S, Boor P, Wiedemann S, Kunter U, Haubold U, van Roeyen CR, Eitner F, Kawachi H, Starling G, Alvarez E, Smithson G, Floege J: Antagonism of PDGF-D by human antibody CR002 prevents renal scarring in experimental glomerulonephritis. J Am Soc Nephrol 17: 1054–1062, 2006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.