

Summary
Robustness in biology is the stability of phenotype under diverse genetic and/or environmental perturbations. The circadian clock has the remarkable stability of period and phase which—unlike other biological oscillators—is maintained over a wide range of conditions. Here we show that the high fidelity of the circadian system stems from robust degradation of the clock protein, PERIOD. We show that PERIOD degradation is regulated by balanced ubiquitination/deubiquitination, and disruption of the balance can destabilize the clock. In mice with loss-of-function mutation of the E3 ligase gene β-Trcp2, the balance of PERIOD degradation is perturbed, and the clock becomes dramatically unstable, presenting a unique behavioral phenotype unlike other circadian mutant animal models. We believe our data provide a molecular explanation for how circadian phases such as the wake-sleep onset times can become unstable in humans and present a unique mouse model to study human circadian disorders with unstable circadian rhythm phases.
eTOC Blurb
In certain human sleep disorders, wake-sleep onset times shift daily in an unpredictable manner. D'Alessandro et al. develop a unique mouse model that emulates the human sleep disorders and provide a molecular explanation for unstable wake-sleep rhythms.
Introduction
Daily rhythms in mammalian physiology and behavior, such as wake-sleep cycles, are controlled by a molecular circuit called the circadian clock [1-3]. Circadian rhythms are extremely reliable, with little cycle-to-cycle variation under constant conditions such as constant darkness [4]. Because the circadian clock is a molecular circuit that functions cell autonomously, rhythms can be measured at cellular, tissue, and whole organismal levels [5, 6]. At the organismal level, rhythms can be measured in real time, noninvasively, by monitoring locomotor activity rhythms [7, 8]. The period and phase (e.g., wake-up time or activity onset) of locomotor rhythms in animals are highly stable and reliable over months, enabling prediction of future phases accurately based on measurements in previous cycles.
The molecular backbone of the circadian clock is a transcriptional negative feedback loop with interacting positive and negative elements [9, 10]. CLOCK (or NPAS2) and BMAL1 are the positive elements, activating transcription of many downstream genes, including behavior-regulating genes and the main negative elements, Period (Per, including Per1 and Per2) and Cryptochrome (Cry, including Cry1 and Cry2), whose products form an auto-inhibitory complex [9, 10]. Among all essential clock components, PER is the stoichiometrically rate-limiting component; high amplitude oscillations in PER levels are essential for a robust molecular clock, indicating that duration (period) and phase of the circadian cycle are primarily determined by PER oscillations [11-14].
If robust and consistent PER rhythms are the basis for the high fidelity of the system, then the temporal abundance of PER should be precisely regulated through timely synthesis and degradation of PER protein. Key regulation seems to occur at the posttranslational level because although Per transcription is regulated at genetic and epigenetic levels [15-18], these mechanisms are not essential for generating 24-hr rhythmicity nor for the fidelity of the clock: transgenic expression of a Per-coding sequence from an inducible promoter without any endogenous transcriptional regulation can still produce robust circadian rhythms in Per1/2 double knockout mice [19]. Other studies in mammals as well as Drosophila show that posttranslational regulation of PER associated with degradation works as a circadian timer determining period and phase; PER phosphorylation kinetics, balanced by kinases and phosphatases and associated with degradation, can affect circadian period and phase in a predictable manner [20, 21]. Furthermore, synthetic oscillators built on transcriptional feedback loops but lacking the natural clock's posttranslational regulation are unstable and unpredictable in period and phase from cycle to cycle, even though they may successfully generate self-sustaining oscillations [22-24]. In contrast, the period of the endogenous clock is almost identical across diverse mammalian cell types (including both primary cultured cells and cancer-derived cell lines) [5, 25]. Since the circadian period would be critically influenced by PER degradation kinetics, it is highly likely that the regulation of PER degradation is conserved in these cells. This also suggests that PER degradation should be similar across cell types and cellular environments, unlike most other proteasome substrates such as p53 and β-Catenin (β-Cat), whose half-lives change dramatically under different conditions and in different cell types [26, 27]. Taken together, understanding how PER is precisely and robustly regulated at the posttranslational level is essential to explain why circadian rhythms are more precise and more noise-resistant in vivo than other biological oscillations [28, 29], including synthetic ones.
The Ubiquitin Proteasome System (UPS), which mediates rapid and specific degradation of a target protein through a specific E3 ubiquitin ligase or ligases [30], has been implicated in the circadian clock mechanisms of multiple organisms, including mammals, Drosophila, and Neurospora [31-36]. In mammals, genetic and biochemical studies demonstrated that CRY protein is targeted for proteasomal degradation specifically by the E3 ubiquitin ligases FBXL3 and FBXL21 [37-41]. However, loss-of-function mutations in these two E3 ligase genes did not disrupt molecular and behavioral circadian rhythms dramatically compared to phenotypes caused by mutations in other essential clock genes. By design principles, the circadian clock would be most dramatically affected by disruption in degradation of the rate-limiting component, PER. However, we lack in vivo data on how PER is degraded and how disruption in PER degradation affects circadian behavior. Although E3 ubiquitin ligases β-TRCP1 and 2 have previously been suggested to play a role in PER degradation, β-Trcp1 knockout mice have no circadian phenotype [33], indicating that the paralog β-Trcp2 is redundant in PER degradation or that other E3 ligases are involved.
Here we report that PER degradation is uniquely mediated by balanced ubiquitination and deubiquitination involving at least two different E3 ligases and a specific deubiquitinase, USP14. Genetic and pharmacological disruption in ubiquitination and deubiquitination of PER severely altered clock function. High fidelity of PER degradation is ensured by excess β-TRCP such that even a dramatic reduction in β-TRCP levels does not affect the half-life of PER and the clock. However, when levels of the E3 ligase are reduced below a saturation level relative to the substrate, the fidelity of degradation kinetics is disrupted, resulting in dramatic variability in circadian rhythms from cycle to cycle. Thus β-Trcp double-mutant mice exhibit highly unstable behavioral rhythms, with variable activity onset times almost on a daily basis. By using a mathematical model, we found that such unstable circadian rhythms in β-Trcp mutant mice stem from loss of nonlinear degradation of PER, which has been validated experimentally.
Results
Dynamic ubiquitination and deubiquitination of PER in vivo
If regulation of PER degradation is as central to the circadian clock as we hypothesize, then PER should have a short half-life. To test this hypothesis, we determined and compared the half-lives of PER and the other major clock proteins following cycloheximide (CHX) treatment. To minimize CHX cytotoxicity and effects other than the inhibition of translation, and thus accurately calculate endogenous half-lives of clock proteins, a lowest effective CHX concentration was determined by measuring degradation rates of PER2-LUC in real time. The results indicated that CHX concentration can be lowered to less than 1/8 of what other studies used to inhibit translation [41, 42], while retaining similar efficacy (Figure S1A). When mouse embryonic fibroblasts (MEFs) were treated with this dose, levels of endogenous CLOCK, BMAL1, and CRYs were reduced only slightly over the course of 12 hrs (Figure 1A). However, PER levels were dramatically reduced over the same time course, demonstrating that PER1 and 2 have the shortest half-lives among clock proteins, which is also consistent with previous findings [19] (Figure 1A).
Figure 1. PER degradation is finely tuned by reversible ubiquitination.
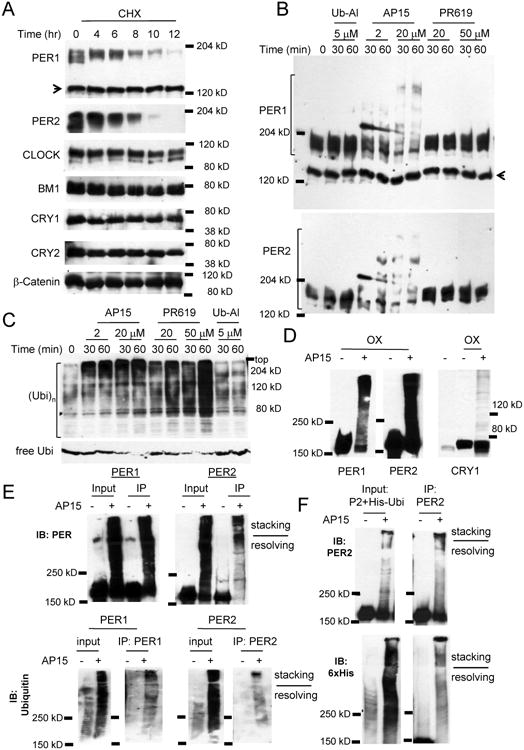
(A) PER1 and 2 have the shortest half-lives among essential clock proteins. Per2Luc MEFs were treated with CHX and harvested at the indicated times. The arrow indicates a nonspecific band in the PER1 blot. These results were replicated in three independent experiments. (B) PER is robustly ubiquitinated when cells are treated with the DUB inhibitor, b-AP15, but not other DUB inhibitors. The PER1 blot was stripped for the PER2 blot. The arrow indicates a nonspecific band. (C) The drugs induced dramatically elevated ubiquitination in total proteins. Note that treatment with b-AP15 or PR-619 almost depleted free ubiquitin (Ubi). Two different Ubi blots are shown. (D) Overexpressed PER proteins are also rapidly ubiquitinated. PER and CRY were overexpressed more than 20-fold above endogenous counterparts in MEFs. (E) The slow migrating PER species are polyubiquitinated PERs. (F) PER2 is ubiquitinated when cotransfected with 6xHis-Ubi. PER2 and 6xHis-Ubi were coexpressed in HEK293a cells. See also Figures S1 and S2A-C.
In vitro studies have suggested that PER can be ubiquitinated by specific E3 ligases such as β-TRCP1/2 [31]. However, in cell culture models, polyubiquitinated PER is not detectable after treating cells with proteasome inhibitors, while polyubiquitinated species of β-Cat, a known β-TRCP substrate, were readily visible under the same conditions [43]. We hypothesized that polyubiquitinated PER, unlike β-Cat, cannot accumulate in vivo in the presence of the proteasome inhibitors because ubiquitinated PER species are subjected to rapid deubiquitination if they are not degraded by the proteasome. If polyubiquitinated PER species are subjected to rapid trimming activity by non-integral deubiquitinases (DUBs) [44, 45], those species would become detectable if the relevant DUBs are inhibited. When MEFs were subject to several broad-spectrum or specific DUB inhibitors, slow migrating PER1 and 2 species appeared only when treated with b-AP15, a specific inhibitor to the non-integral DUBs, USP14 and UCH37 [46] (Figures 1B, S1B and S1C). None of the other major clock proteins were substantially affected by the DUB inhibitors, even though total ubiquitination levels of cellular protein were markedly increased by b-AP15 and some of the other DUB inhibitors (Figures 1C and S1B). For example, treatment with a pan-inhibitor PR-619 induced a dramatic increase in ubiquitinated protein levels and depleted free ubiquitin (Figure 1C). The slow migrating PER species, presumably polyubiquitinated PER, rapidly appeared and became predominant under b-AP15 treatment, even when PER was overexpressed more than 20-fold above endogenous levels (Figures 1D and S2A), suggesting that PER is an intrinsically good substrate for ubiquitination. Free ubiquitin seems to be a limiting component in this ubiquitination since free ubiquitin is almost depleted after 30 minutes of treatment with b-AP15 (Figure S2B). When MEFs overexpressing other clock proteins were treated with b-AP15, only CRY1 was apparently ubiquitinated in a subtle manner (Figure 1D). The slow migrating PER species were indeed due to polyubiquitination (Figure 1E). Furthermore, when PER2 and His-Ubiquitin were transiently co-expressed and treated with b-AP15 in 293 cells, levels of His-Ubi-PER2 increased dramatically compared to control samples without b-AP15 treatment (Figure 1F). Rapid ubiquitination of PER by b-AP15 treatment also occurred in U2OS cells (Figure S2C) suggesting that the system for the dynamic ubiquitination and deubiquitination is conserved across different cell types, and this may be responsible for the specific and invariable half-life of PER and high fidelity of the circadian system.
To identify specific DUBs that can deubiquitinate polyubiquitinated PER proteins, polyubiquitinated PER1 species were immune-purified and incubated with various purified DUBs including USP14 and UCH37. In these initial experiments, none of the DUBs (not even USP14) seemed to affect the substrate, even though some exhibited deubiquitinase activity towards a positive control substrate, Ub-rhodamine 110 (Figures S2D and S2E). Since it was shown that some of DUBs including USP14 depend on the 26S proteasome for normal activity [47, 48], we tested if USP14 mixed with purified 26S proteasome can deubiquitinate and/or degrade the substrate. Moderately polyubiquitinated PER1 species disappeared when exposed to the mixture of 26S and USP14, probably due to deubiquitination followed by degradation (Figure 2A). The 26S proteasome alone also induced apparent deubiquitination, though at a much slower rate. We believe trimming of distal ubiquitination by USP14 facilitated proteasomal degradation by the 26S proteasome because heavily branched ubiquitin chains prevent 26S proteasome-associated Rpn11 from accessing the base of polyubiquitin chains. It is likely that PER degradation typically occurs well before it reaches this extreme polyubiquitination because highly polyubiquitinated PER species accumulated with time rather than decreased under b-AP15 treatment (Figures S2A and S2B). If USP14 is indeed a DUB for ubiquitinated PER in vivo, the half-life of PER and speed of the clock would be regulated by USP14 because the ubiquitination rate of PER would affect degradation kinetics of PER. When PER was coexpressed with wild-type (wt) or a dominant-negative mutant (dn) USP14 in 293 cells [49], PER1 levels were up- and down-regulated, respectively (Figure 2B). PER2 levels were down-regulated when co-expressed with the dn-USP14 (Figure 2B and S2F).
Figure 2. USP14 is a DUB for PER.
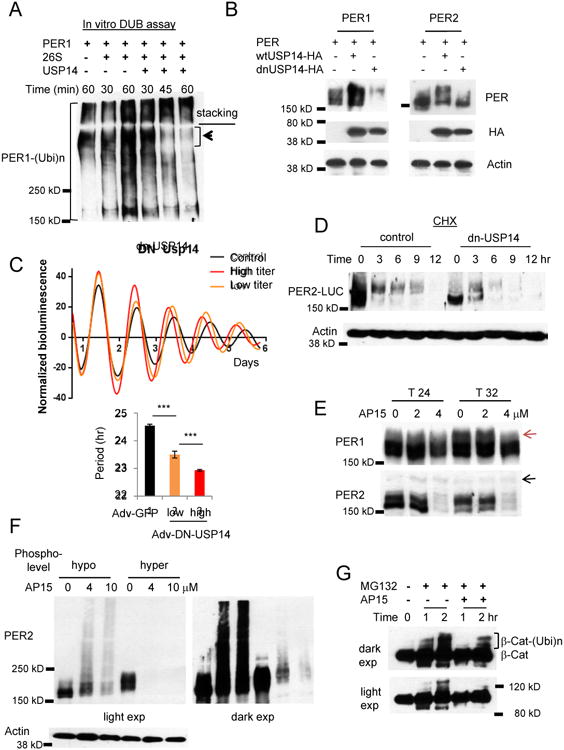
(A) In vitro deubiquitination assays are consistent with the model that polyubiquitinated PER1 is deubiquitinated and degraded by a 26S proteasome-USP14 complex. Note that PER1 species at the top of the resolving gel disappeared when co-incubated with the complex. (B) PER is destabilized when USP14 activity is inhibited. Per1 or Per2 was co-transfected with wt Usp14 or a dnUsp14 in HEK293a cells. (C) The circadian clock is accelerated when dnUSP14 is expressed. Per2Luc MEFs were infected with two different titers of dnUsp14-adenovirus. Data in the lower graph are mean+/-SEM (n=8 each). Representative of three experiments. (D) Hypophosphorylated PER2 species are predominant and rapidly degraded in dnUSP14-overexpressing cells. Representative of two experiments. (E) Nuclear PER species are predominantly affected by the deubiquitinase inhibitor drug b-AP15. Note that hyperphosphorylated PER1 species (indicated by the red arrow) accumulate at T32 only in control and 2 μM-treated cells, but not in 4 μM-treated cells. The black arrow indicated a nonspecific band. T24 and T32 hours represent times after a 2-hr serum shock to synchronize cell rhythms. (F) The sensitivity of hyperphosphorylated PER species is reproduced with overexpressed PER. Hypo- and hyperphosphorylated PER2 was inducibly overexpressed in Rosa-TA; tetO-Per2 MEFs. (G) Ubiquitination of β-Cat is delayed in b-AP15 treated cells. See also Figures S2D-G.
To test how knockdown of USP14 activity affects the endogenous clock, the dnUSP14 was expressed in Per2Luc MEFs. The circadian period was shortened in a dnUSP14 dose-responsive manner because PER degradation was accelerated by the dnUSP14 (Figures 2C, 2D and S2G), indicating that USP14 is indeed a clock-relevant DUB. A low dose of b-AP15 affected only certain PER species that are found in the nucleus. Subcellular fractionation studies showed that only hyperphosphorylated PER1 but both hypo- and hyperphosphorylated PER2 species are found in the nucleus [11]. Only these nuclear PER species disappeared while hypophosphorylated PER1 species were intact under the low dose treatment (Figure 2E), suggesting that nuclear PER proteins are preferentially subjected to ubiquitination and proteasomal degradation. Consistent with this idea, when hyperphosphorylated PER2 species were transiently expressed, they were degraded much faster than non- to hypophosphorylated PER2 by b-AP15 treatment (Figure 2F). Our data suggest that β-Cat is a much less efficient substrate for ubiquitination than PER. If free ubiquitin is limiting and β-Cat is not a preferred substrate for ubiquitination, b-AP15 treatment would delay ubiquitination of these poor substrates. Indeed, β-Cat ubiquitination was significantly delayed in b-AP15-treated cells compared to control cells (Figure 2G). Taken together, these data suggest that PER is a priority substrate for ubiquitination and deubiquitination in cells where free ubiquitin is limiting, which ensures consistent degradation over a wide range of cellular conditions. Therefore, unlike β-Cat, de/ubiquitination of PER and its half-life would be much less affected by stochastic or even stressful variation in the cellular environment.
β-TRCP1 and 2 are essential and partially redundant for the clock
β-Trcp was initially implicated in PER degradation based on homology with Drosophila slimb and on subsequent in vitro studies [31-34, 36]. However, β-Trcp1 mutant mice did not show a significant change in circadian behavior [33] (Figures S3A and S3B). We believe this is due to redundancy between two paralogues, β-Trcp1 and β-Trcp2. In our in vitro binding assays, β-TRCP2 does bind to PER, and does so more efficiently in the presence of CK1δ, as shown previously [36] (Figure S3C). Since constitutive β-Trcp2 deletion could lead to embryonic lethality, we generated a conditional β-Trcp2 mutant mouse to circumvent this issue using the cre-loxP technology (Figure S4). During the course of our study, Ben-Neriah and colleagues demonstrated that β-Trcp2 is indeed essential for development [50]. While β-Trcp1 knockout (ko) mice do not exhibit any discernible defects other than male sterility [51], our constitutive β-Trcp2 ko mice (like Ben-Neriah's) showed embryonic lethality: no homozygous β-Trcp2 ko offspring were generated from the mating of heterozygote parents (β-Trcp2+/knockout first) (n > 40 pups). This difference could be due to differential spatiotemporal expression between the two paralogues or a dramatic difference in expression levels. In MEFs, mRNA levels of β-Trcp2 were 9-fold higher than those of β-Trcp1 (Figure S4D). Thus it may be that β-Trcp1 ko mice have a normal clock because the clock mechanism can continue to rely on the much more abundant β-Trcp2. Accordingly, unlike β-Trcp1 mutant mice, β-Trcp2 mutant mice exhibited a dramatic circadian phenotype that is distinctively different from other existing clock mutant mice (Figure 3). Phase angle (activity onset) changed constantly once deletion of β-Trcp2 was induced. The majority of the mutant mice were still rhythmic but an accurate calculation of period was not possible due to the unstable phase angle (Figures 3A-D and S5A) (n=11). Some of the mutant mice became arrhythmic after period was lengthened (Figure 3A) (n=2). As shown with five different β-Trcp2 mutant mice in Figure 3, no two mice exhibited similar patterns in the phase angle change: phase angle changed abruptly and randomly in all mice. Although in some cases phase angle changed on a daily basis, in most cases, the phase angle seemed stable for several (∼7) days after a sudden shift. We used these days of relative stability to calculate the circadian periods of rhythmic β-Trcp2 mutant mice (n=11). These quantitative data clearly demonstrate that circadian period is generally lengthened and period variability increases dramatically once β-Trcp2 is deleted. After the period is lengthened or the mice rendered arrhythmic, the mutant mice were re-entrained normally by a 12 hr-light:12 hr-dark cycle (LD), indicating that the light input pathway is intact (Figure S5B). Bioluminescence rhythms from mutant MEFs also showed similarly unstable and low amplitude rhythms (Figures 3E and 3F). In addition, the variance in the duration of upswing and downswing increased dramatically after tamoxifen treatment (Figure 3G).
Figure 3. Behavioral rhythms are unstable in β-Trcp2 mutant mice.
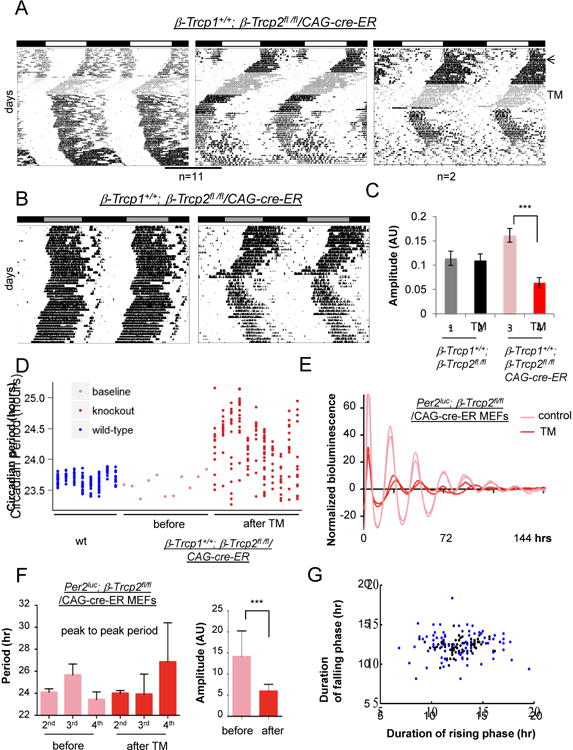
(A) Deletion of β-Trcp2 leads to unstable (11 mice) or arrhythmic (2 mice) behavior. The arrow indicates when a 12 hr light: 12 hr dark (LD) cycle changed to constant darkness (DD), and the shaded area indicates when tamoxifen-containing food was given (Figure S4). (B) Behavioral phenotypes of two more β-Trcp2 mutant mice are shown. The actograms were measured in DD after 2 weeks of tamoxifen treatment. (C) Quantification of amplitude in β-Trcp2 mutant mice. Amplitude was dramatically reduced in β-Trcp2fl/fl/CAG-cre-ER mutant mice (n=11) compared to control mice, β-Trcp2fl/fl without the cre-ER transgene. Tamoxifen treatment in the control β-Trcp2fl/fl mice did not change either period or amplitude (n=9). Period = 23.58+/-0.32 (before) vs. 23.52+/-0.51 (after) (mean+/-STD) (p>0.05). Representative actograms for the control mice are shown in Figure S5A. (D) Quantification of period in β-Trcp2 mutant mice. Since it is not possible to calculate long-term periods and statistical values due to unstable phase angle in all individual mutant mice, short-term periods were calculated over a short duration where phase angle is stable (∼ 1 week) in individual β-Trcp2 mutant mice for ∼ 3 months (red dots) (n=11). Eleven columns represent 11 individual mutant mice. Baseline period before tamoxifen treatment was stable and measured for two weeks (pink dots). Similarly short-term periods were also measured in C57BL6/J over two months (blue dots) (n=8). (E, F, G) Unstable rhythms and reduced amplitude are recapitulated in cultured β-Trcp2 mutant cells. Bioluminescence rhythms were measured from Per2Luc; β-Trcp2fl/fl/CAG-cre-ER MEFs before and after tamoxifen treatment. Period was calculated on the 2nd, 3rd and 4th cycle. n=16 each. Black and blue dots represent before and after TM treatment, respectively. n=12 each in (G). See also Figures S3-5.
β-Trcp1 is partially redundant with β-Trcp2; β-Trcp1 and 2 double-mutant mice showed more severe phenotypes than β-Trcp2 single-mutant mice (Figures 4A-C). The phenotype was proportional to dosage of the β-Trcp genes. Mice with only one allele of β-Trcp1 (β-Trcp1+/-; β-Trcp2-/-) showed consistently longer periods than those with two (β-Trcp1+/+; β-Trcp2-/-), and died within a month of β-Trcp2 gene deletion (n>5). Complete double-mutant mice β-Trcp1-/-; β-Trcp2-/- were lethal within 2 weeks after deletion and showed arrhythmicity before they died (Figure 4A) (n>10). The double-mutant mice were active until a day before their death and even exhibited increased wheel running activity compared to baseline prior to gene deletion. Since wheel running requires high levels of motor coordination, these data suggest that deletion of both β-Trcp genes do not induce broad pathological dysfunction in the brain. Double heterozygotes and mice with only one allele of β-Trcp2 did not show unstable phase angle but exhibited slightly longer periods (Figure 4C). In peripheral tissues, the molecular clock was completely disrupted in double-mutant mice (Figures 4D and 4E). Unlike dPER in slimb-mutant Drosophila, PER1 and PER2 were constitutively hyperphosphorylated and their levels were not dramatically upregulated. Consistent with previous findings [12], CLOCK and BMAL1 were constitutively upregulated in β-Trcp double mutant cells. As expected based on β-Trcp1 studies [33], β-Trcp2 mRNA did not oscillate in peripheral tissues (Figure S6A).
Figure 4. Behavioral rhythms are disrupted in β-Trcp1/2 mutant mice in a gene-dosage dependent manner.
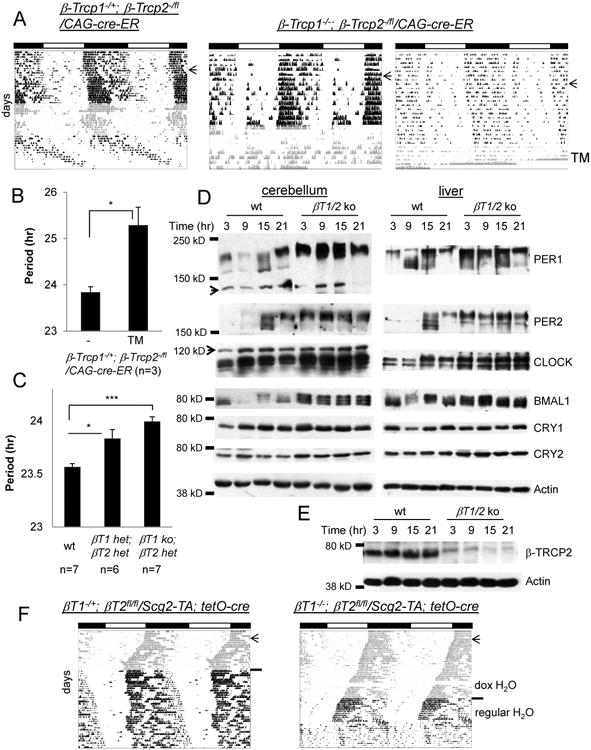
(A) Representative actograms from β-Trcp1+/-; β-Trcp2 fl/f / CAG-CreER-1 (n=3) and β-Trcp1 -/- ; β-Trcp2 fl/fl / CAG-CreER-1 (n=10) mutant mice. The arrow and shaded area indicate the LD to DD transfer and TM treatment, respectively. Locomotor activity was measured until the mutant mice died. (B) Quantification of period before and after TM treatment. (C) β-Trcp1+/-; β-Trcp2-/+ and β-Trcp1-/-; β-Trcp2-/+ mutant mice exhibit slightly longer periods than matching wt mice. The β-Trcp2 null allele (β-Trcp2knockout-First) was used to generate the mutant mice. (D) Peripheral clocks are arrhythmic in β-Trcp1/2 double mutant mice. Wt and ko PER1 results were spliced from the same blots. (E) β-TRCP2 is almost absent 5 days after TM treatment. The mutant liver tissues from (D) were used. (F) Brain-specific β-Trcp2 mutant mice survived longer but exhibited similarly disrupted behavioral rhythms. Doxycycline (dox) in drinking water suppresses recombination in these mice [12].
To avoid the lethality associated with whole-body knockout and thus to monitor activity from double mutant mice for a longer period, we generated neuron-specific mutant mice using Secretogranin2 (Scg2, a neuronal promoter)-TA and tetO-cre transgenes [12]. Neuron-specific β-Trcp double mutant mice survived much longer and also showed similarly unstable phase angle (β–Trcp1+/-; β–Trcp2fl/fl/Scg2-TA; tetO-cre) or arrhythmicity (β–Trcp1-/-; β–Trcp2fl/fl/Scg2-TA; tetO-cre), supporting our hypothesis that unstable phase angle (wake/sleep cycles) is due to a defective clock in the pacemaker neurons rather than disrupted general physiology (Figure 4F).
PER degradation is regulated by multiple E3 ligases
To tease out how PER proteasomal degradation and the clock controlled pathways are altered by β-Trcp deficiency, we measured how PER posttranslational regulation and the transcriptome are altered in β-Trcp mutant MEFs. As in peripheral tissues in mice, PER1 and 2 were constitutively expressed in double mutant MEFs (Figure 5A). A canonical β-TRCP substrate, β-Cat, could not be ubiquitinated and its levels were elevated in β-Trcp1/2 knockout cells (Figure 5B). Consistent with hyperphosphorylated status of PER in β-Trcp1/2 mutant cells, levels of nuclear PER1 and 2 were higher than those in control cells, suggesting that hyperphosphorylated nuclear PER proteins are subjected to proteasomal degradation by β-TRCP (Figure 5C). Proteasomal targeting of nuclear PER by β-TRCP is supported by the subcellular location of endogenous β-TRCP2, which is predominantly perinuclear with mild nuclear presence (Figures 5D and S6B). High levels of constitutive nuclear PER and increased CRY levels (Fig 5C and S6C) suggested constitutive feedback inhibition of CLOCK:BMAL1. Indeed, expression of most of the direct clock-controlled genes (CCGs) were altered in a manner consistent with higher inhibition of CLOCK:BMAL1 (Figure 5E). In addition, deletion of β-Trcp1 and 2 affected gene expression in various signaling pathways including the Wnt pathway (Table S1). As expected from stabilized β-Cat, the Wnt pathway seems to be upregulated (Figure 5E). For example, expression of a major feedback inhibitor and transcriptional target of the Wnt pathway, Wnt inhibitor factor 1 (Wif1), increased dramatically, while a Wnt pathway activator, Wnt16, was dramatically downregulated [52].
Figure 5. Circadian and Wnt pathways are disrupted in β-Trcp1/2 double mutant MEFs.
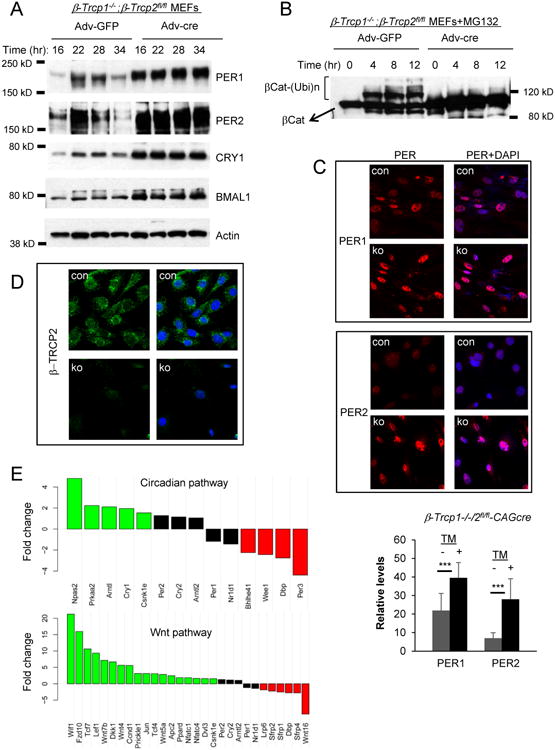
(A) The molecular clock is disrupted in the mutant cells. Cells were harvested at the indicated times after a 2-hr serum shock. Recombination was induced by adenovirus expressing cre recombinase as described previously [20]. (B) The canonical substrate of β-TRCP, β-Cat cannot be ubiquitinated in the mutant cells. (C) PER and CRY are predominantly nuclear in the mutant cells. See Figure S6C for CRY1. Nuclear PER levels are higher in the mutant cells than in control cells. p < 0.001 (unpaired Student's t test). Mean+/-STD. Scale bar = 25μm. (D) β-TRCP2 is mostly perinuclear with the weak nuclear staining. Scale bar = 25μm. A representative single cell is shown in Figure S6C. (E) Genes involved in the circadian and Wnt pathways are dysregulated. Green and red bars indicate significantly up and down-regulated genes, respectively (FDR-adjusted p <0.05). The whole transcriptome is compared between control and double mutant MEFs in Table S1. See also Figure S6.
When stability of PER was measured with CHX treatment, stability of both PER1 and 2 increased significantly with deletion of β-Trcp1 and 2 genes (Figure 6A). Consistent with our prediction, hyperphosphorylated PER species were persistently present after CHX treatment in the mutant cells, suggesting that hyperphosphorylated PER species are preferred targets of β-TRCP. However, those species were still degraded albeit at a slower rate, suggesting that PER is targeted for proteasomal degradation by other E3 ligase(s) in addition to β-TRCP. Indeed, accelerated endogenous PER degradation occurred by b-AP15 treatment in β-Trcp1/2 knockout cells (Figure 6B). Polyubiquitination of PER1 and 2 was also as robust in mutant cells under b-AP15 treatment as in control cells, further supporting that multiple E3 ligases for PER exist (Figure 6B). The levels of β-TRCP seem to be in a great excess relative to the levels of endogenous PER because behavioral rhythms and the half-life of PER are only slightly altered even in β-Trcp2 mutant mice which have lost 90% of total β-Trcp expression. Assuming that β-TRCP1 and 2 are equally potent, these data indicate that proteasomal targeting of PER by excess β-TRCP ensures a maximum rate of degradation always, even when β-TRCP levels or activity fluctuates dramatically.
Figure 6. PER is stabilized in β-Trcp1/2 double mutant cells.
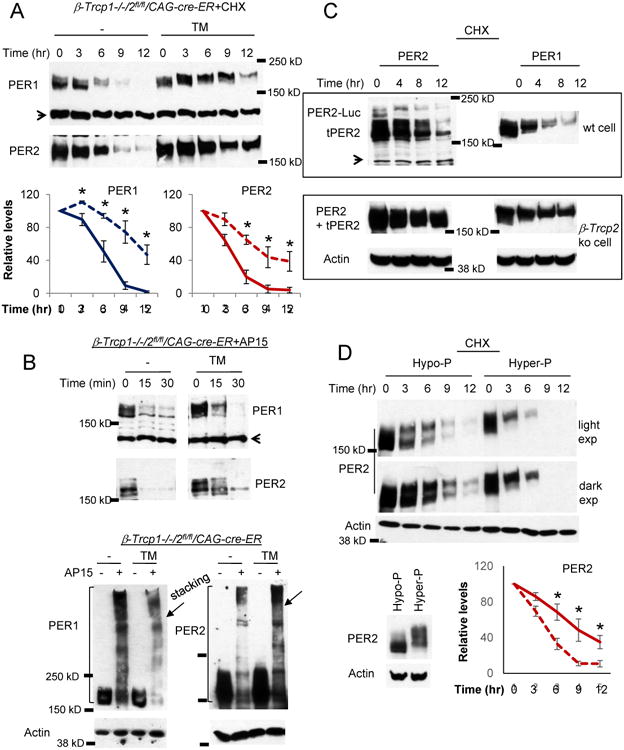
(A) PER stability was measured after CHX treatment. Data are mean+/-SEM, n=3 each. (B) PER can be robustly ubiquitinated in the β-Trcp1/2 double mutant cells. PER degradation and ubiquitination were induced by b-AP15 treatment as in control cells. The arrow in the bottom blots indicates the boundary between stacking and resolving gel. (C) β-TRCP is in excess to PER. Per2Luc homozygous knockin MEFs were used for the wt control, but wt Per2 allele (+/+) was used for β-Trcp2 ko cells. Note that endogenous and exogenous PER2 proteins are different in size in wt control cells but the same in the mutant cells. (D) Hyperphosphorylated PER is more unstable. Comparable amounts of hypo-and hyperphosphorylated PER2 was expressed in the inducible Per2 MEFs before CHX treatment. The solid and broken lines represent hypo- and hyper-phosphorylated PER2 samples, respectively. Mean+/-SEM, n=3.
If β-TRCP abundance is indeed much in excess to PER, degradation of moderately overexpressed PER in wt cells would not be affected because β-TRCP is still in excess. However, the degradation kinetics of overexpressed PER would be greatly slowed in β-Trcp2 mutant cells because the amount of β-TRCP1 in the mutant cell would be barely enough to degrade even endogenous levels of PER, as expected from the behavioral data. Our results were consistent with this expectation: when PER2 was moderately overexpressed, degradation of both endogenous and transgenic PER2 (tPER2) occurred at a similar rate (Figure 6C), but when PER2 was similarly overexpressed in β-Trcp2 mutant cells, PER2 degradation was dramatically slowed, similar to endogenous PER in β-Trcp1/2 double-mutant cells. Endogenous PER1 followed the same pattern.
PER proteins are progressively phosphorylated as they accumulate and disappear [11]. Our data so far suggest that the main role of β-TRCP is to target hyperphosphorylated PER species for proteasomal degradation in a robust and fast manner on top of basal degradation mediated by an unknown mechanism. In other words, PER half-life is progressively shortened as it is more and more phosphorylated because these species are additionally targeted for degradation by β-TRCP, leading to exponential degradation in the later stage of the circadian cycle. To test if hyperphosphorylated PER species are indeed more unstable, half-life was compared between hypo- and hyperphosphorylated PER2 using an inducible transgenic Per2 system [19]. When PER2 was comparably expressed, hyperphosphorylated species were degraded dramatically faster than hypophosphorylated ones (Figure 6D) further supporting that PER phosphorylation is a prerequisite for β-TRCP-mediated ubiquitination and degradation.
The nonlinear degradation of PER is critical for the stability of the circadian clock
In both laboratory experiments and mathematical models, it has been well demonstrated that certain molecular alterations can speed up, slow down, or completely disrupt the circadian clock [3, 41, 53]. However, it has not been shown how the clock and circadian rhythms can be unstable as they are in β-Trcp2 mutant mice. Our mouse experiments suggest that impairment of PER proteasomal degradation can disrupt the stability of the clock. To better understand the underlying design principles of the circadian clock, in silico simulation was performed using a mathematical model for the circadian oscillator that includes proteasomal degradation (Figure 7).
Figure 7. The circadian clock becomes unstable in β-Trcp mutant cells in silico.

(A) Diagram of the modified Kim-Forger model, where Per mRNA (“M”) is translated to PER protein (“C”) in the cytoplasm. The nuclear translocated PER protein (“R”) represses transcriptional activity of the activator (“A”). Here, all parameters and variables are dimensionless (see Method for details). (B) To simulate β-Trcp gene dosage decrease, Kd for PER-proteasome increases in the model. 105 oscillations of stochastic simulation are analyzed. Here, the half-life is defined as the time to reach the 50% of the initial concentration of PER protein. Furthermore, the period, amplitude, half-life are normalized to the value of 1 for WT model. Here, Kd= 0.1, 0.132, 0.167, 0.2, 0.234, 0.275, 0.315, 0.355, 0.4, 0.45, 0.497, to generate half-life values similar to those seen in the experimental data (see Figure. 6A). (C) Trajectories of deterministic simulations (dashed line) and stochastic simulations (solid line). As Kd increases, oscillations in the presence of stochasticity become more variable and finally become completely disrupted. Here, Kd= 0.1, 0.234 and 1, for wt, β-Trcp2-/- and β-Trcp1/2-/-, respectively, to generate half-life values similar to those seen in the experimental data (Fig. 6A). (D) Transient PER2 half-life is gradually shortened in wt cells, but not in mutant cells. Transient half-life of PER2-LUC was calculated from bioluminescence rhythms in wt, β-Trcp2 and β-Trcp1/2 mutant MEFs, after translation is stopped by CHX. N=4 each. See also Figure S7.
We have previously used the Kim-Forger circadian model, which describes the core transcriptional-translational negative feedback loop of the circadian clock [14, 54], but uses a simple linear degradation process for PER. Recent studies have shown that a Michaelis-Menten type of degradation can be used to simulate the biological proteasomal degradation involving multiple components such as ubiquitin; E1, E2, and E3 enzymes; deubiquitinases; and the proteasome [55, 56]. Thus, to incorporate β-TRCP-mediated PER proteasomal degradation into the model, we modified the model (Figure 7A) by replacing the simple linear degradation equation with a Michaelis-Menten equation:
where [R] is the concentration of the substrate protein (PER). The maximal degradation rate, Vmax and half-maximal constant, Kd of the degradation are determined by the combination of the concentrations of E1, E2, E3 ligases, deubiquitinase, and proteasome [56].
Xu and Qu showed that the decrease of the E3 ligase considerably increases Kd, but has little effect on the value of Vmax [56]. Thus, to simulate β-Trcp2 or β-Trcp1/2 mutant clock cells, in which the levels of the E3 ligase β-TRCP are decreased, we increased Kd but fixed the value of Vmax (Figure 7B). Furthermore, to investigate how cellular “noise” affects period and amplitude when Kd changes, we performed a stochastic simulation using the Gillespie algorithm. For 105 stochastic oscillations, we calculated peak-to-peak periods and amplitude of each cycle. As the value of Kd increases, the average period from cycle to cycle increased only modestly, but amplitude decreased dramatically (Figures 7B and 7C) consistent with the phenotype in β-Trcp2 mutant cells (Figures 3 and 4). The most striking outcome of this in silico simulation was the dramatic instability of the oscillator, which is not proportional to the increase in period. This is exactly what we have observed in β-Trcp mutant mice. The period was lengthened only modestly before it becomes arrhythmic as the dosage of β-Trcp decreases. In certain other clock-mutant mice, circadian period can be lengthened to greater extremes while maintaining robust rhythms [38, 57]; thus the arrhythmic behavior in β-Trcp mutant mice cannot be explained by period lengthening. It is rather caused by increased instability or noise of the oscillator due to compromised nonlinear degradation. If Kd increases to a certain level, leading to a 2.5-fold half-life increase in PER stability, rhythms are completely disrupted as observed in β-Trcp1/2 double mutant cells (Figure 7C). On the other hand, varying Vmax leads to a larger change in period but a smaller change in amplitude and period stability than simulations with varying Kd (Figure S7A). Similar patterns were also observed when different circadian clock models were used [54, 58, 59] (Figure S7B).
In our model, when Kd becomes much larger than [R], PER degradation occurs linearly because . Thus, as Kd increases due to β-Trcp mutations, a non-linear degradation process is changed into a linear degradation process. Previous modeling studies have also reported that the nonlinear degradation is critical for robustness of rhythms [60-62].
When Per2Luc β-Trcp wt MEFs were treated with CHX, the transient half-life of PER2-LUC decreased as PER2-LUC levels decreased gradually with time, probably because PER becomes more and more ubiquitinated by β-TRCP (Figure 7D). However, this nonlinear degradation profile was compromised in β-Trcp2 mutant cells, consistent with the model prediction. Although the overall half-life changed mildly in β-Trcp1/2 mutant cells, rhythms are completely compromised. Taken together, our data strongly support the hypothesis that robust PER degradation in a saturated and nonlinear manner is essential for the high fidelity and stability of the circadian oscillator.
Discussion
While the transcriptional negative feedback loop based on PER and CRY is required to generate circadian rhythms, it does not appear enough to explain the exceptional stability of the rhythms, considering the fact that the majority of synthetic oscillators based on transcriptional negative feedback loops lack such stability. This represents a fundamental gap in our understanding of the circadian system. The present study provides a mechanistic understanding of this key property of the clock, along with supporting in vivo and in silico data. Our proposed mechanism for the stability and fidelity of the clock is also tied to how the clock is self-sustained. Mounting evidence supports that PER is “the state variable” in the clock system.
It seems a common theme that PER is regulated by reversible biochemical reactions; PER phosphorylation state is regulated by both kinases and phosphatases [20]. In this study, we have shown that PER ubiquitination is also reversible. Rapid reversible biochemical reactions of PER are seemingly futile, especially considering the long time scale of PER activity in the negative feedback loop. However, we believe this would be the most effective way to build a slow molecular oscillator that can resist variations and fluctuations in the cellular environment. Essential parameters such as time delay, nonlinearity and robustness can be generated simultaneously from the dynamics of reversible opposing reactions: the time delay occurs because the opposing reactions slow down the net change on the substrate (PER). Robustness is maintained over a wide range of cellular environmental fluctuations, such as temperature, if both forward and reverse enzymes are affected at a similar rate, keeping the relative ratio of activity the same. Even if environmental fluctuations affect the reaction only in one direction, modest fluctuations would not affect the reaction rate significantly because all these reactions occur at a saturation level (i.e., enzyme ≫ substrate), which would be maintained whether enzyme levels/activities increase or decrease as long as they are above the saturation level. As shown in this study, in association with the phosphotimer [20, 21], β-TRCP regulation of PER degradation generates a nonlinear degradation of the inhibitor ensuring a robust derepression of the feedback inhibition followed by a robust rising phase in the next cycle.
Inhibition of USP14 activity by a dominant negative USP14 mutant accelerated the oscillator (Figure 2C). We believe that slow degradation of PER by USP14 at the right phase of the feedback loop is essential for the time delay in derepression of circadian transcription as well as for generating nonlinear degradation. It has been long assumed that a time delay before the feedback inhibition is essential for self-sustaining oscillators, although the underlying mechanism is not clearly defined for the circadian clock. Circadian rhythms are strikingly symmetric: e.g., bioluminescence rhythms from PER2-Luciferase consist of a ∼12 hr upswing and ∼12 hr downswing [5]. We believe a symmetric time delay and nonlinearity are present in equal measure on both sides of the curve in order to generate a symmetric oscillator.
β-Trcp has been implicated in the mammalian clock based on homology with slimb in Drosophila clock and in vitro studies as stated above. However, according to our current study, PER ubiquitination can occur without β-TRCP1/2 as PER ubiquitination is largely intact in β-Trcp1/2 double mutant cells. Since F-box ligases recognize phosphodegrons [63], the main role of β-TRCP may be to preferentially target hyperphosphorylated PER for degradation on top of the basal (constant) degradation of PER mediated by other, unknown E3 ligase(s), which leads to nonlinear degradation. Our mathematical modeling predicted that self-sustaining rhythms are unstable or less likely to continue if the β-TRCP step is missing and the nonlinearity is compromised. Deficiency of β-TRCP2 resulted in a small change in circadian period, but dramatic variations of the rhythms from cycle to cycle. In β-Trcp1/2 double mutant cells, PER is still degraded, but rhythms are completely disrupted rather than lengthened because the system is extremely unstable. It has been well demonstrated that nonlinearity is essential for robustness in any gene regulation network and morphogen gradients for pattern formation during development [64, 65].
The wake/sleep cycle is the most salient circadian rhythm in mammals. The wake time (activity onset) is especially tightly coupled to the circadian clock, and thus is frequently used as the marker of phase of circadian rhythms. It is very stable in frequency in most individual animals under constant conditions. The frequency or period can be changed by mutations in clock genes. However, none of the previously characterized clock mutant mice exhibited unstable wake/sleep rhythms as we have now shown in β-Trcp2 mutant mice (e.g., Clockm1Jt and Ovtm mutant mice) [41, 57]. The unstable phenotype is unique among rodent models and emulates certain human circadian sleep disorders. Five different circadian sleep disorders have been recognized depending on the etiology and overt phenotype [66], two of which involve irregular wake/sleep cycles: non-24 hr wake/sleep and irregular wake/sleep disorders. We believe that unstable PER rhythms could be the major underlying cause of irregular wake/sleep cycles in some of the circadian patients. Since it is highly unlikely that both β-Trcp2 alleles are deleted or inactivated in humans, we speculate that the unstable rhythms are more commonly caused by polymorphisms or point mutations in Per genes that affect PER ubiquitination by β-TRCP; this would also disrupt the nonlinear degradation of PER similar to β-Trcp mutation in mice. However, if mutations or polymorphisms including the hPER2 mutation leading to FASPS do not affect Kd and Vmax of PER ubiquitination and degradation, the clock would not be unstable even though the clock can be shortened or lengthened because the timing of ubiquitination may change but not the kinetics.
In short, our study shows that PER is uniquely and extensively ubiquitinated, and this process has a direct consequence on PER degradation, clock function, and behavior. It is imperative to continue to study how PER polyubiquitination is mediated and how this is coupled to proteasomal degradation, as this mechanism may underpin multiple circadian sleep disorders.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Choogon Lee (Choogon.lee@med.fsu.edu).
Experimental Model and Subject Details
Animals
All mice were maintained in a climate-controlled room and used according to the Florida State University Animal Care and Use Committee's guidelines. All experiments involving animals were performed according to approved protocols. We used about equal number of 2-4 months old male and female mice for behavioral studies. Sex differences in behavioral rhythms and cellular clock are very subtle to insignificant [67].
The β-Trcp1 mutant mouse was described previously [51]. The β-Trcp2 mutant mouse harboring the knockout First allele was generated in the C57BL/6N strain at the Knockout Mouse Project Repository (KOMP), at University of California, Davis, in which loxP sites flank exon 4 of the β-Trcp2 (Fbxw11) gene. The heterozygote mutant mice we received from the KOMP were backcrossed into C57BL6/J stain for several generations. These mice were then crossed with a flippase (Flp) driver mouse, Gt (ROSA) 26Sor (tm2(FLP)Sor/J) (Jackson Labs #012930), in order to convert the knockout-first allele to a conditional knockout allele. The resulting offspring were bred with a Cre-driver mouse (CAG-CreEsr-1, Jackson Labs #004682) in order to make a mouse in which β-TRCP2 could be conditionally deleted throughout the body. To generate a brain-specific β-Trcp2 mutant mouse, the β-Trcp2fl/fl mouse was crossed with a Scg2-TA/tetO-cre double transgenic mouse [12]. Scg2-TA was described previously [12]. tetO-cre transgenic mouse was purchased from Jackson Laboratory (#006234).
Mouse Embryonic Fibroblasts (MEFs)
MEFs were prepared from embryos isolated from pregnant female mice, 13 days post-coitum. Embryos were removed, finely minced and treated with 0.25% trypsin and incubated at 37 °C for 30 minutes. The mixture was passed through a fine 100μm membrane, to remove debris, and the resulting cells were maintained at 37 °C, 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum (FBS). β-Trcp2 deletion was induced by 4-hydroxytamoxifen as described previously [20]. Both male and female embryos were used. MEFs originated from different embryos with the same genotype produced same results when they were tested for bioluminescence rhythms.
Other Cell Lines
HEK293a (ThermoFisher #R70507) and U2OS (ATCC HTB-96) cells were obtained from commercial vendors. All cells were maintained at 37 °C, 5% CO2 in DMEM, supplemented with 10% FBS.
Method Details
Genotyping
Different alleles of β-Trcp2 were detected using the following primers.
β-Trcp2 Knockout First
Common loxP: gagatggcgcaacgcaattaat (fw)
Fbxw11-SR2: caaacgttaacaggctggagagatgg (rev)
Amplicon: 670 bp
β-Trcp2Flox and wt alleles
Fbxw11E3Fw: gccctgctttatcattggtatgtatgt
Fbxw11E4Rev: cacagatgacgttccattactg
Amplicon: wt ∼600 bp; mutant 860 bp
β-Trcp2Flox and β-Trcp2- alleles
FRT/loxPFw: gaaagtataggaacttcgtcgaga
Fbxw11-SR2: caaacgttaacaggctggagagatgg (rev)
Fbxw11E4Rev: cacagatgacgttccattactg
Amplicon: flox 560 bp; ko 640 bp
PCR condition: 50-100 ng genomic DNA was set up with dNTP (0.5mM), primers and DNA polymerase (NEB, M0530) in a 20 ul reaction and subjected to the following thermal cycle: 94 C for 30 sec; 35 cycles of 94 C for 30s, 60 C for 45s, 72 C for 45s; 72 C for 10 min.
Analysis of circadian behavioral rhythms and other data
All mice were individually housed in wheel-running cages, with free access to food and water. Wheel-running activity was recorded and analyzed using established software platforms (the Stanford Software System or ClockLab from Actimetrics). In Fig S4B, a 30 min light pulse (∼500 lux) was given to mice at either ZT15 or ZT21 in the last LD cycle. Phase shifts were calculated as described previously [68]. Initially, all animals were placed in a 12h light:12h dark (LD) cycle, for at least 7 days. Mice were then transferred to constant darkness (DD), for at least two weeks, to measure baseline activity. β-Trcp2 deletion was induced in the whole body in β-Trcp2fl/fl/CAG-CreEsr-1 mutant mice by feeding them tamoxifen-containing chow (Harlan TD.130859; 0.4g tamoxifen/kg food) for two weeks. The wt control and β-Trcp2fl/fl control without the CAG-CreEsr-1 transgene in Fig 3 received the same treatment. LD entrainment and baseline behavioral data are not shown in Fig 3B. We showed that this tamoxifen treatment does not induce circadian disruption in wt and floxed mutant mice without the cre transgene [69].
After two weeks, any remaining tamoxifen chow was removed and replaced with regular mouse chow for the remainder of the recordings. For the β-Trcp1/2 brain-specific mutant mice, the breeding mothers were given ad libitum access to drinking water that contained 20μg/mL doxycycline and 5% sucrose, to maintain suppression of the tetO-cre transgene expression in utero. Pups were also maintained on 20μg/mL doxycycline, in 5% sucrose water, throughout the weaning period, the one-week LD and 2-6 weeks DD baseline periods. After measuring baseline in DD, the bottles were switched to regular water to allow for transgene expression and subsequent deletion of β-Trcp2, in a brain-specific manner.
Antibodies
Antibodies to clock proteins were generated against recombinant clock proteins expressed and have been previously reported [11, 70]. CLK-1-GP, BM1-2-GP, C1-GP (CRY1), C2-GP (CRY2), CK1δ-GP and CK1ε-GP antibodies were used at 1:1,000 dilution in 5% milk–Tris-buffered saline containing 0.05% Tween 20 solution. New anti-PER1 and 2 antibodies were generated using the same recombinant proteins described previously [11]. The antibodies were generated by Cocalico Biologicals Inc. (Reamstown, PA, USA). These antibodies were tested and compared with the original antibodies. The best antisera were selected and affinity-purified. To detect PER1 and 2, PER1-GP62 and PER2-GP58 were used (Both were raised in guinea pigs). Endogenous PER proteins detected by these antibodies showed the same oscillation pattern in abundance and phosphorylation as the original ones (PER1-GP and PER2-GP). Antibodies to β-TRCP1 and β-TRCP2 were raised against amino acids 1-130 of β-TRCP1 and amino acids 1-96 of β-TRCP2, respectively. The DNA sequence encoding the β-TRCP1 peptide was cloned into BamHI and HindIII sites of pET23b vector and the DNA sequence for the β-TRCP2 peptide was cloned into BamHI and EcoRI sites of the vector. Antibodies were generated by Cocalico Biologicals Inc. (Reamstown, PA, USA). Representative antiserum to each peptide was affinity purified and designated β1-GP4 for β-TRCP1 and β2-GP15 for β-TRCP2 (Both were raised in guinea pigs). Rabbit anti-ACTIN antibody (Sigma, A5060) and used at 1:2,000. The ubiquitin antibody (Cell Signaling Technologies, #3933) was used at a 1:1,000 dilution. The beta-Catenin antibody (BD Biosciences, #610153) was used at a 1:3,000 dilution. Anti-HA and Flag antibodies were purchased from Sigma (#11583816001, #F3165) and anti-His antibodies from Santa Cruz (#53073).
Adenoviral vectors and plasmids
The construction of the recombinant adenoviral vectors encoding wt-USP14 (pAdTrack-wtUSP14) and a dominant negative (dn)-USP14 (pAdTrack-dnUSP14) followed the procedure of He et al. [71]. Briefly, a wt-USP14 plasmid was purchased from Addgene (#22569). The coding sequence was sub-cloned into a pcDNA 3.1 vector, with the addition of a 5′ SalI restriction site, an HA-tag and a Kozak sequence, and a terminal 3′ NotI restriction site. The sequence between the SalI and NotI sites was subcloned into the pAdTrack-CMV vector. The resultant wt-USP14 pAdTrack was used in virus production. The dn construct was produced from the WT-USP14 in pcDNA3.1, through the introduction of a C114A mutation (Lee et al., 2010). Primers were designed using NEB Basechanger: F1 TGGTAACACTGCCTACATGAATGCCAC and R1 AGGTTTGTCAATCCACATG (DN mutated bases are in bold). Mutagenesis was performed by the FSU Biology Cloning facility, using a Q5 Site-Directed Mutagenesis kit (NEB). After confirmation of the mutation, the construct was sub-cloned into the pAdTrack-CMV vector, using the 5′ SalI and 3′ NotI restriction sites. These shuttle vectors were subsequently cut with PmeI for linearization and then transformed into the E.coli BJ5183 strain, together with the pAdEasy adenoviral backbone vector, to generate complete adenoviral vectors through in vivo recombination. Generation and purification of the recombinant adenovirus into packaging cells (HEK293a, ATCC) was also performed as described previously [71]. Proper titers to achieve >95% infection efficiency, of the purified virus, were assessed by counting green fluorescent protein (GFP)-expressing cells, in culture plates, infected with different concentrations of adenoviruses [12]. The shuttle vectors were used for transfection experiments in Fig 2B. To overexpress PER1, PER2, BMAL1 and CRY1, adenoviral vectors expressing these proteins were used as described previously [12]. Full length cDNA encoding β-Trcp2 were subcloned into pcDNA3.1/V5 using KpnI/XhoI sites after adding Flag tag at the N-terminus. pcDNA-His-Ubiquitin plasmid was generated by Robert Tomko laboratory (Florida State University). pcDNA-Per1, Per2 and CK1δ plasmids were described previously [12].
Cell culture and bioluminescence recording
For bioluminescence recordings, all cells contained the PER2-LUC reporter [5]. Cells were plated into 24-well plates to be approximately 90% confluent 24 hours prior to the start of the experiment. Immediately before the start of the experiment, cells were given a two-hour serum shock with 50% horse serum in DMEM, washed with phosphate buffered saline (PBS) and fresh DMEM supplemented with 1% FBS, 7.5mM sodium bicarbonate, 10 mM HEPES, 25 U/ml penicillin and 25 μg/ml streptomycin and 0.1mM luciferin was added. The plates were sealed with cellophane tape and placed into a Lumicycle (Actimetrics, Wilmette, IL). For bioluminescence experiments, the results were reproduced in at least two independent experiments. For the bioluminescence experiment with dn-USP14, PER2-LUC MEFs were treated with 1:500 and 1:1000 dilutions of purified DN-USP14 adenovirus one day prior to being set up on the Lumicycle.
For immunoblots, cells were seeded in 60-mm dishes to be 90% confluent 24 hours prior to the experiment. On the following day, respective drugs were added the wells of the plates after a two-hour serum shock with 50% horse serum in DMEM. These plates were sealed with cellophane tape and placed into a Lumicycle luminometer (Actimetrics, Wilmette, IL). For immunoblots with MEFs, cells were seeded in 60-mm dishes to be 90% confluent on the day of the experiment. The cells were treated, as described within figure legends, and collected 24 h later. For cycloheximide treatment, 8 μg/ml was added to cells 24 h after serum shock and cells were collected at specified times after the treatment.
Drug treatments in cells
Cells were seeded in 60-mm dishes to be 90% confluent 24 hours prior to the experiment. For Fig 1B, C, 5μM Ub-aldehyde (UBP-Bio F3100), 2μM and 20μM b-AP15 (UBP-Bio F2100), or 20μM, 50μM PR619 (UBP-Bio F2110) was added to wt MEFs for 30 or 60 minutes. E-64 (Sigma E3132), P5091 (UBP-Bio F4110) and MG132 (UBP-Bio F1100) were used at 10, 20 and 40 mM, respectively. In other experiments, b-AP15 was added at 10μM unless otherwise stated. Control cells received the same volume of DMSO. For cycloheximide (CHX) treatment, 8 μg/ml was added to cells.
Transfection, Immunoblotting and Immunoprecipitation
In Fig 2B, Per1, Per2, WT-USP14 and DN-USP14 plasmids were transfected into HEK293a cells using Qiagen's PolyFect Transfection Reagent (Qiagen). Briefly, the PolyFect reagent was mixed with 1μg plasmid DNA and combined with 150μL of DMEM. The solution was mixed briefly and incubated at room temperature for 10 minutes. The mixture was added to the cell medium and incubated for 48 hours before harvest. The transfection in Fig S5 was done similarly using Per1, Per2, CK1δ and β-Trcp2 plasmids.
The cells in 6 cm dishes were harvested and flash-frozen on dry ice. Protein extraction and immunoblotting were performed as previously described [19]. Briefly, tissues or cells were homogenized at 4°C in 10 volumes of extraction buffer (EB) (0.4M NaCl, 20mM HEPES (pH 7.5), 1mM EDTA, 5mM NaF, 1 mM dithiothreitol, 0.3% Triton X-100, 5% glycerol, 0.25mM phenylmethylsulfonyl fluoride, 10mg of aprotinin per ml, 5mg of leupeptin per ml, 1mg of pepstatin A per ml). Homogenates were cleared by centrifugation 12 min, 12,000g at 4°C. Supernatants were mixed with 2× sample buffer and boiled. Proteins were separated by electrophoresis through SDS polyacrylamide gels and then transferred to nitrocellulose membranes. Membranes were blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.05% Tween-20, incubated with primary antibodies overnight followed by incubation with secondary antibodies for 1 h. The blots were developed using an enhanced chemiluminescence substrate (WestFemto, ThermoFisher Scientific).
Immunoprecipitation was performed as described previously [11]. Briefly, protein extracts were prepared as described above. 10% of the initial protein extract was saved for the input. 20μL Protein G sepharose 4 fast flow beads (GE Healthcare) per reaction was equilibrated with 500μL of EB for 15 minutes on a rotating wheel. The beads were centrifuged at 3000 rpm for fifteen seconds and the supernatant was removed. This wash step was repeated three additional times. After the final wash, two volumes of EB were added to the beads. To pre-clear the extracts, 10μL of the equilibrated bead solution were added to the extracts and incubated for 30 minutes on a rotating wheel at 4°C. The samples were centrifuged at 12,000 rpm for 5 minutes at 4°C and then the pre-cleared extract was transferred to a fresh tube. Separately, 10μL per reaction of equilibrated beads were added to 400μL of EB. Then 0.1μg of affinity-purified antibody per reaction was added to the tube. This mixture was incubated at room temperate for one hour on a rotating wheel. The samples were centrifuged at 3000 rpm for fifteen seconds and the supernatant was removed. Next, the protein extract was added into the tube containing the antibody-protein sepharose beads and incubated at 4°C on a rotating wheel for four hours. The tubes were centrifuged at 3000 rpm for fifteen seconds, and the supernatant was removed. 1mL of EB was added to the tube and incubated on a rotating wheel at 4°C for twenty minutes. The samples were centrifuged at 3000 rpm for 15 seconds and the supernatant was removed. This was step was repeated 5 more times to completely wash the beads. After the final wash, the majority of the EB was removed and 30μL of 1X sample buffer was added. The samples were boiled at 95°C for 3 minutes.
Deubiquitination assay
Polyubiquitinated PER1 was immune-purified from b-AP15 treated MEFs overexpressing PER1 (10 μM, 30 min) as described above. A small fraction was mixed with 2X sample buffer and saved for input. The remaining immune complex was used for DUB assays. DUBs and Bovine 26S proteasome were purchased from UBPBio, #J6110 and A1200 (Aurora, CO, USA). DUB assays were performed according to the manufacturer's protocol (J6110) by mixing a fraction of purified polyubiquitinated PER1 with individual enzymes. When DUB assays were performed in the presence of 26S proteasome, 2mM ATP was added (Fig 2A). The reactions were stopped by adding 2× sample buffer followed by boiling for 3 min at 94 C. The DUB assay using Ubi-rhodamine110 was done according to the manufacturer's protocol (J6110).
RNA-sequencing and analysis
Tamoxifen-treated β-Trcp1-/-; β-Trcp2fl/fl/CAG-creER and control wt MEFs were cultured to ∼100% confluency in 15 cm dishes (n=3 each) and subjected to 200nM 4-hydroxytamoxifen (TM) treatment. The tamoxifen-treated cells were harvested on day 4 after the treatment. Cells were harvested using 1× PBS and a cell scraper, then centrifuged for 5 minutes at 3000 revolutions per minute (rpm) at 4°C. The PBS was removed and the cell pellets were flash-frozen on dry ice. The RNA was extracted using Trizol (Invitrogen) according to the manufacturer's instructions. The resulting aqueous phase was removed and transferred to a new tube. One volume of 70% ethanol was added, and this solution was placed onto an RNeasy column (Qiagen) and the RNA-containing was purified according to the manufacturer's instructions. The RNA was finally eluted with 50μL of nuclease-free water. The sample concentrations were measured using a Nanodrop (Thermo Fisher) and frozen at -80°C until processing. Synthetic RNA strands were added to 5μg of RNA from each sample as spike-ins to assess the performance of each library (ERCC ExFold RNA Spike-In Mixes [Ambion]). The mRNA was purified with a NEB Next mRNA magnetic isolation module (NEB, catalog). cDNA libraries were generated from 50ng of isolated mRNA using a NEBnext Ultra mRNA library preparation kit for Illumina sequencers (NEB), and a unique 6-nucleotide index was incorporated to each library (NEB). The concentration of each cDNA library was estimated with KAPA-PCR (KAPA Biosystems), and average fragment length was determined with a bioanalyzer dsDNA kit (Agilent Technologies). 8nM of each cDNA library was pooled into one of three cDNA libraries. Each of the three cDNA libraries underwent additional quality-control analysis via bioanalyzer and KAPA-PCR. 10pM of each cDNA library pool was sequenced on an Illumina HiSeq 2500 at the Translational Science Laboratory at the Florida State University College of Medicine.
Tissue collection and quantitative RT–PCR
Tissue samples from β-Trcp1-/- ; β-Trcp2fl/fl / Cag-CreESR-1 transgenic mice were collected at 4-hour intervals in DD as soon as the animals displayed arrhythmic behavior, which normally occurs within 5-6 days after tamoxifen treatment. Matching wt tissues were collected at the same time.
RT-PCR was performed with RNA purified as described previously and using the following primers:
β-Actin F1 5′-ATGGGTCAGAAGGACTCCTATGTGGG-3′, R1 5′-GGCC ACACGCAGCTCATTGTAGAAGG-3′.
β-Trcp2 F1 5′-GCCCTGCTTTATCATTGGTATGTATGT-3′, R1 5′- CACAGATGACGTTCCATTACTG-3′,
Immunocytochemistry
TM-treated β-Trcp1-/- ; β-Trcp2fl/fl / Cag-CreESR-1 MEFs and untreated control cells were plated on glass cover slips in six-well plates and grown to 70% confluency. The cells were fixed on day 5 after the TN treatment with 4% paraformaldehyde (PFA) in 1× PBS for ten minutes on a shaker at room temperature. The PFA was then washed off with 1× PBS three times for five minutes per wash. The cells were then blocked in 5% fetal bovine serum (FBS) supplemented with 0.5% TritonX-100 in PBS for 30 minutes at room temperature. The cells were incubated in 1:300 dilutions of PER1-GP, PER2-GP, C1-GP or βT2-GP15 antibody overnight, at 4°C, on a shaker. The following day, the primary antibodies were removed and the cells were washed in 1× PBS 3 times for 5 minutes per wash. A Texas Red-conjugated, goat-anti Guinea Pig IgG (H+L) secondary antibody (Thermo Scientific #PA1-28595) or FITC-labeled anti-Guinea Pig secondary antibody for β-TRCP2 (Jackson ImmunoResearch # 706-095-148) was added to the samples, at 1:300 concentration, for two hours at room temperature with shaking. The secondary antibody was removed and cells were washed with 1× PBS 3 times for 5 minutes per wash. Vectashield antifade (Vector Labs) mounting medium with DAPI (H-1200) was added to the coverslips. The coverslips were placed on microscope slides, and the edges were sealed. The images were obtained using a confocal fluorescent microscope.
Quantification and Statistical Analysis
Locomotor activity analysis (Fig. 3, 4, S3, S6)
Wheel-running activity was recorded and analyzed using the Stanford Software System (Santa Cruz, CA, USA) (Fig. S3) or ClockLab (Actimetrics, Wilmette, IL, USA) (all other actograms: Fig. 3, 4, S6). The free-running period was calculated using a χ2-periodogram with 6-minute resolution and the relative power (amplitude), of the circadian spectrum, was determined from a normalized Fast Fourier transform, using a Blackman-Harris window (ClockLab). Significance levels (e.g., period differences between WT and β-Trcp mutant mice) were determined by Student's t test between the two groups except Fig S1A where one-way ANOVA was used. The significance levels were represented as follows: * when p<0.05, ** when p<0.01 and *** when p<0.001.
Bioluminescence rhythm analysis (Fig. 2, 3, S1)
Real-time levels, period, and other features of the bioluminescence rhythms of PerLuc MEFs were evaluated using the Lumicycle software (Actimetrics). Significance levels were determined by Student's t test between two groups. The significance levels were represented as follows: * when p<0.05, ** when p<0.01 and *** when p<0.001.
RNA/cDNA sequence analysis (Fig. 5, S5)
Reads were trimmed of low-quality (-q 20) and adapter sequences at the 3′ end with cutadapt v1.9. Trimmed reads were aligned to the mm10 reference genome [72] with hisat v2.0.3 using default parameters and an index for the reference genome built with transcripts from the gencode comprehensive gene annotation of the mouse genome (GRCm38) version M1 1 for reference chromosome regions. Reads were assigned to transcripts using featureCounts v1.5.0-p3 and differential gene expression analysis was performed with Bioconductor's edgeR package version 3.12.1 using default parameters. Reads with alignment quality <20 were filtered with samtools 1.3.1 and genome-wide coverage of blocks of alignments was calculated using bedtools 2.25, and normalized to reads per million. Genome-wide coverage was used to generate tracks for the UCSC genome browser [73].
PER stability analysis (Fig. 6D)
Our inducible Per2 MEFs (Rosa-rtTA; tetO-Per2) (tet ON system) have been described previously [19]. Hypophosphorylated PER2 was generated by treating the inducible Per2 cells with a low dose of Dox (0.5 ug/ml) for 6 hrs while hyperphosphorylated PER2 was generated by incubating the cells with a maximum dose of Dox (2 ug/ml) for 4 hrs followed by wash-off and further incubation without Dox for 6 hrs. Since transcription stops after Dox is removed, existing PER2 would be continuously phosphorylated without adding de novo synthesized PER2 during the 6 hour period. Relevant bands on the PER immunoblots were quantified using standard densitometry as described previously [11]. Immunostaining intensity was plotted as the mean +/− SEM of 3 replicates.
Immunocytochemistry: fluorescence quantification (Fig. 5C)
ImageJ (v1.49, NIH) was used for fluorescence intensity quantification in Fig 5C. Briefly, an outline was drawn around the nucleus of each cell to be measured, and an identical region was placed in an adjacent area without fluorescent signal for background subtraction. The net average fluorescence intensity was calculated for each experimental group. Statistical analysis was performed using Student's t test in the GraphPad Prism 5.
Mathematical modeling (Fig 7)
The mathematical model in Fig. 7A:
The modified Kim-Forger model [14] [59] [54], where repressor (R) nonlinearly degrades, is described with the following equation:
Where describes the proteasomal degradation of repressor via reversible ubiquitination [55, 56]. Scaling of M, C, and R normalizes all production rates (αi), and with the assumption that βM=βC, non-dimensionalization of time also normalizes the clearance rates (βM=βC) as described in [14, 54]. This leads to the non-dimensionalized model with four dimensionless free parameters, A=0.0659, K=10, Kd =10-1, and Vmax=0.08:
where we kept the notation of variables and parameters from the original model for the simplicity. For stochastic simulations of the model, the Gillespie algorithm is used [74]. We assumed volume Ω=10,000 to convert the concentration to the number of molecules and derive propensity functions so that the simulated C. V. of periods is similar to the experimental measurements (Fig. 3D-F). Furthermore, with this choice of Ω, the number of PER molecules becomes ∼2,000, which is a similar order of magnitude with the experiment-based estimates of PER in liver cells [75]. We assumed that binding and unbinding between repressors and activators, and reactions underlying proteasomal degradation of the repressor occur much faster than other reactions. Due to such large timescale separation, the non-elementary propensity functions (i.e. f(R) and Michaels-Menten function) can lead to accurate stochastic simulations [74].
In the mammalian circadian clock, PER forms a large inhibitory complex including CRY, CK1δ/ε, and NuRD to sequester and repress the BMAL/CLOCK activator complex [12, 76, 77]. While the roles and interactions of all the components of the inhibitory complex are not completely understood, PER is rate-limiting and appears to play the most critical role (e.g. PER, but not CRY, overexpression completely disrupts the circadian rhythms [12]). Thus, rather than attempting to model every component of the whole complex, we assume, for simplicity, that R in the model represents the total concentration of PER in the nucleus.
For the Goodwin type model (Fig. S7B), we used following Kim et al [59].
Transient half-life analysis (Fig. 7D)
We estimated the transient half-life of PER2 as follows. To ensure majority of PER2 is hyperphosphorylated, we treated cells with CHX when PER2-LUC is at peak. Then, the decay curves of PER2-LUC were divided into segments of 1hr with a sliding window, which is moved at an increment of 10 min. Each segment of decay curve is fitted to the exponential decay curve to estimate the transient half-life. When PER2-LUC levels reach around 10% of its initial level after CHX treatment, the data become noisy and unreliable and thus were not included.
Data and Software Availability
Table S1. Transcriptome comparison between wt and β-Trcp1/2 mutant MEFs: https://data.mendeley.com/datasets/j6527m9jw2/draft?a=2e00a744-4521-4545-8a7b-41a161647a29
Supplementary Material
Table S1. Transcriptome comparison between wt and β-Trcp1/2 mutant MEFs: https://data.mendeley.com/datasets/j6527m9jw2/draft?a=2e00a744-4521-4545-8a7b-41a161647a29
Highlights.
-Nonlinear degradation of PER is required for the robustness of circadian rhythms.
-PER degradation is regulated by a balance between ubiquitination and deubiquitination.
-Disrupting this balance causes irregular wake-sleep cycles in mice.
Acknowledgments
We thank Wayne Cheng and Paul Yi for technical assistance, Robert Tomko for helpful discussion and His-Ubiquitin plasmid, Ruth Didier for confocal microscopy during the project. We thank Dennis Chang for assistance with manuscript preparation. This work was partially supported by NIH grant NS-099813 (C.L.) and a bridge grant from the Department of Biomedical Sciences, FSU (C.L.), grants from the NIH (R01-GM057587 and R01-CA076584) to M.P., and Korea Advanced Institute of Science and Technology Research Allowance Grant G04150020, National Research Foundation of Korea Grant N01160447, and T. J. Park Science Fellowship of POSCO (to J.K.K.). M.P. is an Investigator in the Howard Hughes Medical Institute.
Footnotes
Author Contributions: C.L, M.D, J.K. and S.B. planned the experiments. J.K. performed mathematical modeling. Z.J. performed imaging experiments. M.P. provided β-Trcp1 mutant mice and performed in vitro ubiquitination experiments. R.C. and R.N. performed RNA-seq experiment. K.K and D.V. performed RNA-seq data analysis. M.D., S.B., J.W. and C.L. performed all other experiments. C.L, M.D., S.B. and J.K. wrote the manuscript. All authors discussed the results and commented on the manuscript.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24:90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bass J, Lazar MA. Circadian time signatures of fitness and disease. Science. 2016;354:994–999. doi: 10.1126/science.aah4965. [DOI] [PubMed] [Google Scholar]
- 3.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz WJ, Zimmerman P. Circadian timekeeping in BALB/c and C57BL/6 inbred mouse strains. J Neurosci. 1990;10:3685–3694. doi: 10.1523/JNEUROSCI.10-11-03685.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, et al. PERIOD2∷LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 7.Low-Zeddies SS, Takahashi JS. Chimera analysis of the Clock mutation in mice shows that complex cellular integration determines circadian behavior. Cell. 2001;105:25–42. doi: 10.1016/s0092-8674(01)00294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwadate R, Satoh Y, Watanabe Y, Kawai H, Kudo N, Kawashima Y, Mashino T, Mitsumoto A. Impairment of heme biosynthesis induces short circadian period in body temperature rhythms in mice. Am J Physiol Regul Integr Comp Physiol. 2012;303:R8–18. doi: 10.1152/ajpregu.00019.2011. [DOI] [PubMed] [Google Scholar]
- 9.Lowrey PL, Takahashi JS. Genetics of circadian rhythms in Mammalian model organisms. Advances in genetics. 2011;74:175–230. doi: 10.1016/B978-0-12-387690-4.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annual review of physiology. 2010;72:517–549. doi: 10.1146/annurev-physiol-021909-135821. [DOI] [PubMed] [Google Scholar]
- 11.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 12.Chen R, Schirmer A, Lee Y, Lee H, Kumar V, Yoo SH, Takahashi JS, Lee C. Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol Cell. 2009;36:417–430. doi: 10.1016/j.molcel.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee Y, Chen R, Lee HM, Lee C. Stoichiometric relationship among clock proteins determines robustness of circadian rhythms. J Biol Chem. 2011;286:7033–7042. doi: 10.1074/jbc.M110.207217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JK, Forger DB. A mechanism for robust circadian timekeeping via stoichiometric balance. Mol Syst Biol. 2012;8:630. doi: 10.1038/msb.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bollinger T, Schibler U. Circadian rhythms - from genes to physiology and disease. Swiss medical weekly. 2014;144:w13984. doi: 10.4414/smw.2014.13984. [DOI] [PubMed] [Google Scholar]
- 16.Etchegaray JP, Lee C, Wade PA, Reppert SM. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature. 2003;421:177–182. doi: 10.1038/nature01314. [DOI] [PubMed] [Google Scholar]
- 17.Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125:497–508. doi: 10.1016/j.cell.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 19.D'Alessandro M, Beesley S, Kim JK, Chen R, Abich E, Cheng W, Yi P, Takahashi JS, Lee C. A tunable artificial circadian clock in clock-defective mice. Nat Commun. 2015;6:8587. doi: 10.1038/ncomms9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HM, Chen R, Kim H, Etchegaray JP, Weaver DR, Lee C. The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc Natl Acad Sci U S A. 2011;108:16451–16456. doi: 10.1073/pnas.1107178108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu JC, Ko HW, Edery I. NEMO/NLK phosphorylates PERIOD to initiate a time-delay phosphorylation circuit that sets circadian clock speed. Cell. 2011;145:357–370. doi: 10.1016/j.cell.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elowitz MB, Leibler S. A synthetic oscillatory network of transcriptional regulators. Nature. 2000;403:335–338. doi: 10.1038/35002125. [DOI] [PubMed] [Google Scholar]
- 23.Stricker J, Cookson S, Bennett MR, Mather WH, Tsimring LS, Hasty J. A fast, robust and tunable synthetic gene oscillator. Nature. 2008;456:516–519. doi: 10.1038/nature07389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tigges M, Marquez-Lago TT, Stelling J, Fussenegger M. A tunable synthetic mammalian oscillator. Nature. 2009;457:309–312. doi: 10.1038/nature07616. [DOI] [PubMed] [Google Scholar]
- 25.Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY, Liu X, Atwood A, Huss JW, 3rd, Janes J, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139:199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McVean M, Xiao H, Isobe K, Pelling JC. Increase in wild-type p53 stability and transactivational activity by the chemopreventive agent apigenin in keratinocytes. Carcinogenesis. 2000;21:633–639. doi: 10.1093/carcin/21.4.633. [DOI] [PubMed] [Google Scholar]
- 27.Salic A, Lee E, Mayer L, Kirschner MW. Control of beta-catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell. 2000;5:523–532. doi: 10.1016/s1097-2765(00)80446-3. [DOI] [PubMed] [Google Scholar]
- 28.Oikonomou C, Cross FR. Frequency control of cell cycle oscillators. Curr Opin Genet Dev. 2010;20:605–612. doi: 10.1016/j.gde.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee B, Song T, Lee K, Kim J, Berggren PO, Ryu SH, Jo J. Insulin modulates the frequency of Ca2+ oscillations in mouse pancreatic islets. PLoS One. 2017;12:e0183569. doi: 10.1371/journal.pone.0183569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomko RJ, Jr, Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. 2013;82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shirogane T, Jin J, Ang XL, Harper JW. SCFbeta -TRCP controls clock-dependent transcription via casein kinase 1 dependent degradation of the mammalian period-1 (PER1) protein. J Biol Chem. 2005 doi: 10.1074/jbc.M502862200. [DOI] [PubMed] [Google Scholar]
- 32.Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, Virshup DM. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol. 2005;25:2795–2807. doi: 10.1128/MCB.25.7.2795-2807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohsaki K, Oishi K, Kozono Y, Nakayama K, Nakayama KI, Ishida N. The role of {beta}-TrCP1 and {beta}-TrCP2 in circadian rhythm generation by mediating degradation of clock protein PER2. Journal of biochemistry. 2008;144:609–618. doi: 10.1093/jb/mvn112. [DOI] [PubMed] [Google Scholar]
- 34.Grima B, Lamouroux A, Chelot E, Papin C, Limbourg-Bouchon B, Rouyer F. The F-box protein slimb controls the levels of clock proteins period and timeless. Nature. 2002;420:178–182. doi: 10.1038/nature01122. [DOI] [PubMed] [Google Scholar]
- 35.He Q, Cheng P, Yang Y, He Q, Yu H, Liu Y. FWD1-mediated degradation of FREQUENCY in Neurospora establishes a conserved mechanism for circadian clock regulation. EMBO J. 2003;22:4421–4430. doi: 10.1093/emboj/cdg425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ko HW, Jiang J, Edery I. Role for Slimb in the degradation of Drosophila Period protein phosphorylated by Doubletime. Nature. 2002;420:673–678. doi: 10.1038/nature01272. [DOI] [PubMed] [Google Scholar]
- 37.Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, Lee C, Takahashi JS. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–1023. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Godinho SI, Maywood ES, Shaw L, Tucci V, Barnard AR, Busino L, Pagano M, Kendall R, Quwailid MM, Romero MR, et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- 39.Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, Draetta GF, Pagano M. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316:900–904. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- 40.Hirano A, Yumimoto K, Tsunematsu R, Matsumoto M, Oyama M, Kozuka-Hata H, Nakagawa T, Lanjakornsiripan D, Nakayama KI, Fukada Y. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell. 2013;152:1106–1118. doi: 10.1016/j.cell.2013.01.054. [DOI] [PubMed] [Google Scholar]
- 41.Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, Kornblum I, Kumar V, Koike N, Xu M, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152:1091–1105. doi: 10.1016/j.cell.2013.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou M, Kim JK, Eng GW, Forger DB, Virshup DM. A Period2 Phosphoswitch Regulates and Temperature Compensates Circadian Period. Mol Cell. 2015;60:77–88. doi: 10.1016/j.molcel.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 43.Lee H, Chen R, Lee Y, Yoo S, Lee C. Essential roles of CKIdelta and CKIepsilon in the mammalian circadian clock. Proc Natl Acad Sci U S A. 2009;106:21359–21364. doi: 10.1073/pnas.0906651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lam YA, Xu W, DeMartino GN, Cohen RE. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature. 1997;385:737–740. doi: 10.1038/385737a0. [DOI] [PubMed] [Google Scholar]
- 45.Hanna J, Hathaway NA, Tone Y, Crosas B, Elsasser S, Kirkpatrick DS, Leggett DS, Gygi SP, King RW, Finley D. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell. 2006;127:99–111. doi: 10.1016/j.cell.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 46.D'Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17:1636–1640. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 47.Crosas B, Hanna J, Kirkpatrick DS, Zhang DP, Tone Y, Hathaway NA, Buecker C, Leggett DS, Schmidt M, King RW, et al. Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell. 2006;127:1401–1413. doi: 10.1016/j.cell.2006.09.051. [DOI] [PubMed] [Google Scholar]
- 48.Yao T, Song L, Xu W, DeMartino GN, Florens L, Swanson SK, Washburn MP, Conaway RC, Conaway JW, Cohen RE. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol. 2006;8:994–1002. doi: 10.1038/ncb1460. [DOI] [PubMed] [Google Scholar]
- 49.Homma T, Ishibashi D, Nakagaki T, Fuse T, Mori T, Satoh K, Atarashi R, Nishida N. Ubiquitin-specific protease 14 modulates degradation of cellular prion protein. Sci Rep. 2015;5:11028. doi: 10.1038/srep11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanarek N, Grivennikov SI, Leshets M, Lasry A, Alkalay I, Horwitz E, Shaul YD, Stachler M, Voronov E, Apte RN, et al. Critical role for IL-1beta in DNA damage-induced mucositis. Proc Natl Acad Sci U S A. 2014;111:E702–711. doi: 10.1073/pnas.1322691111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, Margottin-Goguet F, Jackson PK, Yamasaki L, Pagano M. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell. 2003;4:799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 52.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JK. Protein sequestration versus Hill-type repression in circadian clock models. IET Syst Biol. 2016;10:125–135. doi: 10.1049/iet-syb.2015.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pierce NW, Kleiger G, Shan SO, Deshaies RJ. Detection of sequential polyubiquitylation on a millisecond timescale. Nature. 2009;462:615–619. doi: 10.1038/nature08595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu L, Qu Z. Roles of protein ubiquitination and degradation kinetics in biological oscillations. PLoS One. 2012;7:e34616. doi: 10.1371/journal.pone.0034616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.King DP, Vitaterna MH, Chang AM, Dove WF, Pinto LH, Turek FW, Takahashi JS. The mouse Clock mutation behaves as an antimorph and maps within the W19H deletion, distal of Kit. Genetics. 1997;146:1049–1060. doi: 10.1093/genetics/146.3.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reischl S, Vanselow K, Westermark PO, Thierfelder N, Maier B, Herzel H, Kramer A. Beta-TrCP1-mediated degradation of PERIOD2 is essential for circadian dynamics. J Biol Rhythms. 2007;22:375–386. doi: 10.1177/0748730407303926. [DOI] [PubMed] [Google Scholar]
- 59.Kim JK, Kilpatrick ZP, Bennett MR, Josic K. Molecular mechanisms that regulate the coupled period of the mammalian circadian clock. Biophys J. 2014;106:2071–2081. doi: 10.1016/j.bpj.2014.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Novak B, Tyson JJ. Design principles of biochemical oscillators. Nature reviews. Molecular cell biology. 2008;9:981–991. doi: 10.1038/nrm2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurosawa G, Iwasa Y. Saturation of enzyme kinetics in circadian clock models. J Biol Rhythms. 2002;17:568–577. doi: 10.1177/0748730402238239. [DOI] [PubMed] [Google Scholar]
- 62.Ananthasubramaniam B, Herzog ED, Herzel H. Timing of neuropeptide coupling determines synchrony and entrainment in the mammalian circadian clock. PLoS Comput Biol. 2014;10:e1003565. doi: 10.1371/journal.pcbi.1003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eldar A, Rosin D, Shilo BZ, Barkai N. Self-enhanced ligand degradation underlies robustness of morphogen gradients. Dev Cell. 2003;5:635–646. doi: 10.1016/s1534-5807(03)00292-2. [DOI] [PubMed] [Google Scholar]
- 65.Felix MA, Barkoulas M. Pervasive robustness in biological systems. Nat Rev Genet. 2015;16:483–496. doi: 10.1038/nrg3949. [DOI] [PubMed] [Google Scholar]
- 66.Zhu L, Zee PC. Circadian rhythm sleep disorders. Neurol Clin. 2012;30:1167–1191. doi: 10.1016/j.ncl.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuljis DA, Loh DH, Truong D, Vosko AM, Ong ML, McClusky R, Arnold AP, Colwell CS. Gonadal- and sex-chromosome-dependent sex differences in the circadian system. Endocrinology. 2013;154:1501–1512. doi: 10.1210/en.2012-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen R, Seo DO, Bell E, von Gall C, Lee C. Strong resetting of the mammalian clock by constant light followed by constant darkness. J Neurosci. 2008;28:11839–11847. doi: 10.1523/JNEUROSCI.2191-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen R, D'lessandro M, Lee C. miRNAs are required for generating a time delay critical for the circadian oscillator. Curr Biol. 2013;23:1959–1968. doi: 10.1016/j.cub.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee C, Weaver DR, Reppert SM. Direct association between mouse PERIOD and CKIepsilon is critical for a functioning circadian clock. Mol Cell Biol. 2004;24:584–594. doi: 10.1128/MCB.24.2.584-594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mouse Genome Sequencing C, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 73.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim JK, Josic K, Bennett MR. The relationship between stochastic and deterministic quasi-steady state approximations. BMC Syst Biol. 2015;9:87. doi: 10.1186/s12918-015-0218-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Narumi R, Shimizu Y, Ukai-Tadenuma M, Ode KL, Kanda GN, Shinohara Y, Sato A, Matsumoto K, Ueda HR. Mass spectrometry-based absolute quantification reveals rhythmic variation of mouse circadian clock proteins. Proc Natl Acad Sci U S A. 2016;113:E3461–3467. doi: 10.1073/pnas.1603799113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Padmanabhan K, Robles MS, Westerling T, Weitz CJ. Feedback regulation of transcriptional termination by the mammalian circadian clock PERIOD complex. Science. 2012;337:599–602. doi: 10.1126/science.1221592. [DOI] [PubMed] [Google Scholar]
- 77.Kim JY, Kwak PB, Weitz CJ. Specificity in circadian clock feedback from targeted reconstitution of the NuRD corepressor. Mol Cell. 2014;56:738–748. doi: 10.1016/j.molcel.2014.10.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Transcriptome comparison between wt and β-Trcp1/2 mutant MEFs: https://data.mendeley.com/datasets/j6527m9jw2/draft?a=2e00a744-4521-4545-8a7b-41a161647a29