

Abstract
Type I IFN and nucleic acid-sensing TLRs are both strongly implicated in the pathogenesis of lupus with most patients expressing IFN-induced genes in peripheral blood cells and with TLRs promoting type I IFNs and autoreactive B cells. About a third of SLE patients, however, lack the IFN signature suggesting the possibility of type I IFN-independent mechanisms. Here, we examined the role of type I IFN and TLR trafficking and signaling in a xenobiotic systemic autoimmunity induced by mercury (HgIA). Strikingly, autoAb production in HgIA was not dependent on the type I IFN receptor even in NZB mice that require type I IFN signaling for spontaneous disease, but was dependent on the endosomal TLR transporter UNC93B1 and the endosomal proton transporter, SLC15A4. HgIA also required the AP-3 complex, which transports TLRs from the early endosome to the late endolysosomal compartments. Examination of TLR signaling pathways implicated the canonical NF-κB pathway and the proinflammatory cytokine IL-6 in autoantibody production, but not IRF7. These findings identify HgIA as a novel type I IFN-independent model of systemic autoimmunity and implicate TLR-mediated NF-κB proinflammatory signaling from the late endocytic pathway compartments in autoAb generation.
Keywords: autoimmunity, autoantibodies, animal model, interferons, TLR
Introduction
The α and β type I interferons are pleiotropic cytokines with diverse effects on the immune system through their actions on T and B lymphocytes, macrophages, and dendritic cells (DCs) (1). They are critical mediators of both innate and adaptive immune responses, and are required for the eradication of certain viral and bacterial infections (2, 3). It is therefore not surprising that type I IFNs play a significant role in autoimmunity (4–6). Indeed, lupus and other autoimmune diseases may occur following type I IFN treatment for cancer, hepatitis, and other disorders (7). Moreover, patients with SLE and other autoimmune diseases express genes induced by type I IFNs (“IFN signature”) in peripheral blood cells, that in some studies correlate with disease activity and severity (8–10). However, although virtually all pediatric SLE patients exhibit an IFN signature (11), about 30% of adult SLE patients do not, suggesting there may be other mechanisms or factors that bypass the requirement for IFN.
In contrast, most lupus-prone strains exhibit modest to absent elevations in type I IFN or IFN signature compared to human SLE (12). Yet, IFNα/β receptor 1 (Ifnar1)-deficiency and loss of type I IFN signaling markedly reduces the production of autoAbs and other disease manifestations (13–15) while sustained type I IFN exposure exacerbates autoimmunity (16–18). The exception is the MRL strain, for which Ifnar1-deficiency and IFN-β treatment have the opposite effects on disease (19, 20). IFN-γ (type II IFN) is also important for the development of lupus and required in all spontaneous and induced mouse models examined so far (21–26).
Although many cell types can produce type I IFN, plasmacytoid dendritic (pDC) cells have been identified as the most effective (27), and pDCs have been implicated as major contributors to lupus. Accordingly, deletion of DCs, including pDCs (28), defective pDC development due to IRF8 deficiency (29), impaired pDC function from reduced expression of the transcription factor E2-2 (Tcf4) (30), or early transient depletion of pDCs (31) all reduced lupus. Furthermore, disease is also inhibited in mice deficient for the proton peptide/histidine transporter, solute carrier family 15, member 4 (SLC15A4), in which pDCs are unable to produce type I IFNs in response to endosomal TLRs (TLR3, 7, 9) and B cells are defective in IgG2c class switching (29, 32–35).
The requirement for endosomal TLR signaling in systemic autoimmunity is well established in models of lupus and is consistent with the genetics and pathogenesis of human SLE (4). Notably, mice with nonfunctional UNC93B1 (Unc93b13d/3d, 3d), which regulates trafficking of TLRs from the endoplasmic reticulum (ER) to endolysosomes, do not respond to ligands of endolysosomal TLR3, TLR7, and TLR9 (36) and consequently 3d lupus-prone B6-Faslpr and BXSB mice fail to develop severe disease (37). Endosomal TLR signaling leads to type I IFN production via IRF7 and the induction of proinflammatory cytokines IL-6, IL-12, proIL-1β and TNF-α via NF-κB, MAP kinase, and IRF5 pathways (34, 38, 39). Evidence further suggests that TLR-mediated type I IFN and NF-kB signaling occurs in distinct endosomal compartments (40) and involves both endocytic and phagocytic endosomal pathways (41). The contribution of these TLR signaling endosomal constituents, particularly those related to NF-κB, have yet to be defined for autoimmune disease.
Mercury is a widely distributed environmental and industrial pollutant (42) that in both humans and animals, induces a milder form of autoimmunity including autoAbs and membranous nephropathy (43–46). The characteristics of mercury-induced autoimmunity (HgIA) are similar to SLE and include lymphocyte proliferation, increased class II MHC expression, hypergammaglobulinemia, polyclonal Abs to self-antigens, notably anti-nuclear Abs (ANAs), and to some extent immune complex deposits in mice (47), as well as necrotizing vasculitis in rats (48–52). Furthermore, HgIA requires CD4+ T cells, B and T cell co-stimulatory molecules, IFN-γ, and susceptible MHC and background genes (25, 53–57), which strongly supports the possibility of related or identical pathogenic mechanisms as spontaneous lupus.
In this paper, we dissected the roles of type I IFN and TLR endosomal trafficking and signaling pathways in autoAb production induced by mercury by examining mice with relevant gene defects. Our findings showed that xenobiotic autoimmunity differed from spontaneous lupus in being type I IFN-independent, but was similar in requiring endosomal TLR signaling and endosomal histidine transport. HgIA was also shown to require the AP-3 complex, Nfkb1, and Il6, but not IRF7. Thus, whereas TLR-induced NF-κB signaling is thought to occur in early endosomes (40), our findings indicate that late endosome/endolysosome and not early endosome NF-kB signaling compartments are essential for TLR-mediated HgIA autoAb production.
Material and Methods
Mice
NZB/B1Scr, NZB/B1Scr-Ifnar1−/−, BXSB/Scr, BXSB/Scr-Unc93b13d/3d, C57BL/6J-Inept (B6-Inept), B6-Feeble, B10.S/SgMcdJ and B10.S-Il6−/− were as described (13, 35, 37, 58). B6, B6Pin.C3-Ap3b1pe/J (pearl), B6.Cg-Nfkb1tm1Bal/J, and B6.129S2-Ifnar1tm1Agt/Mmjax mice were obtained from The Jackson Laboratory (Bar Harbor, ME). All strains were maintained at The Scripps Research Institute (TSRI).
Induction of autoimmunity
Male and female mice (6–16 wk-old) were injected with 40 μg HgCl2 (Mallinckrodt Baker Inc., Phillipsburg, NJ) in 100 μl PBS s.c. 2x/week up to 5 weeks; controls received PBS alone (25). The HgCl2 dose is consistent with occupational exposure (59, 60). Procedures were approved by the TSRI Institutional Animal Care and Use Committee and the TSRI Department of Environmental Health & Safety. Unless indicated, experimental groups were an equal mix of male and female mice.
Serology
Anti-nuclear Abs (ANAs) were determined by ELISA per manufacturer’s instructions (Inova Diagnostics, San Diego CA) or indirect immunofluorescence as described (61). Briefly, for the latter, HEp-2 cells on glass slides (Inova) were incubated with serum diluted 1/100 and then 1/200 dilution of Alexa Fluor 488-conjugated goat anti-mouse IgG (H&L) (Molecular Probes, Carlsbad, CA). ANAs were scored for intensity (0–4+ scale) and pattern under blinded conditions. ELISAs were used to quantitate anti-chromatin (61)(62), ENA5 (Sm, RNP, SS-A/Ro, SS-B/La, Scl70) (Inova Diagnostics, San Diego, CA), anti-DNA, serum IgG (Immunology Consultants Laboratory, Inc, Newburg, OR)(61)(63). A mouse reference sera (Bethyl Laboratories, Montgomery, TX) was used to generate the standard curve (800–0.04 ng/ml).
Immunohistology
Sections of kidney and spleen were stained for direct immunofluorescence as previously described (26). Briefly, 4 μm sections were fixed with ethanol, then incubated with serial dilutions of FITC-conjugated goat anti-mouse IgG Abs (Southern Biotechnology, Birmingham, AL). Glomerular deposits were graded by the highest dilution of Ab that could detect a specific fluorescence (end-point titer). Positive titers less than 1:40 were considered background. Vessel wall IgG deposits were graded on a 0–4+ scale (25). Slides were examined under blinded conditions.
TLR agonist and HgCl2 stimulation of cytokine expression
Mice received 100 μgs of polyinosinic-polycytidylic acid [poly(I:C)], 50 μg of R848 (InvivoGen, San Diego CA), or 40 μg HgCl2 in 200 μl PBS s.c. Preliminary experiments determined that maximal cytokine expression occurred 6 hours after poly(I:C) or HgCl2, and 3 hours after R848 injections. These exposure periods were used to measure IFN-α (LumiKine Xpress, InvivoGen, San Diego, CA), TNF-α, and IL-6 (ELISA MAX, BioLegend, San Diego, CA) by ELISA.
Flow cytometry
Single cell suspensions were prepared from spleen by passage through a 70-micron strainer before and after RBC lysis and two washes with FACS buffer (PBS 2% FCS, 0.5 mM EDTA). Cells (106) were incubated with anti-mouse CD16/32 (BD Pharmingen, San Diego, CA) to block Fc receptors and Zombie Red (BioLegend, San Diego, CA) to identify dead cells followed by washing with FACS buffer and staining with the following anti-mouse Abs for specific cell surface markers purchased from BD Pharmingen (San Diego, CA) or BD Horizon (San Diego, CA), CD3e (FITC, 145-2C11), CD4 (PerCP-Cy5.5, RM4-5), CD8a (BV510, clone 53–6.7), and from Biolegend (San Diego, CA), CD55 (PE, clone RIKO-3), and CD44 (BV650, clone IM7). Data was acquired using BD FACS DIVA software and LSRII flow cytometer and analyzed with FlowJo V10 (Ashland, OR).
Statistics
Data are expressed as mean and standard error and analyzed by GraphPad Software V5.02 (San Diego, CA). Unpaired two-tailed Mann-Whitney U test was used for comparisons between PBS and HgCl2 exposure within a strain. For multiple comparisons, one-way ANOVA with Tukey posttest was used for comparing all possible combinations of HgCl2-exposed or PBS groups. Only p values ≤0.05 are shown.
Results
Systemic autoimmunity induced by HgCl2 is type I IFN independent
To determine if type I IFN is required for HgIA, B6-Ifnar1−/− mice lacking the common receptor for all type I IFNs were exposed to HgCl2 and autoAb production was assessed. Strikingly, after 4 weeks, ANA and autoAbs to chromatin, DNA, and ENA5 in HgCl2-exposed Ifnar1-deficient mice were significantly elevated compared to PBS-treated Ifnar−/− mice. The levels of these autoAbs tended to be lower among Ifnar1−/− mice than in wild type B6 mice given HgCl2. However, this only reached statistical significance for anti-ENA5 where levels were low and could be related to lower total IgG concentrations in Ifnar1−/− compared with wild type mice (Figure 1A and Supplemental Figure 1). Increases in both IgG1 and IgG2a anti-chromatin were observed (Supplemental Figure 2) Thus, significant autoAb production induced by HgCl2 in B6 mice occurs in the absence of type I IFN signaling.
Figure 1. Type I IFN is not required for mHgIA in B6 and autoimmune-prone NZB mice.

A) Wild type or Ifnar1-deficient B6 (n=8/group) or B) wild type or Ifnar1-deficient NZB mice (n=3–12/group) were given 40 μg HgCl2 (filled circle) or PBS alone (open circle) s.c. twice a week for 4 weeks and sera was obtained 3 days after the final injection. Total IgG, ANA, and anti-chromatin were determined as described in Materials and Methods. Mean and standard error (horizontal lines) and only p values <0.05 between groups are indicated.
Susceptibility to HgIA is heavily influenced by autoimmune-predisposing genes (64). Therefore, to eliminate the possibility that the less lupus-prone B6 strain might be uniquely type I IFN-independent, we examined the effect of type I IFN deficiency on HgIA in the NZB, a strain we previously showed was highly dependent on type I IFN for spontaneous lupus (13). Remarkably, Ifnar1-deficient NZB mice were similarly susceptible to HgIA as wild type NZB mice with increases in serum IgG, ANA, and anti-chromatin (Figure 1B). All HgCl2-exposed NZB-Ifnar1−/− and wild type mice were positive for homogeneous ANAs. Finally, comparable amounts of glomeruli and vessel wall IgG, IgM, and C3-containing immune deposits were detected after exposure to HgCl2 in Ifnar1−/− and wild type NZB mice (Table I). Thus, HgIA is type I IFN-independent even in a strain that requires type I IFN for spontaneous disease (13).
Table 1.
Immune complex deposition in mercury-exposed Ifnar1−/− and wild type NZB mice.
| IC Deposits | NZB-Ifnar1−/− | NZB |
|---|---|---|
| Glomerular IgG | 4480±640a | 4480±640 |
| (67±13)b | (267±187) | |
| Glomerular IgM | 2.3±0.25 | 2.5±0.29 |
| (2.7±0.3) | (2.0±0.6) | |
| Glomerular C3 | 2565±905 | 3215±635 |
| (4267±853) | (2137±428) | |
| Kidney Vessel IgG | 3.0±0 | 3.0±0 |
| (0±0) | (0±0) | |
| Kidney Vessel C3 | 2.5±0.29 | 2.5±0.29 |
| (0±0) | (0±0) | |
| Spleen Vessel IgG | 2.8±0.25 | 3.0±0 |
| (0±0) | (0±0) | |
| Spleen Vessel C3 | 1.8±0.25 | 2.5±0.29 |
| (0±0) | (0±0) |
Type I IFN-independent autoimmunity is dependent on endosomal TLR trafficking and signaling, but not on IRF7
To dissect the pathogenesis of type I IFN-independent autoimmunity, we next focused on the endosomal TLRs as TLR7 and TLR9 (TLR7/9) are critical for the production of both type I IFNs and autoAbs in spontaneous lupus models (65). Accordingly, we examined the effect of mercury on lupus-prone BXSB mice expressing the Unc93b13d/3d (3d) mutation that blocks endosomal nucleic acid-sensing TLR trafficking and signaling as well as spontaneous lupus (37, 66). In sharp contrast to type I IFN, endosomal TLR signaling was required for induction of hypergammaglobulinemia and autoAbs by HgCl2 (Figure 2A) indicating that trafficking of TLRs from the endoplasmic reticulum to endolysosomal and phagosomal compartments is required for type I IFN independent autoimmunity.
Figure 2. Type I IFN-independent mHgIA requires UNC93B1, but not IRF7.

A) BXSB-3d mice and control BXSB (n=4–6/group) or B) IRF7-deficient B6 (Inept) and control B6 mice (n=8/group) were given HgCl2 (filled circle) or PBS (open circle) and total IgG, ANA, and anti-chromatin determined. Methods and analysis are the same as Figure 1. The same PBS-treated B6 mice are in Figure 1A as wild type, Inept and Ifnar−/− B6 were studied concurrently.
TLR7/9-induced responses, critical for the development of autoAbs, are mediated through the MyD88-TRAF6 complex and activation of the IRF7, canonical NF-kB, MAP kinase (MAPK), and IRF-5 pathways (67). Of these, IRF7 primarily serves to promote the production of type I interferon and therefore we hypothesized that it was likely dispensable for type I-independent autoimmunity. Indeed, IRF7-deficient Inept mice, which fail to produce IFN-α upon TLR agonist injection (35), responded to HgCl2 like B6-Ifnar1−/− mice with no significant differences in total serum IgG, ANA, or anti-chromatin autoAb levels when compared with IRF7-intact wild type mice (Figure 2B). Similar observations were made for anti-DNA and anti-ENA5 responses (Supplemental Figure 1) and for IgG1 and IgG2a anti-chromatin subclasses (Supplemental Figure 2). Thus, the endosomal TLR-induced pathways critical for HgIA can be inferred to involve one or more of the proinflammatory pathways.
Type I IFN-independent autoimmunity requires adaptor protein 3 (AP-3) complex-dependent late endosomes
To further define the TLR-induced pathways required for type IFN-independent autoimmunity we examined pearl mice deficient in the adaptor protein-3 (AP-3) complex because of a mutation in the AP-3β1 subunit. The AP-3 adaptor complex plays an essential role in transporting proteins with an acidic di-leucine motif from the early endosome to late endosomes/lysosomes as well as to lysosome-related organelles, such as melanosomes and platelet dense bodies (68–70). In pDCs, IRF7 activation and consequent type I IFN production requires AP-3-mediated trafficking of TLR7/9 to the late endolysosomal compartments (40) whereas there is controversy about whether AP-3 is needed for proinflammatory cytokine production (35, 40). In contrast, conventional DCs do not require AP-3 for TLR7/9-mediated induction of proinflammatory cytokines (35). These observations suggested that HgCl2 autoimmunity would occur in the absence of AP-3 since it is IRF7- and type I IFN-independent, therefore presumably not dependent on pDCs, which promote lupus by endogenous TLR-dependent production of type I IFN (4). Unexpectedly, however, total IgG and autoAbs were not induced in pearl mice after exposure to mercury clearly demonstrating that trafficking of AP-3 cargos including nucleic acid-sensing TLRs distal to early endosomes is required to drive autoAb production (Figure 3, Supplemental Figure 1). This dependence upon AP-3 also posed the question of what role other components of endosomal TLR trafficking and signaling play in type I IFN independent autoimmunity.
Figure 3. Type I IFN-independent mHgIA requires an intact AP-3 adaptor complex.

Ap3b1-deficient (pearl) (n=8/group) and control B6 (n=8/group) mice were given HgCl2 (filled circle) or PBS (open circle) and total IgG, ANA, and anti-chromatin determined. Methods and analysis are the same as Figure 1. Pearl mice were 60% male and 6–11 weeks old.
Type I IFN-independent autoimmunity is dependent on Slc15a4
The histidine peptide transporter SLC15A4 contains the acidic di-leucine motif recognized by AP-3 and is required for proper acidification of late endosome/lysosome compartments (32, 35). Consequently, it is required for signaling by nucleic acid-sensing TLRs in pDCs, but only partially required in B cells and not needed for TLR signaling in cDCs (32, 35). Importantly, disruption of SLC15A4, either through the feeble mutation (29) or gene deletion (32), abrogates development of autoAbs and other features of autoimmunity in lupus-prone mice. When B6 mice homozygous for the feeble mutation were exposed to mercury, autoAb responses failed to develop and there was only a modest increase in total IgG compared to wild type animals (Figure 4). SCL15A5-deficiency is also associated with a B cell intrinsic preferential reduction in IgG2c (equivalent to IgG2a) class switching (32, 33). Indeed, this was also observed in HgIA for both SLC15A4-deficient (feeble) and AP-3-deficient (pearl) mice as well as type I IFN-deficient (Ifnar1−/−) and IRF7-deficient (Inept) mice (Supplemental Figure 3). These observations reveal that type I IFN-independent HgIA requires essentially the same endosomal TLR trafficking and signaling components as type I-dependent spontaneous autoimmunity.
Figure 4. Feeble mice are resistant to type I IFN-independent mHgIA.

Slc15a4-deficient (feeble) (n=8/group) and control B6 (n=6–9/group) mice were given HgCl2 (filled circle) or PBS (open circle) and total IgG, ANA, and anti-chromatin determined. Methods and analysis are the same as Figure 1. Feeble mice were 12–16 weeks old.
Type I IFN independent autoimmunity is dependent on NF-κB and IL-6
To document the requirement for other TLR-induced pathways (34) in type I IFN-independent HgIA, we examined the canonical NF-kB pathway using Nfkb1-deficient B6 mice (71). Indeed, Nfkb1 deficiency completely blocked the development of hypergammaglobulinemia or autoAbs indicating an absolute requirement for the canonical NF-κB pathway (Figure 5A). This supports an essential role for nucleic acid-sensing TLR-mediated NF-κB signaling in HgIA although other immunological defects associated with Nfkb1 deficiency could also have contributed (71).
Figure 5. HgIA is dependent on the canonical NF-κB pathway and the proinflammatory cytokine, IL-6.

A) B6-Nfkb1−/− (n=8/group) or B) B10.S-Il6−/− (n=8/group) mice and corresponding wild type controls were given HgCl2 (filled circle) or PBS (open circle) and total IgG, ANA, and anti-chromatin determined. Methods and analysis are the same as Figure 1. B6-Nfkb1−/− mice were all female.
To investigate the role of proinflammatory cytokines that could mediate type I IFN independent autoimmunity we next examined IL-6, one of several cytokines dependent on NF-κB-mediated signaling (72). Strikingly, absence of IL-6 in B6 mice exposed to mercury resulted in marked suppression of serum IgG, ANA and anti-chromatin autoAbs similar to mercury-exposed NF-κB p50 deficient mice (Figure 5). Thus, the requirement for endosomal TLRs, NF-κB, and IL-6 supports the argument that type I IFN independent autoimmunity develops via endosomal TLR trafficking and signaling leading to NF-κB activation and proinflammatory cytokine expression.
Soluble TLR agonists are more effective at eliciting proinflammatory cytokines than HgCl2
To understand why HgIA was dependent on a proinflammatory cytokine pathway and not type I IFN, we compared the early in vivo IFN-α and proinflammatory cytokine responses of wild type B6 mice to subcutaneous injections of TLR ligands [poly(IC) and R848] or HgCl2 (Figure 6). As expected, both TLR3 and TLR7 ligands induced the production of IFN-α and proinflammatory cytokines, IL-6 and TNF-α, consistent with activation of both IRF7 and NF-κB TLR signaling pathways (67). In contrast, mercury was a weaker stimulator of systemic cytokine expression than TLR agonists and exhibited a very different cytokine profile. Notably, serum levels of IFN-α and TNF-α were unchanged by HgCl2 exposure, while IL-6 levels were significantly increased above PBS controls. These findings support the importance of the proinflammatory signaling pathway and IL-6 in particular in HgIA.
Figure 6. IFN-α and proinflammatory cytokine production induced by TLR agonists and HgCl2.
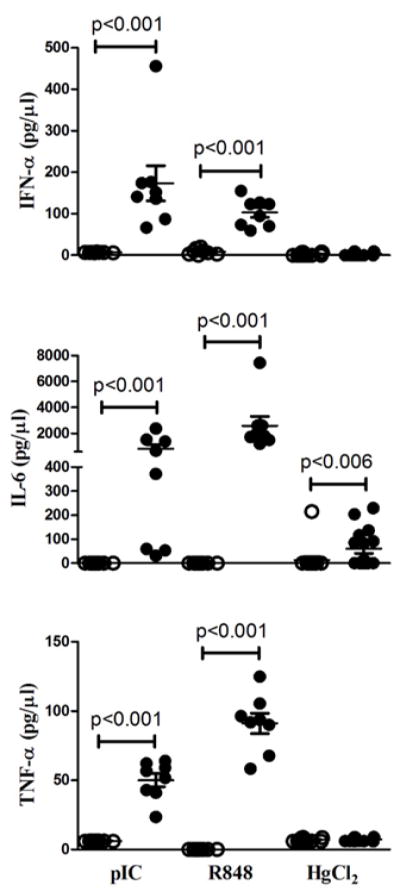
Serum IFN-α (Top panel), IL-6 (Middle panel) and TNF-α (Bottom panel) in B6 (n=8/group) mice injected subcutaneously with 100 μg of poly(IC) for 6 h; 50 μg of R848 for 3 h; or 40 μg HgCl2 for 6 h in 100 μl PBS. The optimal timepoints for each were determined in preliminary experiments. Agonist or HgCl2 (filled circles) and PBS controls (open circles) are indicated. P<0.05 for comparisons between agonist or HgCl2 and PBS groups are shown.
AP-3 is required for T cell activation in HgIA
Endosomal TLR trafficking and signaling are not only important for proinflammatory cytokine production but also contributes to adaptive immunity by promoting MHC class II antigen presentation (73). Indeed, T cell activation is impacted by deficiencies in Unc93b1, Slc15a4 or Ap3b1 indirectly because of reduced TLR-mediated activation of antigen-presenting cells and not because of defective antigen presentation machinery (74–76). As autoAb responses in HgIA are T cell dependent (55, 63) and associated with increases in activated/memory (CD44high CD55low) CD4+ T cell subset (77), we asked if AP-3 deficiency affected T cell activation in mice exposed to HgCl2. Indeed, while HgCl2 exposure in wild type mice increased percentages of activated/memory (CD44high CD55low) CD4+ T cells, in pearl mice, this population was not only reduced prior to HgCl2, but also failed to expand after mercury exposure (Figure 7). This impaired T cell activation is consistent with defective antigen-presentation and could contribute to the lack of HgCl2-induced humoral autoimmune responses.
Figure 7. Activated/memory CD4+ T cells are reduced in HgCl2 exposed pearl mice.
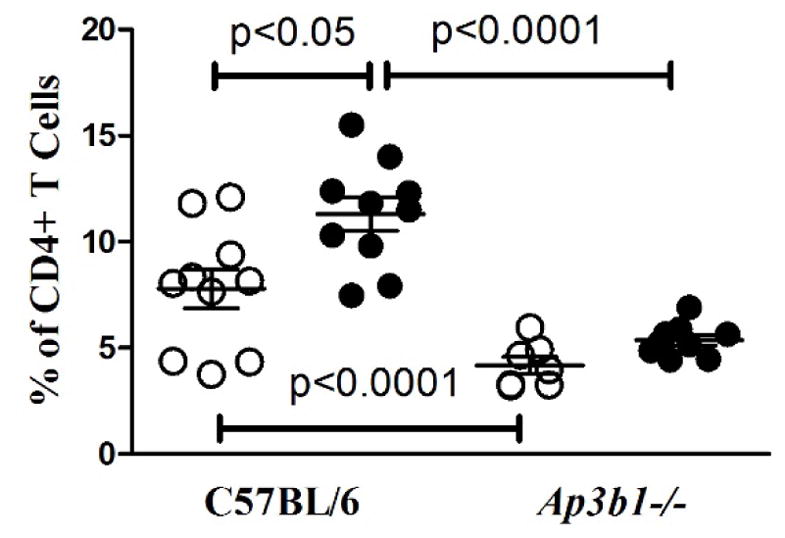
Percentages of activated/memory (CD44high CD55low) CD4+ splenic T cells in control B6 and Ap3b1-deficient (pearl) mice (n=7–10/group) exposed to 40 μg HgCl2 (filled circle) or PBS (open circle) s.c. twice a week for 5 weeks. Activated/memory CD4+ T cells were determined by flow cytometry as described in Materials and Methods. Mice were 60–70% female. P>0.05 for comparisons between PBS- versus HgCl2-treated Ap3b1-deficient (pearl).
Discussion
Using a genetics approach, we have made several novel observations about the roles of type I IFN and TLR trafficking and signaling pathways in the production of autoAbs in xenobiotic autoimmunity. Thus, we found that a) the development of HgIA does not require either the type I IFN receptor or IRF-7, b) autoimmunity is dependent on endosomal trafficking by UNC93B1 and the AP-3 complex, c) deficiency of the endosomal SLC15A4 inhibits HgIA, and d) signaling through the canonical NF-κB pathway and production of the proinflammatory cytokine, IL-6, are both required. Taken together, these findings allow new insights into the pathogenesis of HgIA and the endosomal TLR signaling processes needed to induce autoAbs.
The current paradigm for human idiopathic systemic autoimmune disease argues for a central role for type I IFNs (5), characterized by the IFN “signature” (8, 78), and with special emphasis on the contribution of innate cells, especially pDCs (1, 5, 79). This is supported by animal models of idiopathic (6, 13, 14) and pristane-induced SLE (15) which exhibit severe disease phenotypes and dependence on type I IFN including association of an IFN signature with more rapid onset of disease (80). However, while the IFN gene signature (8, 81) identifies a majority of patients with severe disease, there is a significant subgroup lacking this signature in which milder disease might be due to type I IFN independent mechanisms. Here, we demonstrate that type I IFNs are dispensable for the pathogenesis of HgIA even in the NZB lupus strain for which development of spontaneous disease is type I IFN-dependent. These results provide evidence that the accepted paradigm of a pivotal role for type I IFN in systemic autoimmune disease is not always applicable.
While our studies do not examine how mercury exposure leads to TLR activation and cytokine expression, it is possible that the cell death and proteolysis mediated by mercury releases nucleic acid material that could interact with TLRs (82, 83). Mercury exposure of human neutrophil granulocytes also induces the formation of neutrophil extracellular traps providing another source of TLR agonists (84). These studies are consistent with our observation that mercury exposure induces a localized inflammatory response (85) regulated in part by cathepsin B (61) which is known to be involved in TLR responses (86, 87). Furthermore, the reported finding that exposure of T cells to mercury induces MHC class II-dependent cytokine production and proliferation in vitro only in the presence of antigen-presenting cells (88) is consistent with mercury generating TLR ligands that activate cells to present self-antigens.
Our findings in UNC93B1-deficient mice showed that mercury could not compensate for what appears to be a fundamental requirement for endosomal TLRs in the production of autoAbs to nucleic acid-containing Ags (66). UNC93B1 is required for trafficking nucleic acid-sensing TLRs from the endoplasmic reticulum to the appropriate endolysosomal and phagolysosomal compartments where TLRs are processed, engage ligand, and initiate signaling cascades (89, 90). However, the relative importance of TLR signaling pathways that induce type I IFN or proinflammatory cytokines, and the relevance of specific endocytic/phagocytic compartments in autoimmunity have yet to be addressed. We examined these issues by studying the type I IFN-independent HgIA model in AP-3- and SLC15A4-deficient mice.
Recent studies have identified two distinct UNC93B1-mediated pathways for endosomal TLR engagement depending on the size of the nucleic acid-containing material (41, 91) (Supplemental Figure 3). Accordingly, the engulfment of small nucleic acid-containing material alone or in immune complexes by endocytosis or FcγR interaction, are directed through an endocytic pathway dependent on AP-3. The AP-3 complex sorts and transports protein cargos expressing an acidic di-leucine motif within the endocytic pathway from the early endosome to the lysosome for degradation, and is required for the development of lysosome-related organelles and synaptic vesicles (69). Deficiency of the AP-3 complex blocks the transport of relevant cargos, including TLRs, from early endosomes to late endosomes and subsequent compartments. In contrast, endosomal trafficking and TLR engagement following uptake of large immune particles by FcγR in pDCs or macrophages occur through a microtubule-associated protein 1 light chains 3α (LC3)-associated phagocytosis (LAP) pathway. This pathway is independent of AP-3, and in addition to LC3, involves several autophagy genes including VPS34, Atg5, and Atg7, through a process distinct from canonical autophagy (92). Our findings show that loss of AP-3 is sufficient to block autoAb production thereby implicating a critical role for the endocytic pathway and AP-3-mediated TLR signaling. From this, it can also be inferred that autoAb production is induced in large part by small nucleic acid-containing particles or immune complexes, which are the main type of cargoes taken up by endocytosis.
As noted, a previous study using unfractionated FLT3-derived BM cells reported that TLR-mediated type I IFN signaling was generated in AP-3-dependent late endosome/endolysosome compartments (“IFN endosome”) while proinflammatory cytokine signaling occurred in early (“NF-κB”) endosomes that did not require the AP-3 transport (40). In contrast, another study showed that for purified pDCs both type I IFN and proinflammatory cytokine production required the AP-3 complex while purified conventional DCs did not require AP-3. Our results do not support the finding that most NF-kB and proinflammatory signaling emanate from early endosomes, and instead support the requirement for TLR trafficking to the late endosomes/endolysosomes for both essential proinflammatory signaling and autoimmunity (Supplemental Figure 4).
Endosomal TLRs undergo proteolytic processing by cathepsins and asparagine endopeptidases in acidic conditions, but recently furin was shown to cleave TLR7 at neutral pH in both humans and mice (93). Thus, it was postulated that TLR7 could be functional in early endosomes prior to the development of an acidic environment. Our finding, however, that type I IFN-independent autoimmunity is dependent on AP-3 does not support a major role for early endosomal furin-processed TLR7 in autoimmunity possibly because the optimal binding of nucleic acids to TLR occurs in acidic conditions (90). Another important consideration is how relevant these findings for AP-3 in HgIA are to spontaneous autoimmune disease. In this regard, in preliminary studies, we have confirmed the dependence of AP-3 for spontaneous lupus in B6-lpr mice (data not shown), which suggests that the conclusions here related to AP-3 are more broadly applicable to spontaneous and type I IFN-dependent autoimmunity.
To further dissect the relationship between the endosomal compartments and type I IFN-independent autoimmunity, we also examined feeble mice that are deficient in SLC15A4, an endosomal histidine peptide transporter that contains an AP-3 di-leucine binding motif (35). SLC15A4 is required for type I IFN and proinflammatory cytokine induction by nucleic acid-sensing TLRs in pDCs, but not cDCs, and is not required for TLR9-induced B cell proliferation (35). Importantly, lupus-prone B6-lpr feeble mice exhibit reduced disease, which was ascribed to defective TLR-induced cytokine production, especially type I IFN, in pDCs (29) and later studies also showed that SLC15A4 had a subtle intrinsic defect in B cells that inhibited isotype-switching to IgG2c and autoAb production (32, 33). Furthermore, evidence indicated that this was caused by an alteration in histidine homeostasis that impaired endosomal acidification and activation of TLR7, which then prevented the triggering of a TLR7-mediated mTOR-IRF7-type I IFN circuit required for autoAb production by B cells (32). Our findings in SLC15A4-deficient mice exposed to mercury were similar in that there was a selective reduction of the IgG2a (equivalent to IgG2c) isotype and reduced total IgG autoAbs, which in HgIA is primarily of the IgG1 isotype (25). Thus, our data implicate IRF7-independent TLR signaling in pDC and B cells as being critical for HgIA pathogenesis, but do not support the conclusion that SLC15A4 primarily promotes autoAb production by an IRF7-type I IFN circuit.
B6 mice deficient in Nfkb1, the p50 subunit of NF-κB, have significant defects in B cell function including basal and specific Ab responses (71). The dramatic effects of such a gene deficiency on HgIA induction were clearly demonstrated by the total lack of autoAb response following HgCl2 exposure. This suggested that the requirement for endosomal TLR in type I IFN independent autoimmunity is linked to signaling through the canonical NF-κB pathway. In support of this possibility, we document the contribution of IL-6, consistent with previous studies showing susceptibility to HgIA is associated with increased expression of this cytokine (58). NF-κB contributes to the regulation of IL-6 expression, which is known to play a major role in both idiopathic (94–96) and induced systemic autoimmunity (58, 97). Taken together, these studies suggest that type I IFN-independent autoimmunity involves endosomal TLR trafficking and signaling leading to activation of the canonical NF-κB pathway and proinflammatory cytokine elaboration.
In conclusion, we show that autoAb production induced by a xenobiotic develops through a type I IFN-independent mechanism and further define the central TLR-related endosomal trafficking and signaling pathways required to induce autoimmunity. Targeting of these pathways could provide novel approaches to the treatment of endosomal TLR-mediated autoimmune diseases particularly those that are type I-independent or do not respond to type I IFN inhibition.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health [grants ES007511, ES021464, and ES022625] to KMP and [HL114408] to DHK. This is manuscript #29475 from The Scripps Research Institute.
References
- 1.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–335. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 2.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 3.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 4.Kono DH, Baccala R, Theofilopoulos AN. TLRs and interferons: a central paradigm in autoimmunity. Curr Opin Immunol. 2013;25:720–727. doi: 10.1016/j.coi.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 6.Kiefer K, Oropallo MA, Cancro MP, Marshak-Rothstein A. Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol. 2012;90:498–504. doi: 10.1038/icb.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niewold TB. Interferon alpha-induced lupus: proof of principle. J Clin Rheumatol. 2008;14:131–132. doi: 10.1097/RHU.0b013e318177627d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Obermoser G, Pascual V. The interferon-alpha signature of systemic lupus erythematosus. Lupus. 2010;19:1012–1019. doi: 10.1177/0961203310371161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronnblom L, Eloranta ML. The interferon signature in autoimmune diseases. Curr Opin Rheumatol. 2013;25:248–253. doi: 10.1097/BOR.0b013e32835c7e32. [DOI] [PubMed] [Google Scholar]
- 10.Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, Brohawn P, Kiener PA, Richman L, Fiorentino D, Greenberg SA, Jallal B, Yao Y. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis. 2011;70:2029–2036. doi: 10.1136/ard.2011.150326. [DOI] [PubMed] [Google Scholar]
- 11.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhuang H, Szeto C, Han S, Yang L, Reeves WH. Animal Models of Interferon Signature Positive Lupus. Frontiers in immunology. 2015;6:291. doi: 10.3389/fimmu.2015.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, Theofilopoulos AN. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197:777–788. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braun D, Geraldes P, Demengeot J. Type I Interferon controls the onset and severity of autoimmune manifestations in lpr mice. J Autoimmun. 2003;20:15–25. doi: 10.1016/s0896-8411(02)00109-9. [DOI] [PubMed] [Google Scholar]
- 15.Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Lyons R, Sobel E, Satoh M, Reeves WH. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum. 2007;56:3770–3783. doi: 10.1002/art.23023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J Immunol. 2005;174:2499–2506. doi: 10.4049/jimmunol.174.5.2499. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Bethunaickan R, Huang W, Lodhi U, Solano I, Madaio MP, Davidson A. Interferon-alpha accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum. 2011;63:219–229. doi: 10.1002/art.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fairhurst AM, Mathian A, Connolly JE, Wang A, Gray HF, George TA, Boudreaux CD, Zhou XJ, Li QZ, Koutouzov S, Banchereau J, Wakeland EK. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur J Immunol. 2008;38:1948–1960. doi: 10.1002/eji.200837925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hron JD, Peng SL. Type I IFN protects against murine lupus. J Immunol. 2004;173:2134–2142. doi: 10.4049/jimmunol.173.3.2134. [DOI] [PubMed] [Google Scholar]
- 20.Schwarting A, Paul K, Tschirner S, Menke J, Hansen T, Brenner W, Kelley VR, Relle M, Galle PR. Interferon-beta: a therapeutic for autoimmune lupus in MRL-Faslpr mice. J Am Soc Nephrol. 2005;16:3264–3272. doi: 10.1681/ASN.2004111014. [DOI] [PubMed] [Google Scholar]
- 21.Haas C, Ryffel B, Le Hir M. IFN-gamma is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice. J Immunol. 1997;158:5484–5491. [PubMed] [Google Scholar]
- 22.Lawson BR, Prud’homme GJ, Chang Y, Gardner HA, Kuan J, Kono DH, Theofilopoulos AN. Treatment of murine lupus with cDNA encoding IFN-gammaR/Fc. J Clin Invest. 2000;106:207–215. doi: 10.1172/JCI10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng SL, Moslehi J, Craft J. Roles of interferon-gamma and interleukin-4 in murine lupus. J Clin Invest. 1997;99:1936–1946. doi: 10.1172/JCI119361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacob CO, van der Meide PH, McDevitt HO. In vivo treatment of (NZB X NZW)F1 lupus-like nephritis with monoclonal antibody to gamma interferon. J Exp Med. 1987;166:798–803. doi: 10.1084/jem.166.3.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kono DH, Balomenos D, Pearson DL, Park MS, Hildebrandt B, Hultman P, Pollard KM. The prototypic Th2 autoimmunity induced by mercury is dependent on IFN- gamma and not Th1/Th2 imbalance. J Immunol. 1998;161:234–240. [PubMed] [Google Scholar]
- 26.Pollard KM, Hultman P, Toomey CB, Cauvi DM, Hoffman HM, Hamel JC, Kono DH. Definition of IFN-gamma-related pathways critical for chemically-induced systemic autoimmunity. J Autoimmun. 2012;39:323–331. doi: 10.1016/j.jaut.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011;29:163–183. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33:967–978. doi: 10.1016/j.immuni.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baccala R, Gonzalez-Quintial R, Blasius AL, Rimann I, Ozato K, Kono DH, Beutler B, Theofilopoulos AN. Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc Natl Acad Sci U S A. 2013;110:2940–2945. doi: 10.1073/pnas.1222798110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, D’Agati V, Elkon KB, Reizis B. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med. 2014;211:1969–1976. doi: 10.1084/jem.20132522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, Colonna M. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med. 2014;211:1977–1991. doi: 10.1084/jem.20132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, Furuyama-Tanaka K, Karyu H, Sugiura Y, Shimizu Y, Hosaka T, Goto M, Kato N, Okamura T, Suematsu M, Yokoyama S, Toyama-Sorimachi N. The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity. 2014;41:375–388. doi: 10.1016/j.immuni.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 33.Dosenovic P, Adori M, Adams WC, Pedersen GK, Soldemo M, Beutler B, Karlsson Hedestam GB. Slc15a4 function is required for intact class switch recombination to IgG2c in response to TLR9 stimulation. Immunol Cell Biol. 2015;93:136–146. doi: 10.1038/icb.2014.82. [DOI] [PubMed] [Google Scholar]
- 34.Theofilopoulos AN, Kono DH, Beutler B, Baccala R. Intracellular nucleic Acid sensors and autoimmunity. J Interferon Cytokine Res. 2011;31:867–886. doi: 10.1089/jir.2011.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blasius AL, Arnold CN, Georgel P, Rutschmann S, Xia Y, Lin P, Ross C, Li XH, Smart NG, Beutler B. Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for Toll-like receptor signaling in plasmacytoid dendritic cells. P Natl Acad Sci USA. 2010;107:19973–19978. doi: 10.1073/pnas.1014051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, Beutler B. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nature Immunology. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 37.Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, Arnold CN, Baccala R, Silverman GJ, Beutler BA, Theofilopoulos AN. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A. 2009;106:12061–12066. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 40.Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science. 2010;329:1530–1534. doi: 10.1126/science.1187029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henault J, Martinez J, Riggs JM, Tian J, Mehta P, Clarke L, Sasai M, Latz E, Brinkmann MM, Iwasaki A, Coyle AJ, Kolbeck R, Green DR, Sanjuan MA. Noncanonical Autophagy Is Required for Type I Interferon Secretion in Response to DNA-Immune Complexes. Immunity. 2012 doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida S, Gershwin ME. Autoimmunity and selected environmental factors of disease induction. Semin Arthritis Rheum. 1993;22:399–419. doi: 10.1016/s0049-0172(05)80032-0. [DOI] [PubMed] [Google Scholar]
- 43.Motts JA, Shirley DL, Silbergeld EK, Nyland JF. Novel biomarkers of mercury-induced autoimmune dysfunction: A cross-sectional study in Amazonian Brazil. Environ Res. 2014;132C:12–18. doi: 10.1016/j.envres.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gardner RM, Nyland JF, Silva IA, Ventura AM, de Souza JM, Silbergeld EK. Mercury exposure, serum antinuclear/antinucleolar antibodies, and serum cytokine levels in mining populations in Amazonian Brazil: a cross-sectional study. Environ Res. 2010;110:345–354. doi: 10.1016/j.envres.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li SJ, Zhang SH, Chen HP, Zeng CH, Zheng CX, Li LS, Liu ZH. Mercury-induced membranous nephropathy: clinical and pathological features. Clin J Am Soc Nephrol. 2010;5:439–444. doi: 10.2215/CJN.07571009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pollard KM, Hultman P. Skin-lightening creams are a possible exposure risk for systemic lupus erythematosus: comment on the article by Finckh et al. Arthritis Rheum. 2007;56:1721. doi: 10.1002/art.22560. author reply 1721–1722. [DOI] [PubMed] [Google Scholar]
- 47.Cauvi DM, Hultman P, Pollard KM. Autoimmune Models. In: McQueen CA, editor. Comprehensive Toxicology. Academic Press; Oxford: 2010. pp. 413–438. [Google Scholar]
- 48.Hirsch F, Couderc J, Sapin C, Fournie G, Druet P. Polyclonal effect of HgCl2 in the rat, its possible role in an experimental autoimmune disease. Eur J Immunol. 1982;12:620–625. doi: 10.1002/eji.1830120716. [DOI] [PubMed] [Google Scholar]
- 49.Aten J, Bosman CB, Rozing J, Stijnen T, Hoedemaeker PJ, Weening JJ. Mercuric chloride-induced autoimmunity in the brown Norway rat. Cellular kinetics and major histocompatibility complex antigen expression. Am J Pathol. 1988;133:127–138. [PMC free article] [PubMed] [Google Scholar]
- 50.Pusey CD, Bowman C, Morgan A, Weetman AP, Hartley B, Lockwood CM. Kinetics and pathogenicity of autoantibodies induced by mercuric chloride in the brown Norway rat. Clin Exp Immunol. 1990;81:76–82. doi: 10.1111/j.1365-2249.1990.tb05294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guery JC, Druet E, Glotz D, Hirsch F, Mandet C, De Heer E, Druet P. Specificity and cross-reactive idiotypes of anti-glomerular basement membrane autoantibodies in HgCl2-induced autoimmune glomerulonephritis. Eur J Immunol. 1990;20:93–100. doi: 10.1002/eji.1830200114. [DOI] [PubMed] [Google Scholar]
- 52.Mathieson PW, Thiru S, Oliveira DB. Mercuric chloride-treated brown Norway rats develop widespread tissue injury including necrotizing vasculitis. Lab Invest. 1992;67:121–129. [PubMed] [Google Scholar]
- 53.Biancone L, Andres G, Ahn H, Lim A, Dai C, Noelle R, Yagita H, De Martino C, Stamenkovic I. Distinct regulatory roles of lymphocyte costimulatory pathways on T helper type-2 mediated autoimmune disease. J Exp Med. 1996;183:1473–1481. doi: 10.1084/jem.183.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang Y, Moller G. In vitro effects of HgCl2 on murine lymphocytes. I. Preferable activation of CD4+ T cells in a responder strain. J Immunol. 1995;154:3138–3146. [PubMed] [Google Scholar]
- 55.Hultman P, Johansson U, Dagnaes-Hansen F. Murine mercury-induced autoimmunity: the role of T-helper cells. J Autoimmun. 1995;8:809–823. doi: 10.1016/s0896-8411(95)80019-0. [DOI] [PubMed] [Google Scholar]
- 56.Hultman P, Bell LJ, Enestrom S, Pollard KM. Murine susceptibility to mercury. II. autoantibody profiles and renal immune deposits in hybrid, backcross, and H-2d congenic mice. Clin Immunol Immunopathol. 1993;68:9–20. doi: 10.1006/clin.1993.1088. [DOI] [PubMed] [Google Scholar]
- 57.Kono DH, Park MS, Szydlik A, Haraldsson KM, Kuan JD, Pearson DL, Hultman P, Pollard KM. Resistance to xenobiotic-induced autoimmunity maps to chromosome 1. J Immunol. 2001;167:2396–2403. doi: 10.4049/jimmunol.167.4.2396. [DOI] [PubMed] [Google Scholar]
- 58.Havarinasab S, Pollard KM, Hultman P. Gold- and silver-induced murine autoimmunity--requirement for cytokines and CD28 in murine heavy metal-induced autoimmunity. Clin Exp Immunol. 2009;155:567–576. doi: 10.1111/j.1365-2249.2008.03831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pollard KM, Pearson DL, Hultman P, Deane TN, Lindh U, Kono DH. Xenobiotic acceleration of idiopathic systemic autoimmunity in lupus- prone bxsb mice. Environ Health Perspect. 2001;109:27–33. doi: 10.1289/ehp.0110927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barregard L, Sallsten G, Conradi N. Tissue levels of mercury determined in a deceased worker after occupational exposure. Int Arch Occup Environ Health. 1999;72:169–173. doi: 10.1007/s004200050356. [DOI] [PubMed] [Google Scholar]
- 61.Toomey CB, Cauvi DM, Hamel JC, Ramirez AE, Pollard KM. Cathepsin B regulates the appearance and severity of mercury-induced inflammation and autoimmunity. Toxicol Sci. 2014;142:339–349. doi: 10.1093/toxsci/kfu189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scatizzi JC, Haraldsson MK, Pollard KM, Theofilopoulos AN, Kono DH. The lbw2 locus promotes autoimmune hemolytic anemia. J Immunol. 2012;188:3307–3314. doi: 10.4049/jimmunol.1103561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pollard KM, Arnush M, Hultman P, Kono DH. Costimulation requirements of induced murine systemic autoimmune disease. J Immunol. 2004;173:5880–5887. doi: 10.4049/jimmunol.173.9.5880. [DOI] [PubMed] [Google Scholar]
- 64.Kono DH, Theofilopoulos AN. Genetics of SLE in mice. Springer Semin Immunopathol. 2006;28:83–96. doi: 10.1007/s00281-006-0030-7. [DOI] [PubMed] [Google Scholar]
- 65.Theofilopoulos AN, Gonzalez-Quintial R, Lawson BR, Koh YT, Stern ME, Kono DH, Beutler B, Baccala R. Sensors of the innate immune system: their link to rheumatic diseases. Nat Rev Rheumatol. 2010;6:146–156. doi: 10.1038/nrrheum.2009.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koh YT, Scatizzi JC, Gahan JD, Lawson BR, Baccala R, Pollard KM, Beutler BA, Theofilopoulos AN, Kono DH. Role of nucleic acid-sensing TLRs in diverse autoantibody specificities and anti-nuclear antibody-producing B cells. J Immunol. 2013;190:4982–4990. doi: 10.4049/jimmunol.1202986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 68.Dell’Angelica EC. AP-3-dependent trafficking and disease: the first decade. Curr Opin Cell Biol. 2009;21:552–559. doi: 10.1016/j.ceb.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 69.Marks MS, Heijnen HF, Raposo G. Lysosome-related organelles: unusual compartments become mainstream. Curr Opin Cell Biol. 2013;25:495–505. doi: 10.1016/j.ceb.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee BL, Moon JE, Shu JH, Yuan L, Newman ZR, Schekman R, Barton GM. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife. 2013;2:e00291. doi: 10.7554/eLife.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 72.Brasier AR. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res. 2010;86:211–218. doi: 10.1093/cvr/cvq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mantegazza AR, Magalhaes JG, Amigorena S, Marks MS. Presentation of phagocytosed antigens by MHC class I and II. Traffic. 2013;14:135–152. doi: 10.1111/tra.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mantegazza AR, Guttentag SH, El-Benna J, Sasai M, Iwasaki A, Shen H, Laufer TM, Marks MS. Adaptor protein-3 in dendritic cells facilitates phagosomal toll-like receptor signaling and antigen presentation to CD4(+) T cells. Immunity. 2012;36:782–794. doi: 10.1016/j.immuni.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Janssen E, Tabeta K, Barnes MJ, Rutschmann S, McBride S, Bahjat KS, Schoenberger SP, Theofilopoulos AN, Beutler B, Hoebe K. Efficient T cell activation via a Toll-Interleukin 1 Receptor-independent pathway. Immunity. 2006;24:787–799. doi: 10.1016/j.immuni.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 76.Blasius AL, Krebs P, Sullivan BM, Oldstone MB, Popkin DL. Slc15a4, a gene required for pDC sensing of TLR ligands, is required to control persistent viral infection. PLoS Pathog. 2012;8:e1002915. doi: 10.1371/journal.ppat.1002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cauvi DM, Cauvi G, Pollard KM. Reduced expression of decay-accelerating factor 1 on CD4+ T cells in murine systemic autoimmune disease. Arthritis Rheum. 2007;56:1934–1944. doi: 10.1002/art.22639. [DOI] [PubMed] [Google Scholar]
- 78.Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004;20:539–550. doi: 10.1016/s1074-7613(04)00108-6. [DOI] [PubMed] [Google Scholar]
- 79.Baccala R, Gonzalez-Quintial R, Lawson BR, Stern ME, Kono DH, Beutler B, Theofilopoulos AN. Sensors of the innate immune system: their mode of action. Nat Rev Rheumatol. 2009;5:448–456. doi: 10.1038/nrrheum.2009.136. [DOI] [PubMed] [Google Scholar]
- 80.Bender AT, Wu Y, Cao Q, Ding Y, Oestreicher J, Genest M, Akare S, Ishizaka ST, Mackey MF. Assessment of the translational value of mouse lupus models using clinically relevant biomarkers. Translational research : the journal of laboratory and clinical medicine. 2014;163:515–532. doi: 10.1016/j.trsl.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 81.Ronnblom L, Alm GV, Eloranta ML. The type I interferon system in the development of lupus. Semin Immunol. 2011;23:113–121. doi: 10.1016/j.smim.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 82.Pollard KM, Pearson DL, Bluthner M, Tan EM. Proteolytic cleavage of a self-antigen following xenobiotic-induced cell death produces a fragment with novel immunogenic properties. J Immunol. 2000;165:2263–2270. doi: 10.4049/jimmunol.165.4.2263. [DOI] [PubMed] [Google Scholar]
- 83.Tsokos GC, Lo MS, Reis PC, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. 2016;12:716–730. doi: 10.1038/nrrheum.2016.186. [DOI] [PubMed] [Google Scholar]
- 84.Haase H, Hebel S, Engelhardt G, Rink L. Ethylmercury and Hg2+ induce the formation of neutrophil extracellular traps (NETs) by human neutrophil granulocytes. Arch Toxicol. 2016;90:543–550. doi: 10.1007/s00204-015-1484-y. [DOI] [PubMed] [Google Scholar]
- 85.Cauvi DM, Cauvi G, Toomey CB, Jacquinet E, Pollard KM. Interplay between IFN-gamma and IL-6 impacts the inflammatory response and expression of interferon-regulated genes in environmental-induced autoimmunity. Toxicol Sci. 2017 doi: 10.1093/toxsci/kfx083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garcia-Cattaneo A, Gobert FX, Muller M, Toscano F, Flores M, Lescure A, Del Nery E, Benaroch P. Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc Natl Acad Sci U S A. 2012;109:9053–9058. doi: 10.1073/pnas.1115091109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matsumoto F, Saitoh S, Fukui R, Kobayashi T, Tanimura N, Konno K, Kusumoto Y, Akashi-Takamura S, Miyake K. Cathepsins are required for Toll-like receptor 9 responses. Biochem Biophys Res Commun. 2008;367:693–699. doi: 10.1016/j.bbrc.2007.12.130. [DOI] [PubMed] [Google Scholar]
- 88.Pollard KM, Landberg GP. The in vitro proliferation of murine lymphocytes to mercuric chloride is restricted to mature T cells and is interleukin 1 dependent. Int Immunopharmacol. 2001;1:581–593. doi: 10.1016/s1567-5769(00)00034-5. [DOI] [PubMed] [Google Scholar]
- 89.Pelka K, Shibata T, Miyake K, Latz E. Nucleic acid-sensing TLRs and autoimmunity: novel insights from structural and cell biology. Immunol Rev. 2016;269:60–75. doi: 10.1111/imr.12375. [DOI] [PubMed] [Google Scholar]
- 90.Majer O, Liu B, Barton GM. Nucleic acid-sensing TLRs: trafficking and regulation. Curr Opin Immunol. 2016;44:26–33. doi: 10.1016/j.coi.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blander JM. Designing a Type I Interferon Signaling Phagosome. Immunity. 2012;37:947–949. doi: 10.1016/j.immuni.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 92.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 93.Hipp MM, Shepherd D, Gileadi U, Aichinger MC, Kessler BM, Edelmann MJ, Essalmani R, Seidah NG, Reis e Sousa C, Cerundolo V. Processing of human toll-like receptor 7 by furin-like proprotein convertases is required for its accumulation and activity in endosomes. Immunity. 2013;39:711–721. doi: 10.1016/j.immuni.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brown KD, Claudio E, Siebenlist U. The roles of the classical and alternative nuclear factor-kappaB pathways: potential implications for autoimmunity and rheumatoid arthritis. Arthritis Res Ther. 2008;10:212. doi: 10.1186/ar2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Finck BK, Chan B, Wofsy D. Interleukin 6 promotes murine lupus in NZB/NZW F1 mice. J Clin Invest. 1994;94:585–591. doi: 10.1172/JCI117373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kiberd BA. Interleukin-6 receptor blockage ameliorates murine lupus nephritis. J Am Soc Nephrol. 1993;4:58–61. doi: 10.1681/ASN.V4158. [DOI] [PubMed] [Google Scholar]
- 97.Richards HB, Satoh M, Shaw M, Libert C, Poli V, Reeves WH. Interleukin 6 dependence of anti-DNA antibody production: evidence for two pathways of autoantibody formation in pristane-induced lupus. J Exp Med. 1998;188:985–990. doi: 10.1084/jem.188.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.