

Abstract
Background
Epithelial barrier dysfunction is a central feature in the pathogenesis of allergic disease. Epithelial-to-mesenchymal transition (EMT) has been proposed as one mechanism afflicting barrier in asthma. However, genes and pathways involved in aberrant epithelial-mesenchymal signaling, and their relationship to asthma severity, are poorly understood.
Methods
We used unbiased gene network analysis to evaluate functional convergence in epithelial gene expression signatures across multiple public access transcriptomics datasets of human asthma, followed by text mining to evaluate functional marker relevance of discovered genes. We objectively confirmed these findings in epithelial brushings and primary asthmatic epithelial cells cultured in different biological contexts.
Results
We found a striking suppression of epithelial differentiation in asthma, overrepresented by insufficiency in insulin and Notch signaling, but with the absence of conventional EMT markers. We identified EFNB2, FGFR1, FGFR2, INSR, IRS2, NOTCH2, TLE1, and NTRK2 as novel markers central to dysregulation of epithelial-mesenchymal signaling, but surprisingly overlooked in asthma research. We found that this “core” signature of asthma is shared by mild, moderate, and severe forms of disease, progressing with severity. Loss of epithelial differentiation induced by insulin deprivation in normal human bronchial epithelial cells cultured in organotypic conditions closely approximated gene expression in asthmatic epithelial brushings.
Conclusions
The comparative analysis of publically available transcriptomes demonstrated that epithelial barrier dysfunction in asthma is characterized by persistent underlying de-differentiation program with complex etiology. The lasting alteration of the asthmatic epithelial cell transcriptome implicates regulation involving metabolism and epigenetics, beyond EMT driven by injury and repair in chronic inflammation.
Keywords: asthma, differentiation and remodeling, epithelial-to-mesenchymal transition, epithelium, “omics” and systems biology
Epithelial barrier dysfunction is believed to be a central feature in the pathogenesis of allergic disease. In asthma, the airway epithelium undergoes dramatic changes, characterized by deficient differentiation and repair, loss of junctional and innate defense proteins, modification of extracellular matrix, and sub-epithelial fibrosis, together indicating loss of competent barrier functionality even in mild forms of disease. Dysregulation of epithelial-mesenchymal signaling consistent with epithelial-to-mesenchymal transition (EMT) has been proposed as one of the mechanisms responsible for dysfunction of the epithelial barrier in allergy1-3. Although existence of EMT-like processes has recently been implicated in multiple allergic diseases, including asthma2-5, specific drivers of EMT and their relationship to disease severity is the subject of controversy.
Chronic inflammation in asthma is sufficient to inflict damage on the epithelial barrier and induce repair process, which in part engages EMT as a mechanism to either expand the mesenchymal precursor pool, or induce adjacent epithelial cells to adopt a more migratory phenotype needed to regenerate a functional mature epithelium. Given persistence of chronic inflammation, ongoing and deficient repair would explain the presence of EMT and loss of barrier function. However, it has been increasingly recognized that barrier disruption may extend beyond simple inflammatory insult and even pre-date development of allergic disease. This is supported by the link between loss-of-function in epithelial barrier genes and allergen sensitization6, the altered barrier state of intact non-lesional skin in atopic dermatitis7, and increased trans-epidermal water loss (TEWL) through uninvolved skin of allergic patients8. Intriguingly, primary epithelial cells cultured from asthmatic airways, taken out of their inflammatory context, similarly exhibit a persistent altered “immature” phenotype with altered junctions and defective repair mechanisms9, 10. Collectively, these reports indicate persistence of epithelial dysfunction even after epithelial cells have been removed from the in vivo inflammatory environment. This suggests a transcriptional reprogramming (non-heritable alteration of cell development) of the epithelial phenotype and existence of systemic factors driving EMT and de-differentiation beyond the injury and repair. Reprogramming of epithelial cells towards pro-mesenchymal phenotype has been shown to occur at the level of epigenetics, microRNA regulation, and metabolism11-13. However, the genes, pathways, and other factors involved in promoting this aberrant developmental program in asthma are poorly understood.
Here, we used unbiased biological gene network analysis to revisit gene expression signatures of bronchial epithelial cells from several studies of asthma, with the goal of identifying unity rather than discrepancy in epithelial barrier gene expression patterns across different asthma severities. Comparing epithelial gene expression signatures across multiple independent studies of asthma, we discovered: (1) there is poor consensus representation of classical EMT markers in asthma gene expression signatures; (2) however, collective suppression of epithelial differentiation programs was well represented across multiple studies of asthma; (3) downregulation of insulin signaling/insulin resistance was identified as one of the possible key factors driving de-differentiation and susceptibility to deficient repair; (4) such epithelial de-differentiation program was shared across mild, moderate, and severe asthma, and became more pronounced with increasing disease severity; (5) EFNB2, FGFR1, FGFR2, INSR, IRS2, NOTCH2, TLE1, and NTRK2 were identified and validated as novel genes capturing epithelial-mesenchymal dysregulation in asthma; 6) insulin deficiency in normal human bronchial epithelial cultures grown in organotypic conditions induced loss of epithelial differentiation and approximated gene expression patterns observed in asthmatic epithelial brushing samples. Collectively, our study suggests that the EMT process implicated in asthma pathogenesis is a manifestation of an underlying profound and persistent suppression of epithelial differentiation/developmental programs, likely driven not only by inflammation and repair, but also by changes in systemic factors such as insulin and insufficiency of differentiation stimuli.
Materials and Methods
Microarray studies, data sets, and clinical sample characteristics
Gene Expression Omnibus (GEO), NCBI's publically available repository of bioinformatics datasets, which holds curated MIAME (Minimum Information About a Microarray Experiment) compliant transcriptomics data, was thoroughly queried for all datasets involving asthmatic epithelium. Only datasets supported by peer-reviewed PubMed-indexed publications, with well-documented clinical descriptions of asthma patients and airway sampling procedures, with sufficient samples to generate a robust list of significantly different genes with FDR significance adjustment, and with proper inclusion of healthy control groups, were selected for this study. A description of all considered microarray datasets found involving asthmatic epithelium, and reasoning for inclusion or non-inclusion in this study, can be found in Supplementary Table 1. Based these criteria, eight gene expression microarray datasets representing different independent studies of asthma were downloaded from the repository. Datasets from different asthma severities were intentionally included, for the purpose of identifying unifying factors across different asthmatic subtypes. 7 out of 8 datasets utilized in this study were derived from bronchial brushings of normal vs. asthmatic patients, immediately lysed for RNA extraction upon sampling. To control for contaminating expression of genes from cell types other than epithelium, a dataset of epithelial samples cultured from epithelial brushings (where only adherent cells were passaged >3 times in plates before RNA extraction) was included as an internal control. Gene annotations were updated using technology-corresponding annotation libraries in R. The GEO and PubMed NCBI accession numbers, as well as details of each microarray study, including sample descriptions (brief patient demographics, asthma severity, and epithelial purity), are provided in Table 1. Our workflow for bioinformatics analysis is illustrated in Fig. 1.
Table 1. Details of asthma studies and associated microarray datasets.
| GSE | PMID | Technology/ Platform | Sample size for each group | Asthma severity (FEV1 % predicted) | Age | Sex (M:F) | Race (W/B/O***) | Sample source | Epithelial purity |
|---|---|---|---|---|---|---|---|---|---|
| 18965 | 20110557 | Affymetrix/ Human Genome U133A | Normal: 7, Asthma: 9 | Mild | 8.6±3.5 (M), 8.5±3.0 (F)** | 23:13 | Not provided | Cultured cells | ∼100% epithelial cells |
| 43696 | 24518246 | Agilent/Whole Human Genome Microarray 4×44K | Normal: 20, Asthma: 50 | Mild (90(83-97)) | 29(23-37)* | 7:17 | Healthy: 14/3/3, Asthma: 29/14/7 | Bronchial brushings | >90% epithelial cells |
| 67472 | 25611785 | Affymetrix/ Human Genome U133 Plus 2.0 | Normal: 43, Asthma: 22 | Mild (89±10) | 34±12** | 9:13 | Reported as “similar race/ ethnicity across both groups” in original study | Bronchial brushings | 97±3% epithelial cells |
| 4302 | 17898169 | Affymetrix/ Human Genome U133 Plus 2.0 | Normal: 26, Asthma: 42 | Moderate (87±12) | 36±12** | 17:25 | Not provided | Bronchial brushings | 97±3% epithelial cells |
| 63142 | 25338189 | Agilent/Whole Human Genome Microarray 4×44K | Normal: 27, Asthma: 72 | Moderate (84±16) | 29±10** | 13:24 | Healthy: 18/5/3, Asthma: 43/25/8 | Bronchial brushings | >90% epithelial cells |
| 67472 | 25611785 | Affymetrix/ Human Genome U133 Plus 2.0 | Normal: 43, Asthma: 40 | Moderate (85±13) | 36±12** | 19:21 | Reported as “similar race/ ethnicity across both groups” in original study | Bronchial brushings | 97±3% epithelial cells |
| 43696 | 24518246 | Agilent/Whole Human Genome Microarray 4×44K | Normal: 20, Asthma: 38 | Severe (56(40-71)) | 47(35-55)* | 12:30 | Healthy: 14/3/3, Asthma: 22/14/2 | Bronchial brushings | >90% epithelial cells |
| 63142 | 25338189 | Agilent/Whole Human Genome Microarray 4×44K | Normal: 27, Asthma: 56 | Severe (56±20) | 45±11** | 16:35 | Healthy: 18/5/3, Asthma: 31/14/3 | Bronchial brushings | >90% epithelial cells |
median (with 25th-75th percentiles)
mean±SD
White/Black/Other
Figure 1. Workflow for bioinformatics analysis of publicly available microarray datasets.
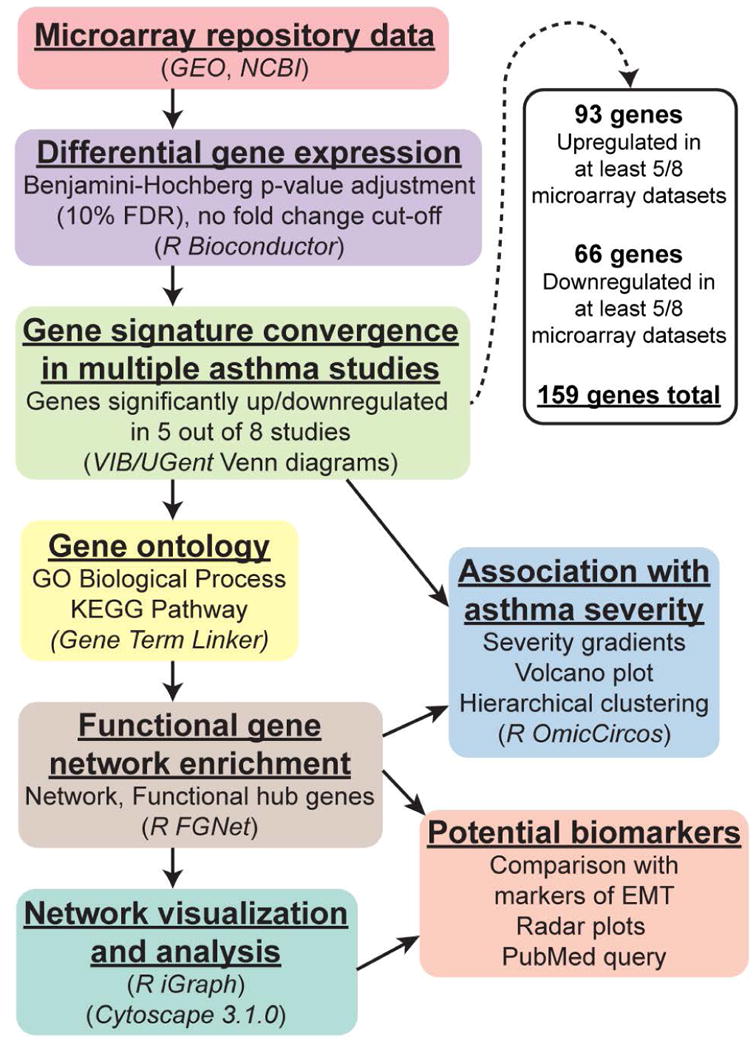
Differential expression
Each individual dataset underwent consistent handling, implementing standard contrast fits on Series Matrix File data uploaded to GEO by the original study groups (preserving original summarization/normalization protocols, to maintain consistency with gene signatures published by original studies), performed in the R computing environment (“limma” package, Bioconductor). For differential gene expression in healthy control vs. disease group comparisons, moderated Benjamini-Hochberg t-tests (with FDR significance adjustment) were used. Only genes significant at an FDR adjustment cut-off of 10% (adjusted p-value < 0.1) were used in subsequent data mining/network analysis. No fold change cut-offs were implemented.
Comparison of gene expression signatures across multiple studies of asthma
To identify genes exhibiting consensus differential expression across different studies of asthma, we identified convergent intersections of gene signatures from multiple studies, using the Bioinformatics & Evolutionary Genomics Venn diagram tool (VIB/UGent, Belgium). Meta-analysis level integration of data in this case was not a feasible approach due to differences in biological hypotheses postulated by different studies (healthy vs. mild, moderate, or severe subject group comparisons) and very different microarray technologies/platforms used across studies (with high punishing effect during cross-normalization). Rather, the analysis focused on straightforward identification of “core” genes and biological functions at the level of outcomes – comparison of gene signatures already identified by independent groups, driven by the principle that if several groups independently report the same gene, this gene is robustly represented in the “core” functional processes in asthma. This approach allowed us to focus on robust and consistent gene expression patterns of epithelial remodeling in asthma, while transcending variation due to disease heterogeneity, experimental differences, technological discrepancy, and patient variability in studies performed by separate research groups. To allow for sufficient number of genes to perform further functional ontology and interactome analysis, we used relaxed stringency criteria to include genes convergent in at least 5 out of 8 studies of asthma. Only genes that exhibited consistent directionality of change (upregulation or downregulation) relative to healthy controls in at least 5 out of 8 studies of asthma (where they showed statistical significance) were included in the consensus. This resulted in a consensus signature of 159 genes (93 upregulated and 66 downregulated). These consensus genes were used in all subsequent analyses, including construction of our functional gene expression network and text mining approach for potential epithelial biomarkers in asthma.
Functional Gene Set Enrichment Analysis
Functional Gene Set Enrichment Analysis (FGSEA) was used to generate functional gene networks and identify “functional hub” genes by meta-grouping of individual gene term sets (referencing GO Biological Process and KEGG Pathways), based on function similarity. The GeneTerm Linker algorithm implemented in the “FGNet” package (R) was used to perform the analysis. GeneTerm Linker is an algorithm for functional annotation of gene signatures that combines a concurrent enrichment analysis (GeneCodis) followed by a non-redundant reciprocal linkage of genes and biological terms (GeneTerm Linker)14. This methodology solves the problem of extreme redundancy and low depth of information in conventional enrichment algorithms, and filters enrichment output results through reciprocal linkage between genes and terms to produce functional metagroups of key biological significance. Parameters set for this analysis included adjusted p-value < 0.05, minimum gene term support of 3. Genes were deemed “functional hub” genes if they belonged to more than one functional meta-group, suggesting a more central role in regulation of biological processes in asthmatic epithelium. Networks generated utilizing this analysis were exported in GLM format for further analysis and visualization, using the “iGraph” package (R).
Pathway analysis, hierarchical clustering, and visualization
Cytoscape 3.2.1 was used for analysis of edge weight, node connectivity, and betweenness within the networks, as well as to visualize functional meta-grouping of genes. The “OmicCircos” package (R) was used to perform hierarchical clustering, and to visualize gradient of severity in normalized fold change of the 159 consensus genes across all studies of asthma, using circular heatmap display.
Gene validation in independent epithelial brushings datasets
One approach we took for validation of our signatures involved interrogation of publically available independent microarray studies of normal vs. asthmatic epithelium that were not initially included in our signature, GSE41861 and GSE64913. Reasons for not including these datasets in our initial analysis were lack of publication in peer-reviewed, PubMed-indexed work (GSE41861) and mixed sampling from peripheral and central bronchi (GSE64913). These studies were technically sound (properly powered and allowed for FDR adjustment), and both datasets were processed identically to the datasets from our comparative analysis. Both new microarray datasets utilized freshly isolated bronchial brushings for their epithelial samples, similar to the source for most of the microarray datasets used in our mining analysis. GSE41861 represented mild, moderate, and severe asthma. GSE64913 included only severe asthmatics in its analysis. To identify genes confirming their expression in these independent cohorts, lists of significantly differentially expressed genes from these studies were blasted against our asthma convergence signatures using Venn analysis.
Gene and functional process validation in primary human bronchial epithelial cell cultures
Another approach taken for validation of our signatures was to perform RNA-Seq analysis on pure primary human bronchial epithelial cells, grown in different biological contexts. Normal human bronchial epithelial cells and diseased human bronchial epithelial cells (asthma) were purchased from Lonza and grown either in submerged conditions, or in fully differentiated organotypic conditions (>3 weeks of culture at air-liquid interface in specialized differentiation media in 0.4 μm Corning Transwell plates, following Lonza's protocol). To simulate the deficiency in insulin signaling, which was implicated as one of the key events in loss of epithelial differentiation via our network analysis, normal human bronchial epithelium was cultured in air-liquid interface in insulin-deficient conditions during differentiation phase of the culture, albeit with sufficient supplementation of other differentiation components (retinoic acid, triiodothyronine, EGF). Importantly, same exact donors in same plating conditions (passage number, plating density) were used for tests in different conditions. When comparing healthy and asthmatic samples, donors were matched by age and sex. All samples were processed simultaneously for downstream analyses.
RNA-Seq sequencing and analysis of epithelial samples
Epithelial cells were immediately lysed using RLT buffer from the Qiagen RNeasy Plus Mini kit (Qiagen). Cell lysates were stored at -80°C until RNA was extracted. RNA isolations were performed using Qiagen RNeasy Plus Mini kit (Qiagen). RNA quality and quantity were measured using Agilent 4200 Tapestation using high Sensitivity RNA ScreenTape System (Agilent Technologies). NEBNext Ultra™ RNA (New England Biolabs, Inc) was used for full-length cDNA synthesis and library preparation. Libraries were pooled, denatured and diluted, resulting in a 2.5 pM DNA solution. PhiX control was spiked at 1%. Libraries were sequenced on an Illumina NextSeq 500 instrument (Illumina Inc), using NextSeq 500 High Output reagent kit (Illumina Inc) (1×75 cycles) with a target read depth of approximate (5-10) million aligned reads per sample. “edgeR” package (R, Bioconductor) was used to analyze sequencing data. Briefly, reads were assessed for quality, aligned to hg19 human reference genome using TopHat algorithm, and normalized and annotated count data were analyzed for differential expression using “deseq2” protocol. All resulting RNA-seq data will be made publically available in NCBI's GEO Omnibus repository.
Results
Convergence of gene expression signatures across different studies of asthma
To identify convergence of gene signatures and functional processes across different asthma studies, we used Venn diagram calculations to identify intersections of healthy control vs. asthma differential gene expression lists from individual studies (using 10% FDR-adjusted significance cut-off, no fold change cut-off). This approach allowed us to focus on robust and consistent gene expression patterns of epithelial remodeling in asthma, while minimizing variation due to experimental differences, technological discrepancy, and patient variability in studies performed by separate research groups. No genes were consistently represented in gene signatures of every single study (8 out of 8 studies). Six genes (CEACAM5, CPA3, INSR, MS4A2, NOX1, SLC16A6) were detected as significantly differentially expressed in 7 out of 8 gene signatures of asthma. 39 genes were shared by 6/8 signatures and 194 genes by 5/8 signatures. To allow for a sufficient number of genes to proceed with functional gene network analysis, we considered the genes shared in 5/8 asthma differential expression signatures, but only selected genes that exhibited a consistent upregulation or downregulation pattern across these studies. This resulted in the consensus list of 159 genes (93 upregulated and 66 downregulated) (Fig. 2). We will refer to these genes as “159 gene asthma consensus signature”. Importantly, a majority of these genes was represented across a range of asthma severity, appearing in studies of mild, moderate, and severe asthma (Fig. 2, 5 and 6). Chromosome mapping of the consensus genes revealed genome-wide distribution, with chromosomes 1, 2, 3, 6, 12, 17, and 19 containing the greatest number of dysregulated genes in asthma (Fig. 2). Interestingly, while 2 genes on the X chromosome showed dysregulation in asthma (GUCY2F and NOX1), not a single Y chromosome gene was affected (Fig. 2). These 159 genes were further subjected to an unbiased gene ontology and functional gene network analysis (with no pre-selection of genes based on their relevance to biological processes of interest) to determine biological significance of this cross-study convergence in the pathogenesis of asthma.
Figure 2. Circular visualization of chromosomal positions, hierarchical clustering, connectivity and severity gradients for all genes in the asthma 159 gene convergence signature.
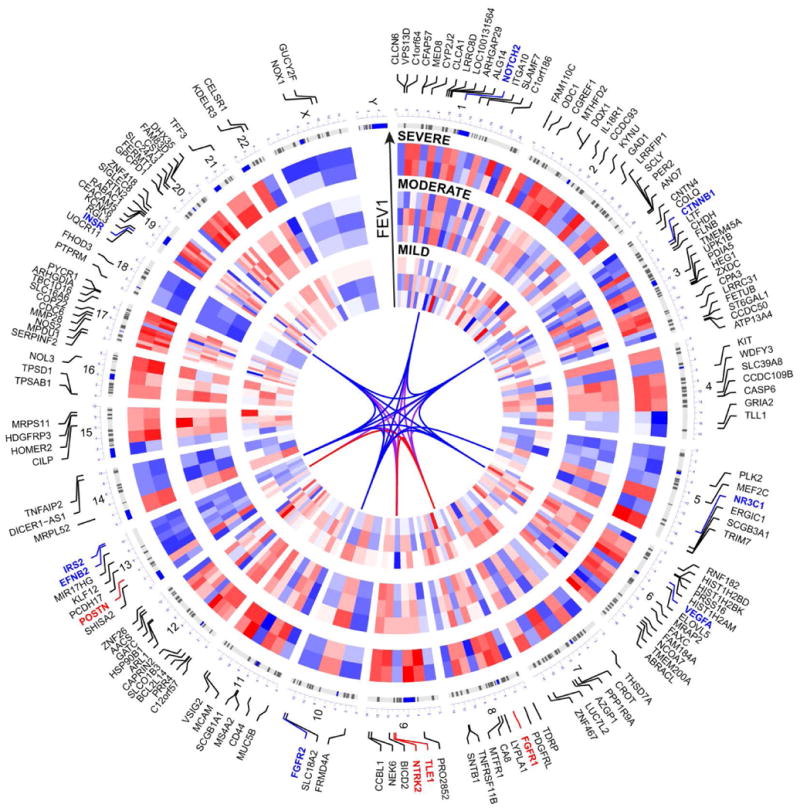
The 8 different microarray datasets are represented by concentric circles radiating outwards from the middle of the circle, with the “mild” asthma studies in the innermost group, the “moderate” asthma studies in the middle group, and the “severe” asthma studies in the outermost group. The specific order of studies, from innermost to outermost, is the same as the order they are listed in Table I. Specific chromosomal location of each gene is shown by lines coming from each gene symbol pointing to a specific position on each chromosome, with cytobands also included. Heatmap weights are based on normalized log2fold changes of gene expression in each microarray dataset; all fold change values were included for every study regardless of significance. Red represents upregulation of gene expression, blue represents downregulation. White spaces are included where a gene was not present on a microarray platform. Key 12 genes representing epithelial de-differentiation program in asthma (further described in Fig. 3) are shown in red and blue (upregulated and downregulated), and line connections (representing highest weight edges from the functional gene network analysis) between these genes are shown in the circle's center.
Figure 5. Epithelial de-differentiation programs in mild, moderate and severe asthma.
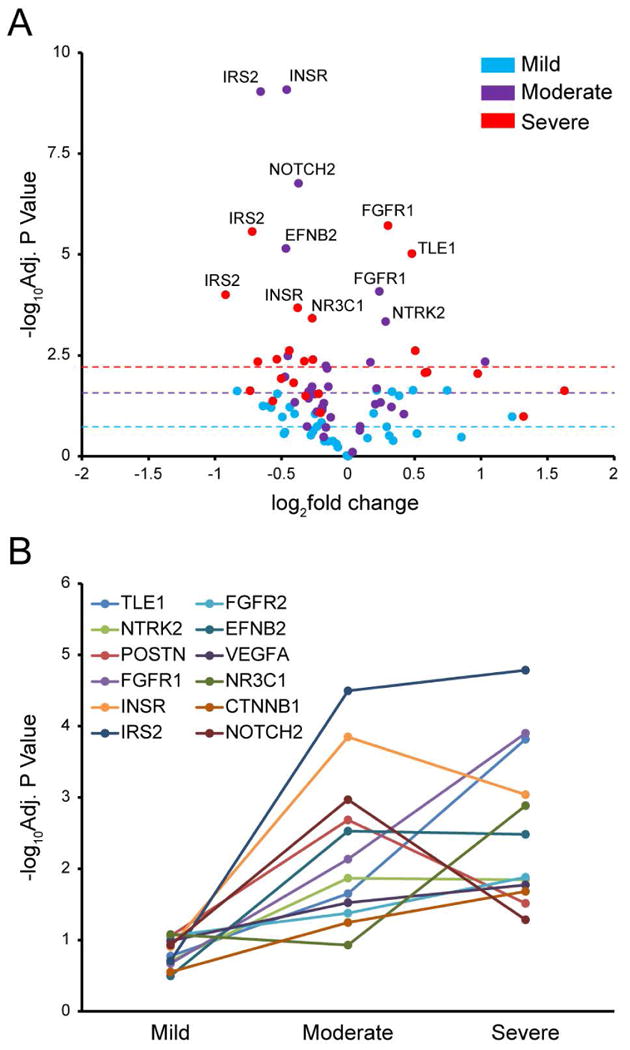
A. Volcano plot showing –log10Adjusted P-value vs. log2fold change for each of the 12 identified key genes (TLE1, NTRK2, POSTN, FGFR1, INSR, IRS2, FGFR2, EFNB2, VEGFA, NR3C1, CTNNB1, and NOTCH2) plotted for each gene in each of the 8 microarray datasets utilized in this study (12 genes × 8 studies = 96 points total). Points from studies of “Mild” asthma are denoted in blue; “Moderate”, purple; “Severe”, red. Medians of all –log10Adjusted P-values for all 12 genes in each severity grouping of array datasets (mild, moderate, severe) are shown with color-coded dotted lines to demonstrate an overall trend of increase in significance corresponding to increase in severity. Data points with the highest values on volcano plot are labeled with corresponding gene names. B. Line chart depicting the change in –log10Adjusted P-value as disease severity increases (based on FEV1), for each of the 12 hub genes. Gene data points in each severity group (mild, moderate, severe) are –log10Adjusted P-values averaged for all studies falling within given severity group.
Figure 6. Validation of 12 epithelial differentiation genes in an independent study of mild, moderate and severe asthma (GSE41861).

Normalized expression values in each sample in GSE41861 (across different asthma severities) for the 12 epithelial differentiation “functional hub” genes identified in our network analysis. ****: p-value < 0.0001, ***: p-value < 0.001, **: p-value < 0.01, *: p-value < 0.05, n.s.: not significant, p-values between 0.05 and 0.15 noted on graphs; moderated Benjamini-Hochberg t-tests (with FDR significance adjustment).
Pathways and functional processes represented by consensus gene expression in asthma
Functional pathway analysis of 159 consensus asthma genes (referencing GO Biological Process and KEGG Pathway databases) revealed a tightly integrated functional network representing several key biological processes and pathways of immediate relevance to the lung and epithelial biology (Fig. 3). Functional gene terms identified through this analysis converged on several meta-groups of biological significance, describing: (1) Lung morphogenesis and epithelial de-differentiation, captured by groupings of the following gene terms: lung development (FGFR2, HEG1, NR3C1), organ morphogenesis (EFNB2, FGFR2, NOTCH2, TLE1), epithelial cell differentiation (FGFR2, VEGFA, UPK1B), cell maturation (CTNNB1, FGFR1, VEGFA), lung-associated mesenchyme development (FGFR1, FGFR2, CTNNB1), positive regulation of mesenchymal cell proliferation (FGFR1, FGFR2, IRS2, VEGFA, CTNNB1), stem cell maintenance (FGFR1, KIT, NOTCH2), and negative regulation of cell growth (CGREF1, CAPRIN2, SCGB3A1); (2) Dysregulation of specific pathways in control of epithelial homeostasis: Notch signaling (NOTCH2, TLE1), ephrin signaling (EFNB2), WNT signaling (CTNNB1), insulin receptor signaling (INSR, IRS2, FGFR1/2), and fibroblast growth factor receptor signaling pathway (FGFR1/2); 3) Dysregulated maintenance of cell junctions and adhesion: homophilic cell adhesion (CELSR1, PCDH17, PTPRM), cell-matrix adhesion (CD44, CTNNB1, ITGA10, POSTN), focal adhesion (CTNNB1, FLNB, ITGA10, VEGFA), and adherens junctions (CTNNB1, FGFR1, INSR, PTPRM); (4) Changes in metabolism and signaling of hormones: response to insulin and glucose stimulus (INSR, IRS2, CASP6), fatty acid metabolic process (AACS, CROT, LYPLA1), phosphoinositide-mediated signaling (CA8, FGFR1, IRS2), arginine and proline metabolism (NOS2, ODC1, PYCR1), response to estradiol stimulus (CASP6, CTNNB1, INSR). For convenience, shorthand annotations for these groups are supplied in color-coded text next to relevant gene clusters in Fig. 3. The highest functional integration in the network (measured by highest weight/connectivity) was found among the following genes: FGFR1, FGFR2, INSR, IRS2, VEGFA, CTNNB1, and NTRK2 (Fig. 3, edges highlighted in crimson color), all implicated as central in maintenance of epithelial homeostasis.
Figure 3. Functional network analysis of consensus asthma gene expression signature.
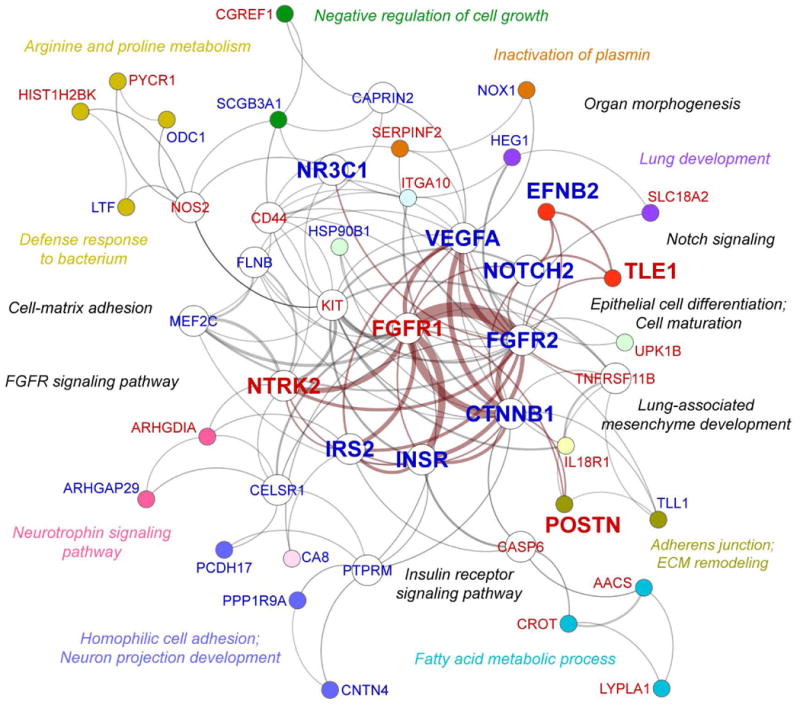
Functional network of 159 genes that were significantly differentially expressed (10% FDR adjusted p-value cutoff) in the same direction (93 upregulated and 66 downregulated) in at least 5 out of 8 asthma microarray datasets shown in Table 1. The functional gene network was generated by meta-grouping of individual gene term sets (referencing GO Biological Process and KEGG Pathways knowledge repositories), based on functional similarity. Each color-defined gene cluster represents a single meta-group defined by functional similarity of biological processes involving these genes, labeled accordingly in matching color adjacent to gene clusters. Genes unique to their corresponding meta-groups are color-coded. Nodes representing “functional hub” genes (belonging to multiple meta-groups) are not color-coded. 12 genes belonging to functional processes representing epithelial morphogenesis and differentiation (as indicated by an unbiased gene term analysis) are displayed in larger fonts and node sizes than other genes. Genes in red were significantly upregulated, while genes in blue were significantly downregulated. Edge (line) thickness is proportional to weight of gene interaction in the network. Connections between hub genes with the highest weight and overall connectivity in the network.
Key genes capturing epithelial de-differentiation program in asthma
Network analysis/FGSEA identified several key “functional hub” genes, which are genes that influence the biology of more than one functional gene term-group, thus playing a central role in regulation of biological processes and pathways represented by the network (Fig. 3, open nodes). As determined by unbiased functional gene network analysis, 12 functionally overrepresented genes (CTNNB1, FGFR1, FGFR2, INSR, IRS2, NOTCH2, NR3C1, NTRK2, VEGFA) and several key downstream signals (EFNB2, POSTN, TLE1) exhibited tight integration and functional association with processes and pathways regulating epithelial morphogenesis and differentiation (Fig. 3, enlarged font size); we will refer to these as our “12 gene epithelial differentiation signature”. To contrast these newly found epithelial homeostasis-related genes with expression of classical markers of epithelial-to-mesenchymal transition, and evaluate their biomarker potential, we illustrated expression of 9 classical EMT markers compared to our 12 key genes capturing the de-differentiaton program across the 8 studies of asthma in a radar plot (Fig. 4A). Among markers of EMT, only CDH2 (N-cadherin), SNAI2 (SLUG), and TWIST1 showed significant differential expression in any asthma study. There was virtually no agreement on expression of these markers across different studies: ZEB1 was significantly differentially expressed in just 1 out of 8 study, SNAI2 and TWIST1 in 2 out of 8 studies, and CDH2 in 3 out of 8 studies (with downregulation rather than upregulation of N-cadherin in 2 of those 3 studies, which is not generally consistent with an EMT phenotype). Compared to descriptors of “epithelial-to-mesenchymal transition”, our key markers of “epithelial de-differentiation” exhibited much greater consensus expression across different studies of asthma. FGFR2, IRS2, NTRK2, and TLE1 exhibited consensus differential expression in 6/8 studies, and insulin receptor (INSR) in 7/8 studies, the rest of our “key” markers appearing in 5/8 studies. To confirm congruity of our 12 gene epithelial differentiation signature to processes of epithelial differentiation and EMT, as well as to determine their relevance and novelty for asthma, we executed PubMed (NCBI) search queries for each individual marker coupled with search terms involving asthma, EMT, and epithelial differentiation (made as inclusive as possible by the inclusion of AND and OR search operators). PubMed searches validated the functional network analysis results by demonstrating that many of the 12 markers described here are important components of the system governing epithelial-mesenchymal homeostasis, but surprisingly unknown or underrepresented in studies of asthma (Fig. 4B). In particular, PubMed searches revealed that the most overlooked processes in the pathogenesis of asthma were downregulation of Notch (NOTCH2, TLE1), insulin signaling (INSR, IRS2, FGFR1, FGFR2) and ephrin signaling (EFNB2), despite their known critical roles in maintenance of epithelial differentiation and competent barrier state. Importantly, as validation of our consensus signature's relevance to current knowledge regarding asthma, these PubMed searches additionally showed many returned results for highly-researched disease markers such as POSTN and CLCA1, also being represented in the consensus signature.
Figure 4. Key genes capturing epithelial de-differentiation program in asthma.
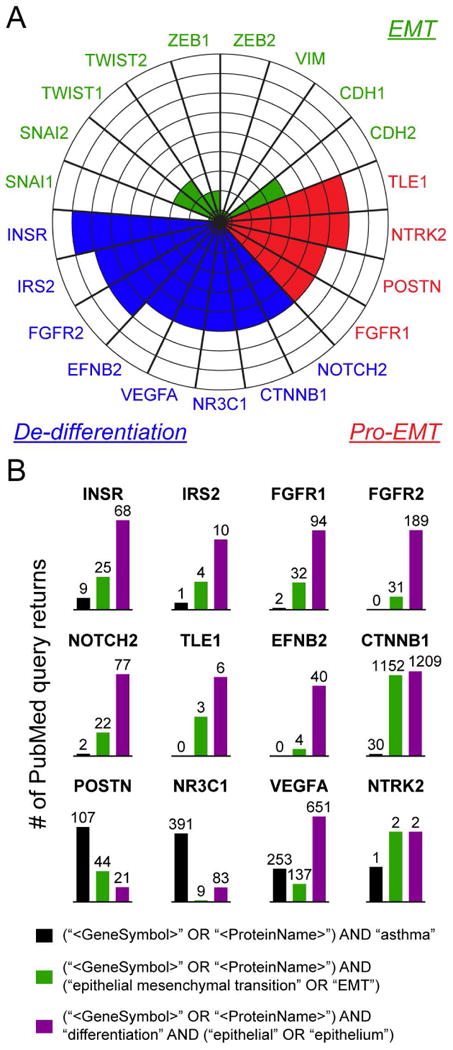
A. Radar plot comparing significant expression of classical EMT markers in the eight microarray datasets with the expression of key genes identified from functional gene network analysis in Fig. 3, in the same microarray datasets. Each sector of the circle represents a different gene, and gene symbols are listed on the outside of the corresponding sector. Green: conventional EMT markers. Red: upregulated key genes from our signature (associated with induction of EMT program). Blue: downregulated hub genes from our signature (representing de-differentiation). Concentric lines within the circle radiating outwards from the center denote number of studies in which a gene occupying any given sector was identified as significantly differentially expressed (i.e., genes highlighted in three concentric circles are differentially expressed in 3 out of 8 asthma studies). B. PubMed results returned for queries involving key genes included in the plot above, coupled with other search terms including asthma, EMT, and epithelial differentiation. Details of each query are shown in the legend; each query first includes the gene name and associated protein name, followed by “asthma”, “EMT”, or “epithelial differentiation” (made as inclusive as possible using AND and OR search operators). The number of studies returned by these PubMed searches is denoted numerically over separate bar graphs for each gene.
Epithelial de-differentiation programs in mild, moderate and severe asthma
Given that the consensus asthma signature of 159 genes is represented in mild, moderate, and severe asthma datasets, we determined whether expression of these genes changes with disease severity. FEV1 (Forced Expiratory Volume) data representing asthma severity for each of the eight studies is shown in Table 1. Hierarchical clustering of these genes by normalized fold change indicated that overall, expression of most genes in the consensus signature increases with severity, represented by its intensification along the FEV1 gradient (Fig. 2). This was also true for our “12 gene epithelial differentiation signature” representing dysregulation of the epithelial differentiation program within this signature (Fig. 2, colored gene labels). Moreover, there was an overall trend for increase in statistical significance of these genes, aligning with severity of different studies of asthma, shown as a volcano plot in Fig. 5A. More detailed interrogation of changing statistical significance of these 12 key genes across different studies of asthma (by assessment of adjusted p-value averaged among each severity grouping) revealed that, on average, significance of this signature increased with severity (Fig. 5B). CTNNB1, FGFR1, FGFR2, IRS2, NR3C1, and VEGFA exhibited gradual increase in significance from mild to moderate to severe asthma. INSR, NOTCH2, and POSTN were most significantly differentially expressed in moderate asthma, while EFNB2 and NTRK2 showed similar levels of statistical significance in moderate and severe asthma (Fig. 5B). In summary, this epithelial de-differentiation program appears to occur to some degree in mild, moderate, and severe asthma. Moreover, expression of key genes representing this process tends to increase with disease severity.
Convergent gene validation in independent epithelial brushing datasets
To validate our convergence gene signatures and functional processes revealed through the data mining analysis, we utilized two different approaches. First, we determined whether these genes appear in other independent publicly available microarray datasets of asthma (GSE41861 and GSE64913) that were not included in the original 8 datasets used in our analysis. The reasoning for not including them in our initial analysis is described in the Methods and Supplementary Table 1. Importantly, these datasets were also performed on freshly lysed bronchial brushing samples, similar to the majority of datasets used to generate our convergence signatures. We specifically assessed the significance of differential expression of our 12 functional hub genes most relevant to epithelia-mesenchymal communication in each of these microarray datasets (Fig. 6 and Supplementary Fig. 1). These datasets both largely validated expression of these 12 genes, and directionality of expression in asthma was consistent with that observed in our mining analysis (e.g., INSR was downregulated in both our convergence signatures and in these new datasets). Importantly, GSE41861 stratifies its asthma patients by mild, moderate, and severe asthma (all simultaneously done in the same study), and this study also confirmed potentiation of differences in expression of genes of interest along the asthma severity gradient (Fig. 6).
Gene and functional process validation in primary human normal and asthmatic bronchial epithelial cell cultures
Another approach we took to validating our functional gene signatures was to perform our own RNA-Seq analysis on cultured human bronchial epithelial cells (normal and asthmatic) in three different biologically-relevant culture contexts: 1) comparing normal vs. asthmatic bronchial epithelial cells in submerged growth conditions; 2) contrasting normal vs. asthmatic bronchial epithelial cells in fully differentiated air-liquid interface conditions, which more realistically approximates the in vivo barrier state of the epithelium, and 3) at the functional process level, comparing normal bronchial epithelial cells in fully differentiated air-liquid interface conditions in complete media (supplemented with all necessary growth factors and hormones) vs. media deficient in insulin, to recapitulate the decrease in insulin signaling that we identified as one of the key de-differentiation events in our convergence asthma network. Importantly, the same donors in the same plating conditions (passage number, plating density) were used for tests in different conditions. When comparing healthy and asthmatic samples, donors were matched by age and sex. All samples were processed simultaneously for RNA-seq. In Fig. 7 (left column), volcano plots show the spread of significantly differentially expressed RNA transcripts in each of these culture conditions. At an adjusted significance level of 0.05, 1912 genes were differentially expressed between healthy and asthmatic epithelium at baseline in submerged culture. In organotypic air-liquid interface conditions, this number increased to 2721 genes. Insulin deficiency during differentiation induced differential expression of 6694 genes in normal bronchial epithelium. We specifically emphasized changes in expression of genes associated with proper epithelial homeostasis and maintenance of differentiation state (markers of mature epithelium, mesenchymal/basal epithelium, “Th2-high” barrier markers, and differentiation process-associated markers (Fig. 7, right column). In all three conditions (submerged asthmatic cells, organotypic asthmatic epithelium, and insulin deficient culture), we found overall loss of differentiation and potentiation of mesenchymal signal. This was specifically supported by downregulation of maturity and differentiation markers MUC5AC (secretory epithelium), EPCAM (epithelial barrier junctions), FGFR2 (loss of differentiation signal central to our convergence networks) and NOTCH2 (Notch developmental signaling pathway), as well as upregulation of mesenchymal/basal markers (VIM, KRT5) and markers of barrier remodeling (POSTN and SERPINB2, classically described as “Th2-high” markers in the literature). Notably, fold change and significance of these changes tended to increase in more biologically-relevant organotypic contexts (note changes in scale in different conditions in Fig. 7), which we additionally emphasized in Fig. 8.
Figure 7. Gene and functional process validation in primary human normal and asthmatic bronchial epithelial cell cultures.
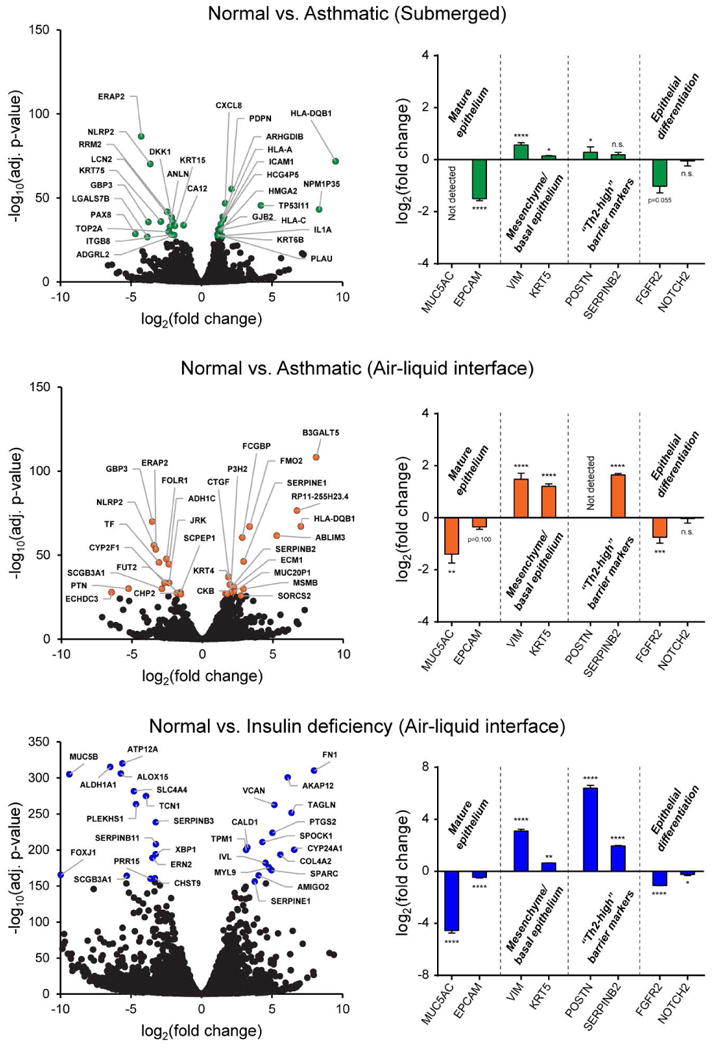
RNA-seq and gene differential analysis was performed for cultured epithelium in three different biological contexts: 1) submerged epithelial cultures, asthmatic vs. normal epithelium comparison (top); 2) organotypic air-liquid interface epithelial cultures, asthmatic vs. normal epithelium comparison (middle); and 3) organotypic air-liquid interface epithelial cultures, with the loss of differentiation induced by insulin deficiency in normal human bronchial epithelial cells (bottom). Volcano plots show gene differential expression under each scenario (left). The top 30 genes by FDR-adjusted p-value in each dataset have been colored and labelled with gene names. Next to each volcano plot (right), gene markers representing loss of differentiation in all three scenarios are shown in bar graphs. These include markers attributable to mature epithelium, mesenchymal/basal epithelium, “Th2-high” barrier markers, and genes associated with active epithelial differentiation. Bar graphs represent log2(fold change) of asthmatic or insulin deficient normal epithelium vs. normal controls, based on normalized RNA transcript counts. The same donors were used in all three scenarios, and asthmatic vs. normal donors were matched by age and sex. ****: p-value < 0.0001, ***: p-value < 0.001, **: p-value < 0.01, *: p-value < 0.05, n.s.: not significant, p-values between 0.05 and 0.10 noted on graphs; moderated Benjamini-Hochberg t-tests (FDR p-value adjustment).
Figure 8. Comparison of validation coverage of asthma convergence signatures in different biological contexts.
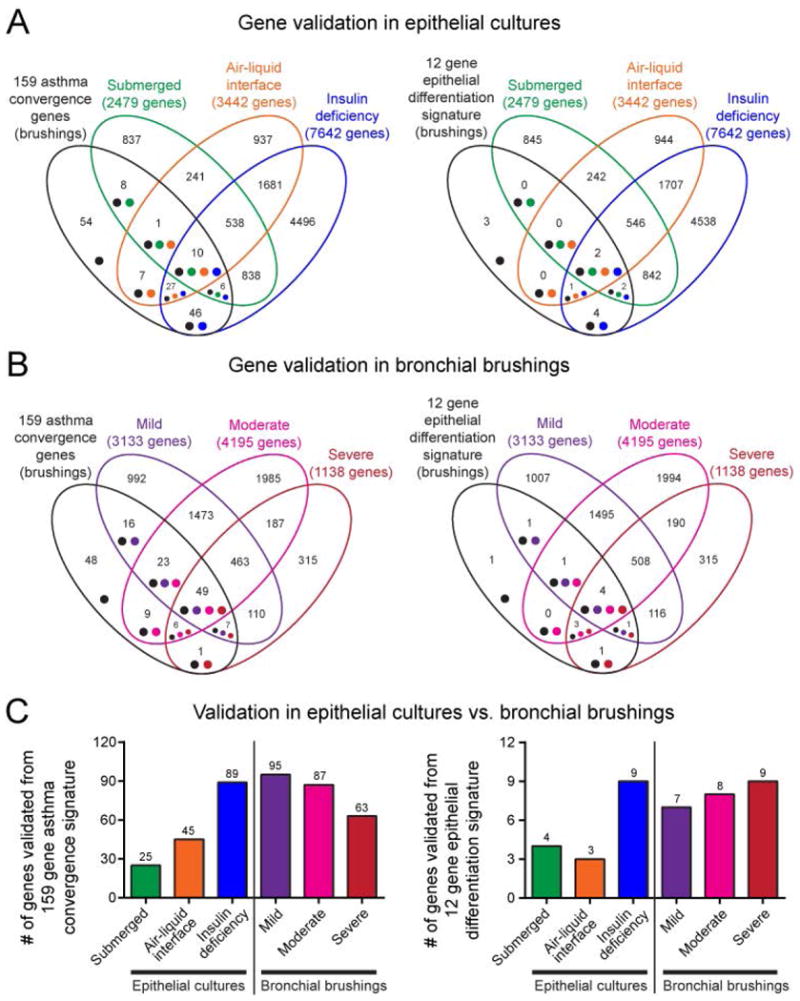
A. Venn diagrams comparing 159 asthma convergence signature (left) and 12 gene epithelial differentiation signature (right) against gene expression in epithelium grown in different biological contexts as presented in Fig. 7. B. Venn diagrams comparing 159 asthma convergence signature (left) and 12 gene epithelial differentiation signature (right) against gene expression in epithelial brushings (from GSE41861), stratified by mild, moderate, and severe asthma, as presented in Fig. 6. C. Summary charts contrasting the strength of convergence signature validation in different biological sources and culture contexts. The number of genes denoted over individual bars in bar graphs matches number of genes in corresponding intersections in the Venn diagrams above.
Strength of validation in different biological contexts
During confirmation steps, it became apparent that strength of validation (number of genes confirmed in independent studies) is significantly dependent on the biological state of the epithelium under investigation. In Venn diagrams in Fig. 8, we show that when compared to either the 159 asthma convergence signature (Fig. 8, left) or the 12 gene epithelial differentiation signature (Fig. 8, right), the highest number of reconfirmed genes was from direct lysates of epithelial brushings (mild, moderate, and severe asthma patient samples) (Fig. 8B). Overall, fewer mined genes were detected in epithelial cultures (Fig. 8A). However, complexity and in vivo modeling potential of epithelial culture mattered the most: loss of epithelial differentiation simulated by insulin deficiency in cultured organotypic epithelium resulted in a high number of validated “asthma convergence genes”, comparable to the signal from epithelial brushings from subjects with mild, moderate and severe asthma. However, submerged monolayer epithelial culture conditions resulted in the poorest number of detected convergence genes. Gene signature coverage patterns across biological states were similar for 12 gene epithelial differentiation signature as well (Fig. 8, right). Relative numbers of validated genes in different biological contexts are shown in Fig. 8C. In summary, “data mined” genes in asthma convergence signatures represent the complexity of epithelial-mesenchymal communication, which can be measured or modeled best only in biologically relevant, organotypic conditions approximating disease states in vivo.
Discussion
Our study was motivated by the controversy surrounding epithelial-to-mesenchymal transition in asthma. Existence of EMT, potential drivers of this process, and its causal versus consequential place in the pathogenesis of allergic disease are some of the most contested issues in the field15, 16. Access to a shared library of transcriptomic data from the scientific community in web repositories such as Gene Expression Omnibus (GEO, NCBI) provides a unique opportunity to revisit gene expression signatures from multiple independent studies in search of agreement on processes underlying disease pathogenesis17. Understanding convergence among independent studies of one disease, when done in an unbiased manner, represents a powerful approach to the identification of common functional gene expression patterns, while minimizing variation resulting from differences in experimental design and technical details of individual studies. Here, we used gene expression microarray datasets uploaded to GEO NCBI by several independent groups to determine unity in epithelial gene signatures from different studies of asthma and to generate novel hypotheses pertaining to processes driving EMT and epithelial barrier dysfunction.
With the above approach, we found little evidence for differential expression of classical EMT markers in gene signatures of any of these asthma microarray studies. Moreover, in few instances where they were differentially expressed, there was no agreement on expression of these markers among studies, with different studies showing differential expression of different EMT genes. This is not entirely surprising, since EMT in allergic disease is usually reported as a highly localized and transient process, not uniformly occurring along epithelial barriers3. Moreover, many transcriptional drivers of EMT are proteins with short half-lives18, so that their stabilization by local cues can cause rapid changes in gene expression19. Thus, one might not expect to observe consistent differences in the mRNA levels of these factors. Additionally, unlike cancer, where epithelial cells are considered to undergo a full transition to a mesenchymal phenotype in order to migrate to and colonize other sites in the body20, EMT in allergy is more often reported as a mild, “partial” phenotype2. This could also contribute to poor expression of conventional EMT markers in asthma. Despite this absence of robust EMT marker expression, our unbiased bioinformatics approach nonetheless identified a novel suite of potential biomarkers capturing loss of epithelial differentiation and dysregulation of epithelial-mesenchymal signals at the level of gene expression, including EFNB2, FGFR1, FGFR2, INSR, IRS2, NOTCH2, and TLE1. Given their consistent and unidirectional expression across multiple studies of asthma, these key genes could serve as reliable markers for dysregulation of epithelial-mesenchymal signals, consistent with the pro-EMT process implicated in allergic disease. Despite established evidence that these genes play critical roles in regulation of epithelial homeostasis and epithelial-to-mesenchymal communication21-24, most of them have remained overlooked in the field of asthma, assessed by PubMed query method. We were able to prospectively validate expression of these genes in two independent cohorts of asthmatic samples utilizing epithelial brushings and epithelial cultures. Validation was much weaker when these genes were measured in asthmatic vs. normal cultured epithelium in submerged monolayer conditions. The number of validated genes significantly increased when we cultured epithelium in organotypic differentiation conditions, which more realistically represents the biology of stratified epithelial barriers in vivo. This supports the notion that these newly identified asthma genes do not represent static behavior of the epithelium, but rather are functionally relevant markers induced by the need for communication between less and more differentiated cells in barrier settings.
Several key findings emerged from functional network analysis of our 159 gene asthma consensus signature: (1) overall downregulation of morphogenetic and differentiation programs, indicated by downregulation of Notch, insulin, and ephrin signaling (NOTCH2, FGFR2, INSR, IRS2, EFNB2); 2) induction of mesenchymal proliferation and processes supporting epithelial-to-mesenchymal transition (FGFR1, NTRK2, POSTN, TLE1); 3) remodeling and secretion of provisional matrix (POSTN, CD44); (4) downregulation of insulin signaling upstream of FGFR1/2, likely implicating insufficiency of insulin drive and/or insulin resistance (INSR, IRS2, FGFR1, FGFR2); (5) common representation of a de-differentiation program in mild, moderate, and severe asthma, and its increase with disease severity. Of interest, the FGFR1/2 imbalance downstream of insulin receptor signaling had the highest weight in the network. The net effect of upregulated FGFR1 and downregulated FGFR2 is epithelial-mesenchymal imbalance, remodeling, and de-differentiation24, 25, represented by downstream downregulation of Notch signaling (NOTCH2, TLE1), loss of junctions (CTNNB1, PCDH17, CNTN4), and extracellular matrix remodeling (CD44, POSTN). Notch signaling is critical in epithelial differentiation26-28, while TLE1 is elevated during incomplete maturation events and implicated in maintenance of an undifferentiated state of epithelial cells29. EFNB2 is an ephrin with positive roles in epithelial morphogenesis30, 31. POSTN is a marker of Th2-high asthma32, although the mechanism of its expression is not well understood. Several studies link expression of periostin to induction of EMT33-35. Our analysis suggests that increased upregulation of POSTN is an outcome of an overall de-differentiation program, which could represent increased deposition of provisional extracellular matrix as a compensatory drive back towards restoration of mature epithelium36. Evidence for dysregulated Notch signaling in asthma raises the possibility that readily available gamma secretase inhibitors may be worth considering in models of allergic disease.
Despite collective evidence for epithelial de-differentiation program in asthma, one important question remains: which processes drive epithelial-mesenchymal imbalance in asthma? Persistent inflammation and need-for-repair are usual suspects in chronic disease. During repair, epithelial cells have the capacity to revert to mesenchymal cells to expand a precursor pool, or adopt a more migratory phenotype needed to differentiate and replace damaged cells in the epithelial layer37, 38. However, several lines of evidence speak for more profound and less obvious drivers of epithelial dedifferentiation, which may be independent of inflammation and repair. It is becoming clear that disruption of epithelial homeostasis often precedes development of allergic inflammation and disease. This is supported by the link between loss-of-function in epithelial barrier genes and allergen sensitization6, the altered barrier state of intact non-lesional skin in atopic dermatitis7, increased transepidermal water loss (TEWL) through uninvolved skin of allergic patients8, and “immaturity” defects in junctional maintenance and repair in cultured asthmatic epithelium10. However, no mechanism has been proposed so far to explain systemic barrier dysfunction, especially in the absence of inflammation. The results of our hypothesis-generating study strongly implicate the existence of systemic factors underlying epithelial barrier dysfunction in asthma. In particular, our analysis directly implicates aberrant input of hormones regulating energy metabolism. Downregulation of insulin receptor (INSR) and insulin receptor substrate (IRS2), as well as imbalance of insulin downstream signals (downregulation of FGFR2 and upregulation of FGFR1), point to insufficiency of insulin drive and/or insulin resistance. FGF21, a novel hormone partnering with insulin in regulation of glucose homeostasis, also signals through FGFR1 and FGFR239. In support of our findings, previous studies have reported insulin resistance in children and adults with asthma40, 41, and an association of asthma with pre-diabetes and metabolic syndrome42, 43. Moreover, our validation experiments inducing loss of differentiation in epithelial cells by depriving them of insulin (which is long-established as a necessary component in commercial culture of primary epithelium) demonstrate remarkable potential for insulin deficiency to induce epithelial genes and phenotypes consistent with those observed in asthma, including upregulation of “Th2-high” markers and loss of competent barrier state. Metabolic hormones are critical regulators of tissue and epithelial homeostasis via their integration with epithelial developmental pathways (Notch, WNT, Sonic Hedgehog) and differentiation programs44, evidenced by deficiencies in wound healing and epithelial dysfunction in diabetic patients45, 46. Thus, aberrant integration of hormonal signaling could represent a driver of epithelial dysfunction in asthma. Importantly, metabolic aberrations are also linked to epigenetic reprogramming47, 48 and immune response49, 50, suggesting an intriguing possibility that these processes play a role in the origins and predisposition to allergic disease. A persistent phenotype of repair deficiencies in epithelial cultures from asthmatic children further implicates developmental tissue reprogramming and supports this possibility10. Metabolic and endocrine components of asthma pathogenesis are not well understood, and are the subject of current and future studies by our group.
Asthma is a highly heterogeneous disease represented by several clinical and molecular endotypes51. For example, previous gene expression studies have demonstrated existence of Th2-low and Th2-high subtypes of asthma32. In particular, severe asthma resistant to glucocorticoid treatment is thought to involve distinct pathogenic mechanisms and represent a separate endotype of this highly complex disease52. Despite numerous differences in immune and regulatory responses in different forms of asthma, the consensus gene expression program revealed by our analysis implicates the underlying “core” de-differentiation program as a potentially unifying feature of asthma that spans several different endotypes, given its persistence throughout mild, moderate, and severe asthma, as well as in studies performed by several independent groups. Importantly, expression of most genes within our 159 gene asthma consensus signature increases with disease severity, implying that common overarching mechanisms may be shared by all forms of disease, only on different spectrums. Interestingly, our study found that downregulation of the glucocorticoid receptor (GR) (NR3C1 gene) was the most dramatic in severe asthma, while showing comparatively modest downregulation in mild and moderate asthma. Thus, an intriguing possibility is that insufficient levels of GR (relative to mild and moderate asthma) contribute to glucocorticoid treatment resistance in severe asthma. It is also important to recognize that besides its role in the control of inflammation, GR is part of a mechanism regulating tissue and epithelial barrier homeostasis. Inactivation of GR triggers barrier defects, epithelial de-differentiation, and inflammation53-56, which is rationalized by our gene network analysis showing the interconnectivity of NR3C1 dysregulation with processes governing epithelial differentiation (Fig. 3), where dramatic downregulation of NR3C1 in severe asthma coincides with upregulation of FGFR1 and TLE1 – inducers of an “immature” state of the epithelium (Fig. 5).
In summary, our study neither refutes nor supports the existence of EMT in asthma as classically defined by developmental biologists, but instead reinforces the view of an underlying epithelial de-differentiation program that drives epithelial barrier disruption in this complex disease. Our unbiased secondary analysis of gene expression signatures of asthma provided several novel putative markers and pathways that capture the essence of epithelial-mesenchymal dysregulation, which have the potential to be more effective than classical markers of EMT in representing epithelial barrier dysfunction at the gene expression level. Furthermore, our analysis strongly implicates systemic changes in energy metabolism that could have potential to drive loss of epithelial differentiation. In-depth understanding of these mechanisms holds promising future potential for monitoring disruption of epithelial barrier homeostasis and augmentation of epithelial barrier function in prevention or treatment of asthma and other allergic diseases.
Supplementary Material
Supplementary Figure 1. Validation of 12 epithelial differentiation genes in an independent study of severe asthma (GSE64913). Normalized expression values for each sample in GSE64913 (combining central and peripheral bronchial samples) for the 12 epithelial differentiation “functional hub” genes identified in our network analysis. ****: p-value < 0.0001, ***: p-value < 0.001, **: p-value < 0.01, *: p-value < 0.05, n.s.: not significant, p-values between 0.05 and 0.15 noted on chart; moderated Benjamini-Hochberg t-tests (with FDR significance adjustment).
Supplementary Table 1. List of all microarray datasets of asthma available in NCBI GEO repository that utilize sampling of primary bronchial epithelial brushings or cultured epithelial cells. The table has been separated into microarray datasets meeting our criteria for use in data mining analysis vs. those datasets that were not included in our analysis, with the reasons for exclusion indicated.
Acknowledgments
We thank Dr. Steven J. Ackerman (University of Illinois at Chicago, Department of Biochemistry and Molecular Genetics), Dr. William Lowe, Jr. (Northwestern University Feinberg School of Medicine, Division of Endocrinology) and Dr. Robert P. Schleimer (Northwestern University Feinberg School of Medicine, Division of Allergy and Immunology) for thoughtful discussions and assistance in interpretation of data.
Funding: This work was supported by the National Institutes of Health (NIH/NIAID) grants R21AI115055 and R01AI127783 to Dr. Berdnikovs, as well as U19AI106683 (Supplement 2) grant to Drs. Schleimer/Berdnikovs. Additionally, this study was supported by the Ernest S. Bazley Foundation.
Footnotes
Author Contributions: S.B., H.A.V. and L.F.L. conceived and designed the study. S.B., L.F.L. and K.R.A. executed bioinformatics analysis. L.F.L. and L.C.P. performed validation. H.A.V. performed RNA-Seq sequencing. S.B. and L.F.L. analyzed data. S.B. and L.F.L. prepared the figures. S.B., L.F.L., H.A.V. and C.J.G. interpreted the results. The manuscript was written by S.B. and edited by L.F.L. and C.J.G. Final version of the manuscript was approved by S.B. There are no conflicts of interest to declare.
Conflicts of Interest: The authors declare that they have no conflicts of interest.
References
- 1.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. 2014;69:760–5. doi: 10.1136/thoraxjnl-2013-204608. [DOI] [PubMed] [Google Scholar]
- 3.Kagalwalla AF, Akhtar N, Woodruff SA, Rea BA, Masterson JC, Mukkada V, et al. Eosinophilic esophagitis: epithelial mesenchymal transition contributes to esophageal remodeling and reverses with treatment. J Allergy Clin Immunol. 2012;129:1387–96 e7. doi: 10.1016/j.jaci.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol. 2012;12:53–9. doi: 10.1097/ACI.0b013e32834ec6eb. [DOI] [PubMed] [Google Scholar]
- 5.Yang ZC, Yi MJ, Ran N, Wang C, Fu P, Feng XY, et al. Transforming growth factor-beta1 induces bronchial epithelial cells to mesenchymal transition by activating the Snail pathway and promotes airway remodeling in asthma. Mol Med Rep. 2013;8:1663–8. doi: 10.3892/mmr.2013.1728. [DOI] [PubMed] [Google Scholar]
- 6.Weidinger S, Illig T, Baurecht H, Irvine AD, Rodriguez E, Diaz-Lacava A, et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol. 2006;118:214–9. doi: 10.1016/j.jaci.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Suarez-Farinas M, Tintle SJ, Shemer A, Chiricozzi A, Nograles K, Cardinale I, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol. 2011;127:954–64 e1-4. doi: 10.1016/j.jaci.2010.12.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flohr C, Perkin M, Logan K, Marrs T, Radulovic S, Campbell LE, et al. Atopic dermatitis and disease severity are the main risk factors for food sensitization in exclusively breastfed infants. J Invest Dermatol. 2014;134:345–50. doi: 10.1038/jid.2013.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hackett TL, Knight DA. The role of epithelial injury and repair in the origins of asthma. Curr Opin Allergy Clin Immunol. 2007;7:63–8. doi: 10.1097/ACI.0b013e328013d61b. [DOI] [PubMed] [Google Scholar]
- 10.Kicic A, Hallstrand TS, Sutanto EN, Stevens PT, Kobor MS, Taplin C, et al. Decreased fibronectin production significantly contributes to dysregulated repair of asthmatic epithelium. Am J Respir Crit Care Med. 2010;181:889–98. doi: 10.1164/rccm.200907-1071OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol. 2011;18:867–74. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahman SM, Ji X, Zimmerman LJ, Li M, Harris BK, Hoeksema MD, et al. The airway epithelium undergoes metabolic reprogramming in individuals at high risk for lung cancer. JCI Insight. 2016;1:e88814. doi: 10.1172/jci.insight.88814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamouille S, Subramanyam D, Blelloch R, Derynck R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr Opin Cell Biol. 2013;25:200–7. doi: 10.1016/j.ceb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fontanillo C, Nogales-Cadenas R, Pascual-Montano A, De las Rivas J. Functional analysis beyond enrichment: non-redundant reciprocal linkage of genes and biological terms. PLoS One. 2011;6:e24289. doi: 10.1371/journal.pone.0024289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sohal SS, Ward C, Walters EH. Importance of epithelial mesenchymal transition (EMT) in COPD and asthma. Thorax. 2014;69:768. doi: 10.1136/thoraxjnl-2014-205582. [DOI] [PubMed] [Google Scholar]
- 16.Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol. 2014;134:509–20. doi: 10.1016/j.jaci.2014.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riba M, Garcia Manteiga JM, Bosnjak B, Cittaro D, Mikolka P, Le C, et al. Revealing the acute asthma ignorome: characterization and validation of uninvestigated gene networks. Sci Rep. 2016;6:24647. doi: 10.1038/srep24647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz VM, Vinas-Castells R, Garcia de Herreros A. Regulation of the protein stability of EMT transcription factors. Cell Adh Migr. 2014;8:418–28. doi: 10.4161/19336918.2014.969998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15:677–87. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–49. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grego-Bessa J, Diez J, Timmerman L, de la Pompa JL. Notch and epithelial-mesenchyme transition in development and tumor progression: another turn of the screw. Cell Cycle. 2004;3:718–21. [PubMed] [Google Scholar]
- 22.Wilkinson DG. Regulation of cell differentiation by Eph receptor and ephrin signaling. Cell Adh Migr. 2014;8:339–48. doi: 10.4161/19336918.2014.970007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranieri D, Rosato B, Nanni M, Magenta A, Belleudi F, Torrisi MR. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget. 2016;7:5440–60. doi: 10.18632/oncotarget.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomlinson DC, Baxter EW, Loadman PM, Hull MA, Knowles MA. FGFR1-induced epithelial to mesenchymal transition through MAPK/PLCgamma/COX-2-mediated mechanisms. PLoS One. 2012;7:e38972. doi: 10.1371/journal.pone.0038972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gunschmann C, Stachelscheid H, Akyuz MD, Schmitz A, Missero C, Bruning JC, et al. Insulin/IGF-1 controls epidermal morphogenesis via regulation of FoxO-mediated p63 inhibition. Dev Cell. 2013;26:176–87. doi: 10.1016/j.devcel.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Pasolli HA, Williams T, Fuchs E. AP-2 factors act in concert with Notch to orchestrate terminal differentiation in skin epidermis. J Cell Biol. 2008;183:37–48. doi: 10.1083/jcb.200804030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massi D, Panelos J. Notch signaling and the developing skin epidermis. Adv Exp Med Biol. 2012;727:131–41. doi: 10.1007/978-1-4614-0899-4_10. [DOI] [PubMed] [Google Scholar]
- 28.Vooijs M, Liu Z, Kopan R. Notch: architect, landscaper, and guardian of the intestine. Gastroenterology. 2011;141:448–59. doi: 10.1053/j.gastro.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Dehni G, Purcell KJ, Sokolow J, Carcangiu ML, Artavanis-Tsakonas S, et al. Epithelial expression and chromosomal location of human TLE genes: implications for notch signaling and neoplasia. Genomics. 1996;31:58–64. doi: 10.1006/geno.1996.0009. [DOI] [PubMed] [Google Scholar]
- 30.Holder N, Klein R. Eph receptors and ephrins: effectors of morphogenesis. Development. 1999;126:2033–44. doi: 10.1242/dev.126.10.2033. [DOI] [PubMed] [Google Scholar]
- 31.Raft S, Andrade LR, Shao D, Akiyama H, Henkemeyer M, Wu DK. Ephrin-B2 governs morphogenesis of endolymphatic sac and duct epithelia in the mouse inner ear. Dev Biol. 2014;390:51–67. doi: 10.1016/j.ydbio.2014.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–95. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu Q, Tong S, Zhao X, Ding W, Gou Y, Xu K, et al. Periostin Mediates TGF-beta-Induced Epithelial Mesenchymal Transition in Prostate Cancer Cells. Cell Physiol Biochem. 2015;36:799–809. doi: 10.1159/000430139. [DOI] [PubMed] [Google Scholar]
- 34.Yan W, Shao R. Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial-mesenchymal transformation. J Biol Chem. 2006;281:19700–8. doi: 10.1074/jbc.M601856200. [DOI] [PubMed] [Google Scholar]
- 35.Chen M, Zheng SH, Liu Y, Shi J, Qi ST. Periostin activates pathways involved in epithelial-mesenchymal transition in adamantinomatous craniopharyngioma. J Neurol Sci. 2016;360:49–54. doi: 10.1016/j.jns.2015.11.042. [DOI] [PubMed] [Google Scholar]
- 36.Coraux C, Roux J, Jolly T, Birembaut P. Epithelial cell-extracellular matrix interactions and stem cells in airway epithelial regeneration. Proc Am Thorac Soc. 2008;5:689–94. doi: 10.1513/pats.200801-010AW. [DOI] [PubMed] [Google Scholar]
- 37.Herrera MB, Bussolati B, Bruno S, Fonsato V, Romanazzi GM, Camussi G. Mesenchymal stem cells contribute to the renal repair of acute tubular epithelial injury. Int J Mol Med. 2004;14:1035–41. [PubMed] [Google Scholar]
- 38.Barriere G, Fici P, Gallerani G, Fabbri F, Rigaud M. Epithelial Mesenchymal Transition: a double-edged sword. Clin Transl Med. 2015;4:14. doi: 10.1186/s40169-015-0055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emanuelli B, Vienberg SG, Smyth G, Cheng C, Stanford KI, Arumugam M, et al. Interplay between FGF21 and insulin action in the liver regulates metabolism. J Clin Invest. 2014;124:515–27. doi: 10.1172/JCI67353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morishita R, Franco Mdo C, Suano-Souza FI, Sole D, Puccini RF, Strufaldi MW. Body mass index, adipokines and insulin resistance in asthmatic children and adolescents. J Asthma. 2016;53:478–84. doi: 10.3109/02770903.2015.1113544. [DOI] [PubMed] [Google Scholar]
- 41.Thuesen BH, Husemoen LL, Hersoug LG, Pisinger C, Linneberg A. Insulin resistance as a predictor of incident asthma-like symptoms in adults. Clin Exp Allergy. 2009;39:700–7. doi: 10.1111/j.1365-2222.2008.03197.x. [DOI] [PubMed] [Google Scholar]
- 42.Perez MK, Piedimonte G. Metabolic asthma: is there a link between obesity, diabetes, and asthma? Immunol Allergy Clin North Am. 2014;34:777–84. doi: 10.1016/j.iac.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garmendia JV, Moreno D, Garcia AH, De Sanctis JB. Metabolic syndrome and asthma. Recent Pat Endocr Metab Immune Drug Discov. 2014;8:60–6. doi: 10.2174/1872214807666140107151023. [DOI] [PubMed] [Google Scholar]
- 44.Roarty K, Rosen JM. Wnt and mammary stem cells: hormones cannot fly wingless. Curr Opin Pharmacol. 2010;10:643–9. doi: 10.1016/j.coph.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salazar JJ, Ennis WJ, Koh TJ. Diabetes medications: Impact on inflammation and wound healing. J Diabetes Complications. 2016;30:746–52. doi: 10.1016/j.jdiacomp.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullin GE, Eastern JS. Cutaneous signs of thyroid disease. Am Fam Physician. 1986;34:93–8. [PubMed] [Google Scholar]
- 47.Folmes CD, Terzic A. Energy metabolism in the acquisition and maintenance of stemness. Semin Cell Dev Biol. 2016;52:68–75. doi: 10.1016/j.semcdb.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ryall JG, Cliff T, Dalton S, Sartorelli V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell. 2015;17:651–62. doi: 10.1016/j.stem.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, Mauro C. Intermediates of Metabolism: From Bystanders to Signalling Molecules. Trends Biochem Sci. 2016;41:460–71. doi: 10.1016/j.tibs.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Naquet P, Giessner C, Galland F. Metabolic adaptation of tissues to stress releases metabolites influencing innate immunity. Curr Opin Immunol. 2016;38:30–8. doi: 10.1016/j.coi.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 51.Wenzel SE. Complex phenotypes in asthma: current definitions. Pulm Pharmacol Ther. 2013;26:710–5. doi: 10.1016/j.pupt.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 52.Lin TY, Poon AH, Hamid Q. Asthma phenotypes and endotypes. Curr Opin Pulm Med. 2013;19:18–23. doi: 10.1097/MCP.0b013e32835b10ec. [DOI] [PubMed] [Google Scholar]
- 53.Sevilla LM, Latorre V, Sanchis A, Perez P. Epidermal inactivation of the glucocorticoid receptor triggers skin barrier defects and cutaneous inflammation. J Invest Dermatol. 2013;133:361–70. doi: 10.1038/jid.2012.281. [DOI] [PubMed] [Google Scholar]
- 54.Manwani N, Gagnon S, Post M, Joza S, Muglia L, Cornejo S, et al. Reduced viability of mice with lung epithelial-specific knockout of glucocorticoid receptor. Am J Respir Cell Mol Biol. 2010;43:599–606. doi: 10.1165/rcmb.2009-0263OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bayo P, Sanchis A, Bravo A, Cascallana JL, Buder K, Tuckermann J, et al. Glucocorticoid receptor is required for skin barrier competence. Endocrinology. 2008;149:1377–88. doi: 10.1210/en.2007-0814. [DOI] [PubMed] [Google Scholar]
- 56.Chen H, Sun X, Chi R, Li X, Feng J, Wu J, et al. Glucocorticoid dexamethasone regulates the differentiation of mouse conducting airway epithelial progenitor cells. Steroids. 2014;80:44–50. doi: 10.1016/j.steroids.2013.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Validation of 12 epithelial differentiation genes in an independent study of severe asthma (GSE64913). Normalized expression values for each sample in GSE64913 (combining central and peripheral bronchial samples) for the 12 epithelial differentiation “functional hub” genes identified in our network analysis. ****: p-value < 0.0001, ***: p-value < 0.001, **: p-value < 0.01, *: p-value < 0.05, n.s.: not significant, p-values between 0.05 and 0.15 noted on chart; moderated Benjamini-Hochberg t-tests (with FDR significance adjustment).
Supplementary Table 1. List of all microarray datasets of asthma available in NCBI GEO repository that utilize sampling of primary bronchial epithelial brushings or cultured epithelial cells. The table has been separated into microarray datasets meeting our criteria for use in data mining analysis vs. those datasets that were not included in our analysis, with the reasons for exclusion indicated.