

Abstract
Bacteria can cause life-threatening infections, such as pneumonia, meningitis or sepsis. Antibiotic therapy is a mainstay of treatment, although antimicrobial resistance has drastically increased over the years. Unfortunately, safe and effective vaccines against most pathogens have not yet been approved, thus developing alternative treatments is important.
We analyzed the efficiency of FH6-7/Fc, a novel antibacterial immunotherapeutic protein against the gram-positive bacterium Streptococcus pyogenes. This protein is composed of two domains of complement inhibitor human factor H (FH complement control protein modules 6 and 7) that bind to S. pyogenes, linked to the Fc region of IgG (FH6-7/Fc). FH6-7/Fc has previously been shown to enhance complement-dependent killing of and facilitate bacterial clearance in animal models of the gram-negative pathogens, Haemophilus influenzae and Neisseria meningitidis. We hypothesized that activation of complement by FH6-7/Fc on the surface of bacteria gram-positive bacteria such as S. pyogenes will enable professional phagocytes to eliminate the pathogen. We found that FH6-7/Fc alleviated S. pyogenes induced sepsis in a transgenic mouse model expressing human FH (S. pyogenes bind FH in a human-specific manner). Furthermore, FH6-7/Fc, which binds to Protein H and select M proteins, displaced FH from the bacterial surface, enhanced alternative pathway activation and reduced bacterial blood burden by opsonophagocytosis in a C3-dependent manner in an ex vivo human whole-blood model. In conclusion, FH-Fc chimeric proteins could serve as adjunctive treatments against multidrug-resistant bacterial infections.
Introduction
S. pyogenes also known as group A streptococcus (GAS) is a gram-positive bacterium that causes a wide spectrum of diseases. Symptoms range from mild superficial skin infections to invasive and life-threatening disease (1, 2). Worldwide more than 700 million S. pyogenes infections occur annually with at least 663,000 new cases of invasive infections (3). In order to evade the immune system, S. pyogenes binds a variety of serum proteins, including albumin, fibronectin, plasminogen, IgG, complement Factor H (FH) and C4b-binding protein (C4BP) (4–10). S. pyogenes is exclusively a human pathogen, which may in part be related to its ability to inhibit human complement by binding to the complement inhibitors FH and C4BP in a human-specific manner (4, 11, 12). Recently, we showed that recruitment of these human complement inhibitors is crucial for S. pyogenes virulence in the mouse model (13).
The complement system is a pivotal branch of the innate immunity against invading pathogens by marking them for removal following opsonization with C3b (14). Complement is activated by surface-bound antibodies and certain pathogen associated molecular patterns (PAMPs), which initiate the protein cascade to eliminate bacteria, viruses, fungi and non-self cells (15). Complement activation can occur via three pathways, namely the classical, lectin and alternative pathways. All three pathways converge at the level of the C3 convertase, resulting in C3b deposition on the target (16). In addition to C3b deposition that promotes opsonophagocytosis, complement activation also releases anaphylatoxins such as C3a and C5a to stimulate the immune system and initiate a response. The final step of complement activation results in formation of a lytic pore, called the membrane attack complex, which can directly lyse gram negative bacteria and eukaryotic cells (17). Therefore, this powerful, but potentially detrimental cascade has to be tightly regulated to prevent unwanted host cell damage (16). In addition to several surface-bound complement regulators, C4BP and FH are two of the major soluble complement inhibitors, which protect the body’s own cells from unwanted complement activation and killing (18, 19).
Many bacteria have developed evasion strategies, to prevent immune recognition. One strategy, also used by S. pyogenes as discussed above, is to recruit the host’s complement regulatory proteins FH and C4BP to the bacterial surface (20, 21). Ideally, these two complement inhibitors should exclusively protect host cells, but not microbes from complement attack (22). However, bacteria ‘hijack’ these C4BP and FH to their surface (23) to escape opsonization and persist in the host (24). For example, several bacteria including S. pyogenes (25) bind FH via domains 6–7 or 18–20 (reviewed in(26)). Since the complement inhibiting activity of FH is located in domains 1–4, FH bound to surfaces via domains 6–7 or 18–20 can effectively limit complement activation (27).
Since the identification of penicillin nearly 90 years ago (28), infectious diseases have been treated with various antibiotics. Unfortunately, more and more bacteria exhibit resistance against common antibiotics, and frequently against several antibiotic classes simultaneously (29, 30), which may herald a ‘post-antibiotic era’. New approaches for antibacterial therapies are urgently needed (31).
To achieve this, we designed a chimeric protein consisting of domains 6 and 7 of human FH, which are fused to the Fc domain of human IgG1 (FH6-7/hFc). Many pathogens recruit human FH via domains 6 and 7 to inhibit complement activation on their surfaces (32). FH6-7/hFc bound to a pathogen is expected to compete out serum FH from the microbial surface, which would permit uninhibited alternative pathway amplification. Furthermore, the IgG1 Fc domain of the chimeric protein will activate the classical pathway of complement through interaction with C1q of the C1 complex. Finally, surface-bound FH6-7/hFc also can be recognized by Fc receptors (FcγR) on professional phagocytes to initiate opsonophagocytosis. We have previously provided proof-of-concept of efficacy of FH/Fc molecules in the treatment of gram-negative bacterial infections caused by H. influenzae, N. meningitidis and N. gonorrhoeae (33–35). Here we study the efficacy of human FH-Fc chimeric proteins on gram-positive bacterial infections using the example of S. pyogenes as an example.
Material and methods
Bacteria and cell lines
S. pyogenes strains AP1 (S. pyogenes strain 40/58 serotype M1) and AP18 (strain 8/69, serotype M18), both from the WHO Collaborating Centre for Reference and Research on Streptococci, Prague, Czech Republic), MC25 (lacks M1 protein;(36)), BM27.6 (lacks protein H and M1;(37)) and BMJ71 (lacks protein H and M1, SIC, C5a peptidase;(38)) were grown in Todd-Hewitt broth medium (Oxoid) overnight at 37°C, 5% CO2 without shaking. Mutant strains were cultured in medium supplemented with 150μg/ml kanamycin (MC25), 1μg/ml erythromycin (BM27.6) or 5 μg/ml tetracycline (BMJ71). Overnight bacteria cultures were diluted to OD600=0.1 in fresh medium and grown with same conditions until exponential phase growth was reached at OD600=0.3–0.4. After centrifugation and washing with 1× PBS, bacteria were ready to use.
Freestyle CHO-S suspension cells (Life Technologies) were used for expression of fusion proteins. Cells were cultured in FreeStyle™ CHO Expression Medium (Thermo Fisher) supplemented with 8mM L-Glutamine (Hyclone) at 37°C, 8% CO2, 130 rpm shaking.
Proteins and antibodies
Protein M1, protein H and its fragments were expressed in E. coli and purified on human IgG column as described previously ((5, 37, 39)). FH was purified from human plasma. Aggregated human IgG were obtained by heat-aggregation of immunoglobulins (Immuno) at 65°C. Zymosan and cytochalasin D were purchased from Sigma-Aldrich. FH and FH6-7/mFc fusion protein were radioactively labelled using Iodobeads (Pierce) and Na-125I (PerkinElmer Life Science) according to manufacturer’s protocol. Antibodies used for flow cytometric analysis: goat anti-human C9 (CompTech), donkey f(ab)2 anti-goat IgG conjugated to Alexa Fluor 647 (Jackson ImmunoResearch), MRC OX-24 anti-human FH conjugated to Dylight 650 (40), goat anti-mouse IgG conjugated to Alexa Fluor 647 (Invitrogen), goat anti-human IgG conjugated to Alexa Fluor 647 (Invitrogen) goat anti-human IgG conjugated to Alexa Fluor 488 (Invitrogen). Antibodies used for complement deposition assay: rabbit anti-human C4c (Dako), rabbit anti-human C3d (Dako), goat anti-rabbit IgG conjugated to HRP (Dako). The following antibodies used for phagocytosis assay were purchased from BioLegend, if not stated otherwise: anti-human CD56 conjugated to APC/Cy7, anti-human CD15 conjugated to PerCP/Cy5.5, anti-human CD19 conjugated to APC/Cy7, anti-human MHCII conjugated to Alexa Fluor 647, anti-human CD11c conjugated to PE, anti-human CD3 conjugated to APC/Cy7, anti-human CD16 conjugated to PE/Cy7, anti-human CD14 conjugated to PE/Dazzle, Human TruStain FcX, mouse anti-S. pyogenes (AbD Serotec) conjugated to Alexa Fluor 488 and BV405. 10μg/ml of following antibodies were used to block Fcγ receptors (41): anti-human CD16 (Invitrogen, #16-0166-85), anti-human CD32 (LSBio, #LS-C187457) and anti-human CD64 (eBioscience #16-0649-85). Compstatin CP40, which blocks C3 activation, was prepared at the University of Pennsylvania as described previously (42). Antibodies for Western blot analysis: anti human C3a/C3a des-Arg (Hycult) and anti-mouse Ig/HRP (DAKO).
Production of chimeric FH IgG Proteins in CHO-S cells
Chimeric proteins (FH6-7/18-20hIgG1; FH6-7/18-20mIgG2a) were expressed in CHO cells and purified on Protein A/G columns as described previously (34). Briefly, floating FreeStyle™ CHO-S cells (Life Technologies) at a concentration of 1×106 cells/ml were transfected with plasmids encoding fusion protein constructs in FreeStyle™ CHO Expression Medium (Thermo Fisher). Transfected floating FreeStyle™ CHO-S cells were incubated at 37°C and 8% CO2 under shaking at 125–130 rpm for up to 12 days. Every second day cell medium containing the produced chimeric proteins was collected and stored at −20°C for subsequent protein purification. Then, cells were resuspended in fresh FreeStyle™ CHO Expression Medium for further incubation. Proteins within the collected cell supernatant were purified on a protein A/G column (GE Healthcare) at 4°C. The elution of the proteins was performed using glycine and 6M guanidinium chloride. Eluates were transferred into Spectra pore membranes for dialysis in 1× PBS at 4°C with 3 buffer exchanges over 24h. Protein concentration was determined using a Cary 50 bio photometer (Varian).
FH and FH6-7/mFc fusion protein binding to protein H
Purified proteins (protein M1, protein H, protein H fragments and α-1-antytripsin) were diluted in HEPES buffer (50 mM HEPES, 50 mM NaCl) and immobilized in microtiter plate (MaxiSorp BreakApart, NUNC) overnight at 4°C. Next day, plates were washed three times with washing buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20) and unspecific binding was blocked with 5% Difco skim milk (Becton Dickinson) diluted in wash buffer. 125I-FH and 125I-FH6-7/mFc were diluted in HEPES buffer and added in increasing amounts as indicated. After overnight incubation at 4°C, plates were washed using washing buffer and radioactivity was measured using a Wizard2 γ counter (PerkinElmer Life Science).
Flow cytometry analysis of C9 deposition on bacteria
Harvested bacteria S. pyogenes AP1 or BM27.6 were incubated with 10% normal human serum and increasing amounts of FH6-7/hFc or FH18-20/hFc, respectively, diluted in GVB++ buffer (5 mM veronal buffer pH 7.3, 140 mM NaCl, 0.1% gelatin, 1 mM MgCl2, and 5mM CaCl2) for 1h at 37°C, 5% CO2. After incubation, bacteria were washed with 0.5% BSA in 1× PBS and stained for C9 (goat anti-human C9, CompTech) at 4°C for 30 min. Subsequently after washing with 1× PBS, primary antibodies were detected with secondary anti-goat Abs conjugated to AlexaFluor-647 (Jackson ImmunoResearch). Amount of deposited C9 component bound to bacteria surface was measured using Cyflow space flow cytometer (Partec).
Complement deposition
Microtiter plates (MaxiSorp BreakApart, NUNC) were coated with FH6-7/hFc (10 μg/ml), α-1-antytripsin (10 μg/ml) and aggregated human IgG (classical pathway (CP), 5 μg/ml, Kiovig, Baxalta) or zymosan (alternative pathway (AP), 10 μg/ml, Z-4250; Sigma-Aldrich). Plates were incubated overnight at 4°C and washed next day three times with washing buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20). Between every step plates were washed thrice with washing buffer. Further unspecific binding was blocked with 5% Difco skim milk (Becton Dickinson) diluted in wash buffer (classical pathway) or quenching buffer (3% fish gelatin, 50 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20, pH 8; alternative pathway). Next plate was incubated with increasing concentration of normal human serum diluted in GVB++ (5 mM veronal buffer pH 7.3, 140 mM NaCl, 0.1% gelatin, 1 mM MgCl2, and 5 mM CaCl2; CP) or Mg++EGTA (2.5 mM veronal buffer (pH 7.3) containing 70 mM NaCl, 140mM glucose, 0.1% gelatin, 7mM MgCl2, and 10 mM EGTA; AP). Serum incubation time was 20 min for C3b, 15 min for C4b (CP) and 30 min for C3b (AP) at 37°C. After incubation, deposited components were detected with primary anti-C3d (Dako) (CP, AP) and anti-C4d (Dako) (CP) diluted in corresponding blocking buffer. Then, bound primary Abs were detected with secondary anti-rabbit HRP-conjugated Abs. Finally, samples were developed using OPD tablets (Kem-En-Tec) and signal was measured at OD490 (Cary 50 MPR microplate reader, Varian).
Hemolytic assay
For classical pathway activation, 150μl of sheep erythrocytes (Håtunalab AB; corresponding to ≈3.75×108 red blood cells) were washed three times with GVB++ buffer (5 mM veronal buffer pH 7.3, 140 mM NaCl, 0.1% gelatin, 1 mM MgCl2, and 5 mM CaCl2) and incubated with Amboceptor (Dade Behring, 1:500 in GVB++) for 20min at 37°C with 650rpm shaking. After incubation, erythrocytes were washed 3 times and resuspended in GVB++. The suspension volume was adjusted in the way that 10μl from erythrocyte stock lysed in 90μl H2O resulted in OD410= 1–1.2. For alternative pathway respectively, 150μl rabbit erythrocytes (Håtunalab AB) were washed 3 times and suspended in Mg-EGTA buffer (2.5 mM veronal buffer pH 7.3 containing 70 mM NaCl, 140 mM glucose, 0.1% gelatin, 7 mM MgCl2, and 10 mM EGTA). Then, the suspension volume was adjusted in the same way as for sheep erythrocytes. To assess hemolytic activity, sheep erythrocytes were incubated with increasing amounts of FH6-7/hFc diluted in GVB++ buffer for 20 min. After incubation, cells were washed and subsequently mixed with 0.75% NHS to allow complement activation for 10 min. Remaining erythrocytes were pelleted and hemolysis was measured at OD410 (Cary 50 MPR microplate reader, Varian). Similarly, rabbit erythrocytes were incubated with increasing amounts of FH6-7/hFc fusion protein diluted in Mg-EGTA buffer. Subsequently after washing and mixing with 6% NHS, complement was allowed to be activated on erythrocytes for 15 min. Finally, erythrocytes were centrifuged and released hemoglobin was measured at OD410.
All incubations were performed at 37°C and 650 rpm shaking (Thermomixer, Eppendorf). Hemolytic assay using anti-CD59 treated human erythrocytes has been performed as described previously (35).
Binding assay on bacteria using FACS
S. pyogenes strains AP1, MC25, BM27.6, BMJ71 (1×107 CFU) in GVB++ were coincubated with indicated concentrations of chimeric FH proteins in the presence of 10% FCS at 37°C, 5% CO2 for one hour. Bacteria were harvested and washed with 1× PBS. Subsequently, bacteria were stained with Cell Trace Calcein Violet (Molecular Probes) and stained with goat anti-human IgG-Alexa Fluor 488 (Invitrogen; to detect FH-hIgG1 proteins) or goat anti-mouse IgG-Alexa Fluor 647 (Invitrogen; to detect FH-mIgG2a proteins) for 30 minutes. Thereafter, bacteria were washed twice with 1× PBS and resuspended in 1x PBS. Binding of fusion proteins to bacterial surface was measured using cyflow space flow cytometer (Partec) and geometric mean fluorescence intensity (gMFI) was calculated with FlowJo software (Tree Star). To assess serum FH binding, bacteria were similar treated, however incubated in 10% NHS and stained with MRC OX-24 anti-human FH conjugated to Dylight 650. To identify bacteria, cell trace violet positive events were gated and further analyzed.
Whole blood killing assay
FH6-7/hFc was added to lepirudin (Refludan; Celgene) treated human blood in different concentrations before infection with indicated amounts of different S. pyogenes strains. Complement activation was blocked at the level of C3 with 20μM compstatin CP40. Fcγ receptors were blocked with a combination of 10μg/ml each of anti-human CD16, CD32 and CD64 that were added to whole blood and incubated for 20 min at 37 °C before adding bacteria. Following the addition of bacteria, the blood was incubated on an end-over end shaker at 37°C, 5% CO2. At indicated time points, 50μl of the mixture was diluted serially in TBS and plated on blood agar plates. Blood agar plates were incubated over night at 37 °C and 5% CO2 in order to enumerate surviving S. pyogenes.
Phagocytosis assay using Amnis Flowsight
Harvested bacteria were stained with CFSE and subsequently sub-cultured for 2 hours to reduce the CFSE intensity per bacteria. Human blood (200μl) was infected with 1×107 bacteria and incubated for 30 minutes at 37°C and 5% CO2. Next, blood samples were treated with human Trustain Fcx to block Fc receptors for 10 minutes on ice prior to addition of antibodies. After 30 min incubation, red blood cells were lysed using RBC lysis/Fixation solution (Biolegend) with subsequent incubation for 15 minutes at room temperature. After washing with PBS, cells were analyzed using Amnis Flowsight.
Surface plasmon resonance analysis
The affinity between protein H and FH6-7/hFc was analyzed by surface plasmon resonance using a Biacore 2000 (GE Healthcare). FH-IgG (CompTech) was immobilized on CM5 sensor chip (GE Healthcare) using standard amino coupling reaction to reach 2500 resonance units (RU). Protein H diluted to 0.07–25 nM concentrations in running buffer (50 mM HEPES, pH 7.8 containing 100 mM NaCl and 0.005 % Tween-20) was injected at flow of 30 μL/sec for 100 sec. and the dissociation was then followed for 300 sec. The signal from the control surface was subtracted. In all experiments, two consecutive injections of 2 M NaCl, 100 mM HCl were used to remove bound ligands during a regeneration step. The BiaEvaluation 3.0 software (Biacore) was used to determine affinity constants using Langmuir 1:1 interaction model with drifting baseline. The experiment was performed two independent times.
Platelet aggregation assay
Platelet aggregation was analyzed from three independent donors, as recommended by the International Society on Thrombosis and Haemostasis (43). Briefly, plasma was purified from healthy volunteers and platelet-rich (PRP) and platelet-poor plasma (PPP) was prepared. After mixing PPP and PRP together with 2μM ADP (positive control), FH6-7/FH or PBS (negative control), samples were analyzed in a light transmission aggregometer. Additionally, one sample was left untreated before analysis (spontaneous aggregation, additional negative control). Optical density was measured and plotted as percentage. Area under curve analysis revealed the total amount of platelet aggregation.
C3a Western blot
Lepirudin treated plasma was incubated either with 2mg/ml zymosan particles, PBS or 50μg/ml FH6-7/hFc for 1h at 37°C with shaking. Particles were removed and supernatants were applied on a 15% SDS-polyacrylamide gel. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane and subsequently incubated with mouse anti-human C3a followed by an incubation with goat anti-mouse Ig-HRP. The C3a signal was visualized using ECL(Millipore) and detection with a CCD camera (Chemi-Doc, Biorad). Due to a very strong signal in the positive control in the initial experiment, the zymosan-treated sample was diluted 10-fold prior to electrophoresis in the experiment shown.
Animal survival studies
All animals were housed and bred under SPF conditions in the animal facility at the University of Massachusetts Medical School Worcester, USA.
Two hours prior to infection, female and male animals were treated either with 50 μg FH6-7/hFc or 50μg goat IgG intraperitoneally. Subsequently, via lateral tail vein injection animals were infected intravenously with 100 μl bacterial suspensions in PBS containing 1.4×108/ml S. pyogenes AP1. Injections were repeated on day 2, 4 and 6 (50μg protein/animal). All animals were closely monitored for signs of disease for up to eight days; gravely moribund mice were euthanized.
Ethical statement
Use of animals in this study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the Swedish Animal Welfare Act SFS1988:534. Experiments were approved by the Institutional Animal Care and Use Committee at the University of Massachusetts Medical School, Worcester, MA, USA.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0 and 7.0b software. To test for significance, we used either a 1-way or 2-way ANOVA analysis with Bonferroni’s, Dunnets or Tukeys post-test or a Mantel-Cox test as indicated. P<0.05 was considered to be significant.
Results
We analyzed binding of Factor H domains 6 and 7 coupled to human IgG1 Fc (FH6-7/hFc; Fig.1A) to Streptococcus pyogenes AP1 and its isogenic mutants MC25 (lacks the M protein), BM27.6 (lacks protein H) and BMJ71 (lacks both M protein and protein H). FH6-7/hFc bound similarly to the parental wild type strain AP1 and MC25 (Fig.1B). Binding to the mutant BM27.6, as measured by fluorescence intensity, was diminished almost 2-fold compared to the wild-type strain. Bacteria lacking both surface virulence factors, M/H proteins, BMJ71 showed binding barely above background levels. These results indicate that FH6-7/hFc binds to both M protein and protein H on S. pyogenes. Prior work has suggested S. pyogenes also binds FH through domains18–20 (44, 45). We observed binding of FH18-20/hFc to the wild type AP1, and to a lesser extent to MC25 (Fig.1C). Both, BM27.6 and BMJ71 did not bind FH18-20/hFc, suggesting that M protein and protein H may both contribute to binding of the C-terminal domains of FH.
Figure 1. Factor H domains 6–7 but not 18–20 bind to S. pyogenes AP1 proteins H and M.
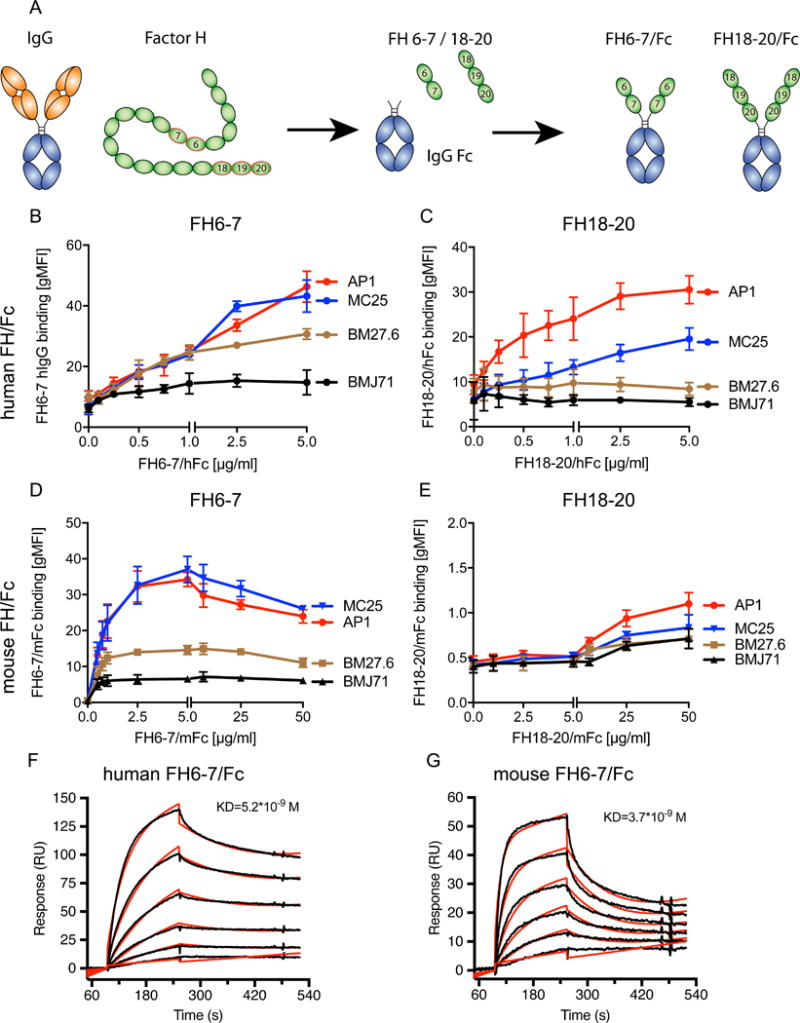
(A) Schematic representation of FH6-7- and FH18-20/Fc. Human FH domains were fused to Fc of either human IgG1 or mouse IgG2a. Binding of the different fusion proteins to S. pyogenes AP1 and isogenic mutants lacking M protein (MC25), protein H (BM27.6) or both M protein and protein H (BMJ71) was tested using flow cytometry. (B) Binding of FH6-7/hFc to S. pyogenes AP1 and isogenic mutants lacking either protein H and/or M protein. (C) FH18-20/hFc binds only to bacteria expressing protein H (AP1 and MC25) but not the two mutants lacking protein H (BM27.6 and BMJ71). (D) FH6-7/mFc binds predominantly to AP1 and MC25, both expressing protein H. (E) FH18-20/mFc does not bind to any of the tested bacteria. Biacore analysis revealed very strong binding between protein H and FH6-7/hFc or FH6-7/mFc with KD=5.2*10−9M (F) and KD=3.7*10−9M (G) respectively. Mean (±SD) from at least 3 independent experiments are shown (B–E).
Since S. pyogenes is known to bind human, but not mouse IgG-Fc (39), we employed FH6-7 and FH18-20 fused to mouse IgG2a to determine if binding of FH6-7/hFc to AP1 is mediated mainly through FH or through human IgG-Fc domains of the fusion proteins tested. As seen with FH6-7/hFc, AP1 and MC25 bound FH6-7/mFc well, whereas BM27.6 and BMJ71 bound reduced amounts or no FH6-7/mFc, respectively, indicating that binding of FH6-7/hFc occurred through the FH domains (Fig.1D). However, we barely detected any binding of FH18-20/mFc to the bacteria (Fig.1E). These results indicate that FH18-20/hFc bound almost exclusively through the human IgG-Fc part of the protein, while FH6-7 presumably binds via both domains: FH6-7 as well as hIgG1-Fc. Using surface plasmon resonance analysis, we estimated the binding affinity of protein H to FH6-7/hFc to be KD=5.2*10−9M (Fig. 1F) and FH6-7/mFc to be KD=3.7*10−9M (Fig.1G), indicating a strong interaction for both chimeric proteins.
FH6-7/hFc competes out serum FH from the bacterial surface and increases complement attack
Next, we tested whether FH fusion proteins were able to compete out serum FH on the surface of S. pyogenes. Therefore, we incubated the bacteria in 5% normal human serum (NHS) that contained increasing concentrations of FH6-7/hFc (Fig.2A). Even 2.5μg/ml FH6-7/hFc was sufficient to completely block FH binding to the bacterial surface of AP1 and MC25. BM27.6 and BMJ71 did not bind any FH from serum. In contrast, FH18-20/hFc did not displace any serum FH from AP1 or MC25 (Fig.2B). Even high concentrations (150μg/ml) of FH18-20/hFc had no effect on serum FH binding (data not shown). These data provide further evidence for binding of FH6-7/hFc via the FH domains, in contrast to binding of FH18-20/hFc via the Fc domain.
Figure 2. FH6-7/hFc competes out serum FH and activates complement.
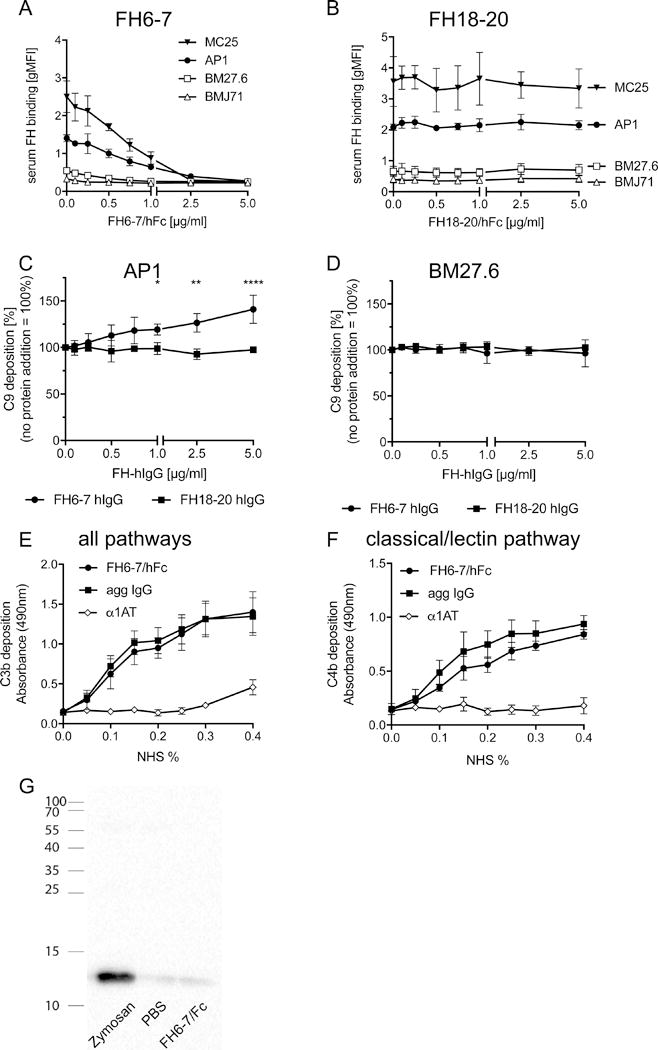
Bacteria were incubated in serum in increasing amounts of FH6-7/hFc (A) or FH18-20/Fc (B) and analyzed for binding of serum FH. (C) Flow cytometry was used to measure surface bound C9, the final step of complement activation. FH18-20 did not increase C9 on AP1. (D) Complement C9 deposition was not affected by FH6-7/hFc or FH18-20/hFc on BM27.6 that does not bind either molecule. FH6-7/Fc immobilized on microtiter wells activates the classical and lectin pathways as measured by C3b (E) and C4b (F) deposition. (G) Plasma was incubated with FH6-7/hFc, PBS or zymosan and analyzed for C3a generation as a sign of complement activation. Mean (±SD) from at least 3 independent experiments are shown. Statistical significance of differences was calculated using a 2-way ANOVA with Dunnett’s post-test (overall p value in C p> 0.0001); *, p<0.05; **, p<0.01 and ****, p<0.0001.
We hypothesized that FH-IgG binding to S. pyogenes would increase complement attack through enhanced classical pathway activation and opsonisation by Fc part of the chimeric protein, and also by preventing FH mediated inhibition of complement activation. To confirm our hypothesis, we analyzed C9 deposition on S. pyogenes AP1 from 10% NHS in the presence of increasing amounts of the chimeric proteins; multiple C9 molecules participate in the formation of membrane attack complex (MAC), which is the final stage in complement activation. We found that FH6-7 significantly increased C9 deposition on AP1 compared to NHS alone (Fig.2C). In contrast, FH18-20/hFc did not increase C9 deposition on the bacterial surface. As expected, neither fusion protein had any impact on BM27.6 (Fig. 2D), since this mutant does not bind any FH domain(s). Consistent with uninhibited complement activation on its surface, high MAC formation occurred on the surface of BM27.6 and was not altered by addition of a fusion protein. To determine if the Fc portion of FH6-7/hFc could activate complement, we coated microtiter plates with the fusion protein, aggregated IgG as a positive control and α1AT as a negative protein control. Indeed, FH6-7/hFc increased both C3b (Fig.2E) as well as C4b deposition (Fig.2F) on the plate similar to aggregated IgG, indicating that FH6-7/hFc activates the alternative and classical pathway. To address the possibility that complement activation in solution may have contributed to complement component deposition on ‘bystander’ bacteria, we incubated plasma for 60 minutes with either FH6-7/hFc, PBS or zymosan particles and measured C3a generation as a measure of complement activation. Plasma samples were subjected to western blot analysis for C3a (Fig.2G). Zymosan strongly activated complement resulting in high amounts of C3a production, while PBS and FH6-7/FH showed similar, but very weak signals. Taken together, FH6-7/hFc activates complement only when bound to the bacterial surface, but not when in solution.
FH binding site on S. pyogenes
Initial binding experiments indicated that protein H and M protein were both ligands for FH6-7 (Fig.1B). To exclude the influence of human IgG-Fc, we used 125I-labelled FH6-7/mFc to test binding of the FH domains to protein H, M1 and α1AT (Fig.3A). FH6-7/mFc bound in a dose dependent manner to protein H, slightly less to M1 but not to the negative control, α1AT.
Figure 3. Identification of binding sites for FH on S. pyogenes AP1.
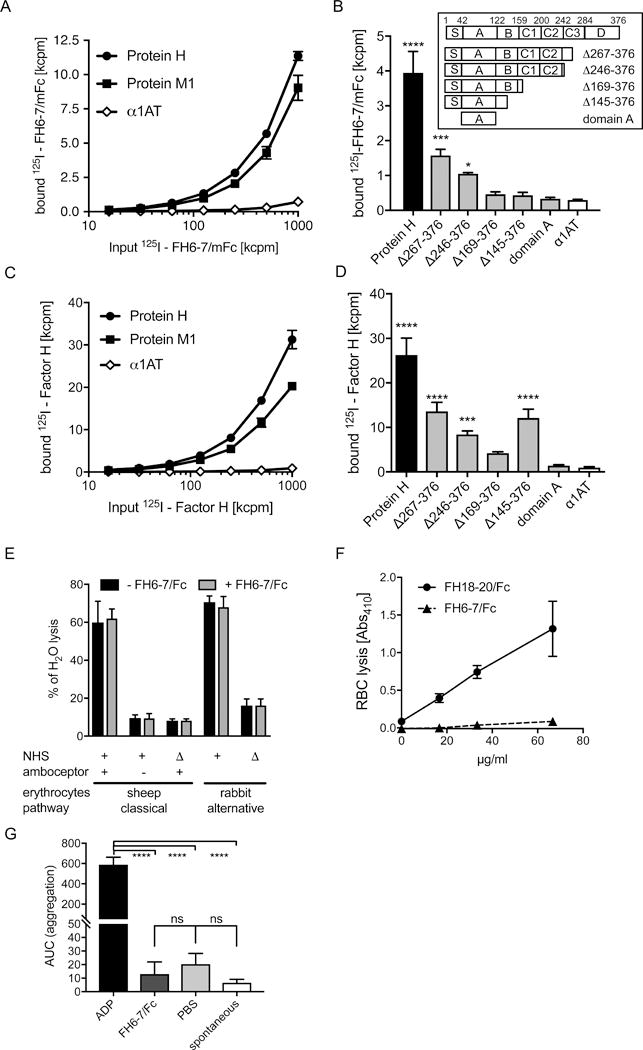
(A) Both protein H and M bind FH6-7/mFc. Increasing amounts of 125I-FH6-7/mFc were incubated with immobilized protein M1, protein H and as a α1AT (negative control). (B) The binding site for FH domains 6–7 is mainly located in domains C3 and D of protein H (see insert). Immobilized full length protein H as well as the indicated fragments of protein H were incubated with 125I-FH6-7/hFc, (C) Full-length FH binds to M protein and protein H. 125I-FH was tested for binding to immobilized proteins H and M1. (D) Full-length FH also binds to protein H via domains C3 and D. FH6-7/. Statistical significance of differences was calculated using a 1-way ANOVA with Dunnets post-test (overall p value in B and D p<0.0001) or Bonferonni’s post-test (overall p value in G p<0.0001); *, p<0.05; **, p<0.01; ***, p<0.001 and ****, p<0.0001.
Since we identified protein H as one of the major ligands for FH6-7/hFc, we aimed to identify the binding site on protein H for FH domains 6 and 7 (Fig.3B). We found, that full length protein H bound most FH6-7/mFc, while protein H fragments ∆267–376 and ∆246–376 (see Fig.3B insert) still bound significantly more FH6-7/mFc than the irrelevant protein control α1AT, yet markedly decreased compared to full length protein H (Fig.3B). Thus, we concluded that the binding site for FH6-7/mFc is located in domains C (mainly C3) and D of protein H.
Similarly, we used 125I-radiolabelled Factor H to compare binding to M1 and protein H. Similar to FH6-7/mFc we found that FH binds both protein H as well as M protein (Fig.3C). Using the protein H fragments, we detected significant binding of 125I-FH binding to full length protein H and fragments ∆267–376 and ∆246–376 as well as ∆145-376. This binding pattern suggests that full length FH binds to domains C and D, as well as domain B of protein H (Fig.3D).
FH6-7 does not affect hemolysis or platelet aggregation
Unwanted displacement of FH from tissues and cells could be detrimental for the body. Therefore, we asked whether FH6-7/hFc affected hemolysis of erythrocytes. To determine whether FH6-7/hFc augmented classical pathway activation, we sensitized sheep erythrocytes with antibodies and subsequently incubated these with FH6-7/hFc. Before addition of serum, cells were washed to assess function of only erythrocyte-bound fusion protein. We did not detect any difference in hemolytic activity at any tested FH6-7/hFc concentration. Similarly, we used non-sensitized sheep erythrocytes to identify if FH6-7/hFc could directly sensitize the erythrocytes. Again, there was no increase in hemolysis compared to background hemolysis (Fig.3E, left side). These data show that FH6-7/hFc had no effect on classical pathway-mediated hemolysis. To analyze the effect of the fusion protein on the alternative pathway, we used rabbit erythrocytes, which were incubated with increasing amounts of FH6-7/hFc. Erythrocytes were washed to remove unbound FH6-7/hFc. As expected, the fusion protein had no influence on the hemolysis of rabbit erythrocytes suggesting that it does not compete with FH in this experimental system (Fig.3E right side).
FH is the major protective molecule on human erythrocytes where CD59 function is blocked, for example with α-CD59 antibodies (46). FH6-7/hFc did not induce hemolysis of α-CD59 treated human RBCs (Fig.3F) at any of the tested concentrations. In contrast, FH18-20/Fc, which is known to induce hemolysis served as a positive control (35). Taken together our experiments show that FH6-7/hFc does not cause complement-dependent lysis of erythrocytes.
To verify that FH6-7/hFc does not affect coagulation, we performed a platelet aggregation assay (Fig.3G). Serum from 3 healthy individuals was analyzed by either adding FH6-7/hFc, PBS as a negative control or ADP as a positive control. A fourth untreated sample was used to measure spontaneous activation. A comparison of the area under the curves of the different reaction mixtures revealed that FH6-7/hFc did not activate platelets beyond levels seen with PBS and ‘spontaneous activation’ tubes.
Bacterial uptake in human blood increases upon addition of FH6-7/hFc
We infected human blood from 5 healthy individuals ex vivo with S. pyogenes AP1 and subsequently stained for professional phagocytes to measure uptake of bacteria. Using Amnis flowsight technology, we discriminated between bacterial uptake and adhesion (Fig.4a). We excluded all cells with extracellular bacteria (Fig.4a ‘excluded cells’ #78784 + #81540) and analyzed then CFSE signal in the PMN population. We observed PMNS without phagocytosed bacteria (Fig.4a #8074 + #33332) as well as PMNS that phagocytosed 1 (#16415), 2 (#40855), 3 (#83010) or more than 3 (#84420) bacteria, judged by the CFSE signal originating from the bacteria.
Figure 4. PMNs show increased uptake of bacteria in the presence of FH6-7/hFc.
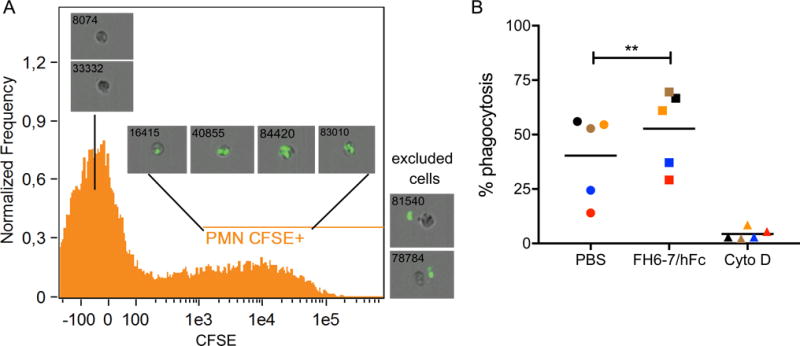
Non-opsonized S. pyogenes AP1 was incubated in human blood in the presence and absence of FH6-7/hFc. Leukocytes were stained and analyzed using Amnis flowsight technology. PMNs with ingested bacteria were discriminated from extracellular bacteria (A). FH6-7/hFc significantly increased phagocytosis rate of bacteria (B). PMNs treated with cytochalasin D served as a negative control. Mean from 5 individual donors (indicated by color) are shown. Statistical significance of differences was calculated using a matched 1-way ANOVA with Tukey’s post-test (overall p value in B p<0.0063); **, p<0.01.
40.4% of all neutrophils in blood ingested S. pyogenes in the absence of FH6-7/hFc (Fig.4B, individual donors color matched). Upon addition of the fusion protein, phagocytosis rate increased in the individual samples between 6.4% and 16.4% to 52% in average. Addition of cytochalasin D completely blocked phagocytosis and confirmed that we had measured bacterial uptake and not merely adhesion to neutrophils.
FH6-7/hFc prevents bacterial growth in blood
To assess the effect of FH6-7/hFc on bacteria during sepsis, we infected human blood with S. pyogenes AP1 in the presence or absence of FH6-7/hFc. Bacterial growth was analyzed over a period of 3h (Fig.5a). In the presence of 50μg/ml FH6-7/hFc (red bars), bacterial burden was reduced by more than 60% within the first 60 minutes. Over the next two hours the bacterial numbers remained stable (static) while untreated bacteria replicated impressively; after 3 hours, the difference between the untreated sample and the samples treated with 50μg/ml FH6-7/hFc was more than 1.5log10. Treatment with 10μg/ml FH6-7/hFc (grey bars) showed significantly reduced bacterial burden compared to untreated controls, but the decrease was not as pronounced as in samples with 50μg/ml (red bars). In fact, almost 1 log10 bacterial growth occurred over 3 h in the presence of the ‘low dose’. Similarly, blood infected with S. pyogenes AP18 and treated with FH6-7/hFc showed similar reduction in bacterial burden over time. The effect was even more pronounced using low level (10 μg/ml) FH6-7/hFc. The difference between treated and untreated blood was about 2 log10 bacteria/ml at 2 or 3 h post addition of bacteria (Fig.5B). As expected, bacteria unable to bind FH6-7/hFc were not affected and CFU counts were similar in treated and untreated samples at all time points (Fig.5C–D) This shows, that FH6-7/hFc is effective only against bacteria which bind FH via domains 6–7.
Figure 5. Complement is important for FH6-7/hFc to reduce bacterial blood burden.
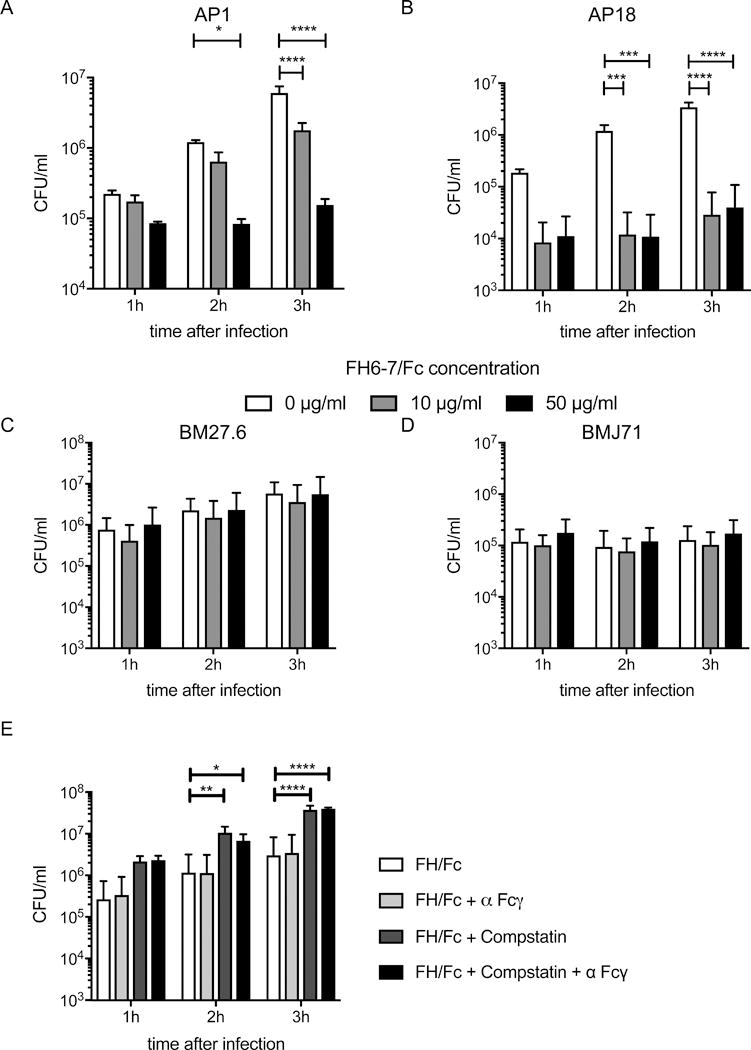
Human blood was infected with 2 × 105 CFU/ml S. pyogenes AP1(A) or AP18 (B), 2 × 106 CFU/ml S. pyogenes BM27.6 (C) or 2 × 107 CFU/ml S. pyogenes BMJ71 (D) treated with indicated amounts of FH6-7/hFc and incubated for up to 3h. FH6-7/hFc significantly reduces the number of bacteria compared to untreated samples for both bacterial strains binding the fusion protein (AP1(A) and AP18 (B)). FH6-7/hFc had no influence on S. pyogenes BM27.6 (C) or BMJ71 (D), since both strains do not bind the fusion proteins. (E) Human blood treated with 20μg/ml FH6-7Fc was infected with 2 × 105 CFU/ml S. pyogenes AP1. Prior to infection, samples were pretreated with compstatin and/or α-FcγRs as indicated. Mean (±SD) from at least 3 independent experiments are shown. Statistical significance of differences was calculated using 2-way ANOVA with Dunnett’s post-test (overall p value in A, B and E p<0.0001, C p=0.8247 and D p=0.4248) *, p<0.05; **, p<0.01; ***, p<0.001 and ****, p<0.0001.
Complement activation is required for opsonophagocytic killing by FH6-7/hFc
We aimed to delineate the mechanism whereby FH6-7/hFc increases uptake and killing of group A streptococci. Since phagocytosis is mainly mediated either via Fcγ receptors or complement opsonization, we performed a blood killing assay in the presence of FH6-7/hFc where Fcγ receptor (FcγR), complement C3 activation, or both were blocked (Fig.5E). Blocking opsonization with C3 fragments using compstatin led to a significant increase of bacterial survival (~1 log10 decrease in CFU at each time point tested). Surprisingly, blocking Fcγ receptors alone did not change bacterial blood burden, suggesting that complement activation was essential for FH6-7/hFc-mediated opsonophagocytic killing in whole human blood.
FH6-7/hFc alleviates experimental S. pyogenes sepsis in mice
To show in vivo efficiency of the fusion protein, we infected human FH tg animals with 1.4×107 S. pyogenes AP1. S. pyogenes binds only human, but not mouse FH. Therefore, use of human FH tg mice provided a complement-inhibiting barrier that FH6-7/hFc may encounter in humans. Animals treated with 50μg FH6-7/hFc every other day showed significantly reduced mortality compared to the control group receiving control goat IgG (Fig.6). Four out of 10 animals treated with the fusion protein survived the infection, while all animals receiving control goat IgG died within 5 days. These data provide evidence that FH6-7/hFc reduces bacterial burden in blood and increases survival of S. pyogenes infected animals. Taken together, our data suggests that FH6-7/hFc blocks FH binding to bacteria, leading to increased complement deposition, enhanced phagocytosis and impaired bacterial survival.
Figure 6. FH6-7/hFc increases survival in experimental S. pyogenes sepsis.
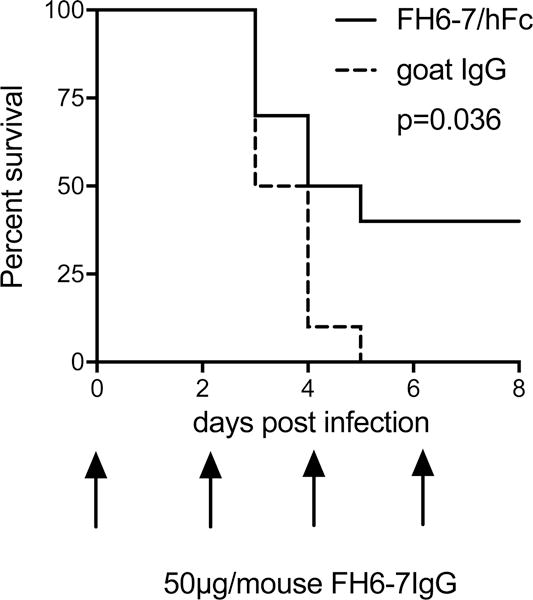
FH6-7/hFc treated human FH transgenic mice show reduced mortality in an experimental i.v. S. pyogenes sepsis model. Human FH transgenic mice (n=10/group) were treated with either FH6-7/hFc or goat IgG (control that does not bind S. pyogenes) i.p. with the dosing schedule shown on the X-axis. Statistical significance of differences was calculated using a Mantel-Cox analysis.
Discussion
Here we demonstrate efficacy of a FH-IgG fusion protein against Streptococcus pyogenes infections, thus expanding their utility against Gram-positive bacteria that bind FH. FH-Fc chimeric proteins could be further developed as a new class of antibacterial proteins, which activate complement in order to opsonize their targets for elimination by phagocytes or, in case of gram-negative bacteria, directly lyse them by MAC.
FH6-7/hFc has already been proven efficacious against Neisseria meningitidis (34), and Haemophilus influenzae (33), while a derivative of FH18-20/Fc that contains a D to G mutation at position 1119 in domain 19 was effective against Neisseria gonorrhoeae (35). Of note, all 3 bacterial species belong to the group of Gram-negative bacteria, which are generally susceptible to complement mediated lysis (17, 47). To achieve MAC-mediated lysis of Gram-negative bacteria, the chimeric FH-Fc needs to bind to bacteria in order to displace FH from the bacterial surface and or activate the classical pathway through Fc. However, complement alone cannot directly lyse Gram-positive bacteria due to their thick cell wall (48). Instead complement-mediated opsonisation of the bacteria with iC3b is crucial for efficient phagocytosis. We showed that the chimeric protein bound to the bacterial surface and competed out FH, increased complement activation, led to a significant increase of phagocytosis by PMNs, reduced bacterial burden in human blood ex vivo and significantly improved survival of mice in a S. pyogenes sepsis model.
Many pathogens have developed the ability to recruit complement inhibitors such as FH and C4BP to their surface to evade complement (reviewed in(20, 32)). FH and C4BP tightly regulate complement activation by accelerating the decay of the C3 convertase and serving as co-factors for factor I-mediated inactivation of C3b (49–52). Complement activation is dampened on pathogens ‘coated’ with these inhibitors and thus are protected from opsonophagocytosis (44). Blocking the binding of complement inhibitors renders the bacteria again susceptible to complement and would allow the immune system to eliminate the invaders. FH-IgG chimeric protein act by competing out serum FH bound to those bacteria and also activate the classical pathway through its Fc region (33–35).
Similar to at least 10 other bacterial species (26), most strains of S. pyogenes recruit serum FH through domains 6 and 7 to their surface in order to protect themselves from complement attack (53, 54). The regions in FH that interact with microbes (domains 5–7 and 19–20) are distinct from the domains responsible for complement inhibition (domains 1–4). Thus, FH6-7/hFc lacks any complement inhibiting activity, prevents binding of functional host FH to the pathogen and also activates complement through Fc. Given that several pathogens bind FH through similar region in FH, it raises the possibility that FH6-7/hFc may possess activity against several other microbes (32).
To our surprise, we found that in addition to protein H, the M1 protein from S. pyogenes AP1 also bound FH6-7-IgG. Protein H, but not the M1 protein, has been described to bind FH (13, 55, 56). Interestingly, FH seems to have more than one binding site: domains C and D, similar to FH6-7/Fc and presumably the N-terminal part of domain B. Strain AP1 used in this study expresses both protein H and M1, while the previous study employed a different M1 expressing strain, S. pyogenes SF370. It is possible that differences in the M proteins between the strains may account for differences in FH binding to these S. pyogenes strains.
S. pyogenes is known to bind human, but not mouse, IgG (39, 57). Thus, we used FH/Fc constructs that contained mouse IgG-Fc in this study to show that binding of FH6-7/hFc occurred specifically via the FH domains. In contrast FH18-20/hFc bound S. pyogenes exclusively through human IgG domains, thus explaining why FH18-20 did not compete out serum FH and did not enhance complement activation on the bacteria. While we cannot exclude the possibility that a proportion of FH6-7/hFc may also bind to the bacteria via the human IgG part, we have shown that even a low amount (≈50nM) of FH6-7/hFc is able to effectively compete out FH completely.
Another advantage of FH6-7/hFc is that drug resistance, if it were to occur, would lead to a loss of binding of endogenous FH and FH like protein 1 (FHL-1) that both possess complement inhibiting function and thereby protect the bacteria. Loss of FH/FHL-1 binding would lead to a considerable decrease of bacterial fitness in vivo because the resulting increase in C3 fragment deposition would lead to bacterial clearance by phagocytes (13, 53).
In comparison to Gram-negative organisms, Gram-positive bacteria cannot be lysed through complement mediated pore formation, although MAC can still be detected on their surface (58). Although the role of MAC associated with Gram-positive bacteria is unclear, a recent study showed that MAC can be transferred from the surface of an opsonized particle to the macrophage plasma membrane and initiate the NLRP3 inflammasome resulting in caspase-1 activation and IL-1β and IL-18 secretion (59).
Therefore, the mode of action of FH6-7/hFc on S. pyogenes must differ from the previous described mechanisms for Neisseria and Haemophilus. If bacteria cannot be directly lysed by complement, professional phagocytes must clear the microbes employing either extracellular killing mechanisms or intracellular degradation after phagocytosis. We showed that FH/Fc increased opsonisation and following opsonophagocytosis. This was presumably caused on the one hand by displacement of FH and concomitant loss of protection from complement deposition and on the other hand the additional activation of the classical complement pathway through the IgG1-Fc domains of our chimeric protein (60). In addition to C3b and iC3b, the Fc domain of the IgG1 backbone engages FcγR on professional phagocytes (61). FcγR engagement alone does not mediate killing of S. pyogenes, while complement driven opsonization is the most important pathway to clear the bacteria (62). Using compstatin CP40 we confirmed this result in untreated human full blood as well as in the presence of FH6-7/Fc despite FH6-7/Fc binding to bacteria via FH domains and presenting the hIgG1-Fc, blocking.
Fcγ receptors (CD16, 32 and 64) had no influence on S. pyogenes killing. Compstatin in turn promoted bacterial survival by blocking complement activation at the level of C3. Of note, although compstatin prevents C3b deposition on the bacteria, at the same time it allows for increased C4b deposition (63), which may serve as an opsonin by binding CR1 (64). The potential relevance of this enhanced C4b deposition is still not clear. Non-complement bactericidal effects of serum, such as transferrin, antimicrobial peptides and enzymes also contribute to bacterial killing. Further work is needed to delineate the exact mechanism of action of FH6-7/Fc on gram positive bacteria. Taken together, the FH6-7/hFc mediates opsonophagocytosis through 2 mechanisms: i) activating complement via C1q-IgG1 interaction and ii) blocking FH-dependent complement inhibition (see Fig 2 in (26)).
It is urgent to develop novel approaches to cope with antibiotic resistant bacterial infections, which pose a major threat to human health globally. The Centers for Disease Control as well as the WHO have listed 15 pathogens as concerning, serious or urgent threats which need close monitoring, preventive activities or even urgent and aggressive actions to counteract their antibiotic resistance threat (29, 65). Among those 15 different bacterial species, 9 are known to bind FH, of which at least 7 employ domains 5–7 for binding (26, 32). Since FH6-7/hFc especially targets those binding sites, we believe that the chimeric protein could be a valuable adjunctive immunotherapeutic against antibiotic resistant bacterial pathogens. While FH/Fc fusion proteins have shown promise in pre-clinical models, we acknowledge that further work needs to be undertaken to establish its safety and stability for use in humans.
Figure 7. Graphical summary mechanism of action of FH-IgG against S. pyogenes.
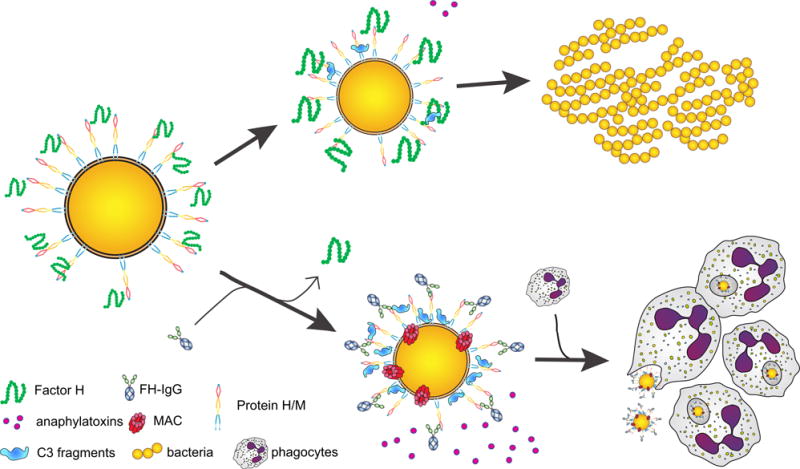
Bacteria recruit complement inhibitors to their surface and thus dampen complement activation. There is also less anaphylatoxin release, which combined with low C3 fragment deposition on the bacteria, does not adequately recruit or engage phagocytes, which results in bacterial replication and disease (upper panel). FH-IgG however diverts full-length, functional FH from the bacterial surface. The resulting complement activation releases abundant amounts of anaphylatoxins that attract professional phagocytes, which engage and engulf bacteria through surface-bound C3 fragments (lower panel). The Fc part of the molecule also activates complement and engages phagocytes via Fcγ receptors, which results in downstream signaling and bacterial killing in phagolysosomes.
Acknowledgments
We thank Nancy Nowak and Bo Zhang for expert help with breeding and caring for mice. Michal Magda is a student of Biotechnology at University of Rzeszow, Poland.
Grant Support: Swedish Research Council (2016-01142), the Swedish Government Funds for Clinical research (ALF), The Torsten Söderberg Foundation, Royal Physiografic Society of Lund and Foundations of Crafoord, Knut and Alice Wallenberg, Lars Hierta Memorial, Österlund, Tore Nilsson, Gustav V 80-years anniversary and grants AI114790, AI118161, AI132296, AI129106 (to S.R.) and AI 068730 (to J.D.L.) from the National Institutes of Health/National Institutes for Allergy and Infectious Diseases.
Abbreviations
- C4BP
C4b binding protein
- FH
Factor H
- mFc
mouse Fc
- hFc
human Fc
References
- 1.Bisno AL, Stevens DL. Streptococcal infections of skin and soft tissues. N Engl J Med. 1996;334:240–245. doi: 10.1056/NEJM199601253340407. [DOI] [PubMed] [Google Scholar]
- 2.Nowak R. Flesh-eating bacteria: not new, but still worrisome. Science. 1994;264:1665. doi: 10.1126/science.8209244. [DOI] [PubMed] [Google Scholar]
- 3.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 4.Horstmann RD, Sievertsen HJ, Knobloch J, Fischetti VA. Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc Natl Acad Sci U S A. 1988;85:1657–1661. doi: 10.1073/pnas.85.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frick IM, Akesson P, Cooney J, Sjobring U, Schmidt KH, Gomi H, Hattori S, Tagawa C, Kishimoto F, Bjorck L. Protein H–a surface protein of Streptococcus pyogenes with separate binding sites for IgG and albumin. Mol Microbiol. 1994;12:143–151. doi: 10.1111/j.1365-2958.1994.tb01003.x. [DOI] [PubMed] [Google Scholar]
- 6.Frick IM, Crossin KL, Edelman GM, Bjorck L. Protein H–a bacterial surface protein with affinity for both immunoglobulin and fibronectin type III domains. Embo J. 1995;14:1674–1679. doi: 10.1002/j.1460-2075.1995.tb07156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ermert D, Weckel A, Agarwal V, Frick IM, Bjorck L, Blom AM. Binding of complement inhibitor C4b-binding protein to a highly virulent Streptococcus pyogenes M1 strain is mediated by protein H and enhances adhesion to and invasion of endothelial cells. J Biol Chem. 2013;288:32172–32183. doi: 10.1074/jbc.M113.502955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thern A, Stenberg L, Dahlback B, Lindahl G. Ig-binding surface proteins of Streptococcus pyogenes also bind human C4b-binding protein (C4BP), a regulatory component of the complement system. J Immunol. 1995;154:375–386. [PubMed] [Google Scholar]
- 9.Carlsson F, Sandin C, Lindahl G. Human fibrinogen bound to Streptococcus pyogenes M protein inhibits complement deposition via the classical pathway. Mol Microbiol. 2005;56:28–39. doi: 10.1111/j.1365-2958.2005.04527.x. [DOI] [PubMed] [Google Scholar]
- 10.Ly D, Taylor JM, Tsatsaronis JA, Monteleone MM, Skora AS, Donald CA, Maddocks T, Nizet V, West NP, Ranson M, Walker MJ, McArthur JD, Sanderson-Smith ML. Plasmin(ogen) acquisition by group A Streptococcus protects against C3b-mediated neutrophil killing. J Innate Immun. 2014;6:240–250. doi: 10.1159/000353754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlsson F, Berggard K, Stalhammar-Carlemalm M, Lindahl G. Evasion of phagocytosis through cooperation between two ligand-binding regions in Streptococcus pyogenes M protein. J Exp Med. 2003;198:1057–1068. doi: 10.1084/jem.20030543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnsson E, Thern A, Dahlback B, Heden LO, Wikstrom M, Lindahl G. A highly variable region in members of the streptococcal M protein family binds the human complement regulator C4BP. J Immunol. 1996;157:3021–3029. [PubMed] [Google Scholar]
- 13.Ermert D, Shaughnessy J, Joeris T, Kaplan J, Pang CJ, Kurt-Jones EA, Rice PA, Ram S, Blom AM. Virulence of Group A Streptococci Is Enhanced by Human Complement Inhibitors. PLoS Pathog. 2015;11:e1005043. doi: 10.1371/journal.ppat.1005043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 15.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor PW. Bactericidal and bacteriolytic activity of serum against gram-negative bacteria. Microbiol Rev. 1983;47:46–83. doi: 10.1128/mr.47.1.46-83.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meri S. Self-nonself discrimination by the complement system. FEBS Lett. 2016;590:2418–2434. doi: 10.1002/1873-3468.12284. [DOI] [PubMed] [Google Scholar]
- 19.Blom AM, Villoutreix BO, Dahlback B. Complement inhibitor C4b-binding protein-friend or foe in the innate immune system? Mol Immunol. 2004;40:1333–1346. doi: 10.1016/j.molimm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Ermert D, Blom AM. C4b-binding protein: The good, the bad and the deadly. Novel functions of an old friend. Immunology letters. 2016;169:82–92. doi: 10.1016/j.imlet.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 21.Parente R, Clark SJ, Inforzato A, Day AJ. Complement factor H in host defense and immune evasion. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev. 2016;274:152–171. doi: 10.1111/imr.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hovingh ES, van den Broek B, Jongerius I. Hijacking Complement Regulatory Proteins for Bacterial Immune Evasion. Front Microbiol. 2016;7 doi: 10.3389/fmicb.2016.02004. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kraiczy P, Wurzner R. Complement escape of human pathogenic bacteria by acquisition of complement regulators. Mol Immunol. 2006;43:31–44. doi: 10.1016/j.molimm.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 25.Sharma AK, Pangburn MK. Localization by site-directed mutagenesis of the site in human complement factor H that binds to Streptococcus pyogenes M protein. Infect Immun. 1997;65:484–487. doi: 10.1128/iai.65.2.484-487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ram S, Shaughnessy J, DeOliveira RB, Lewis LA, Gulati S, Rice PA. Utilizing complement evasion strategies to design complement-based antibacterial immunotherapeutics: Lessons from the pathogenic Neisseriae. Immunobiology. 2016;221:1110–1123. doi: 10.1016/j.imbio.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alsenz J, Lambris JD, Schulz TF, Dierich MP. Localization of the complement-component-C3b-binding site and the cofactor activity for factor I in the 38kDa tryptic fragment of factor H. Biochem J. 1984;224:389–398. doi: 10.1042/bj2240389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fleming A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. 1929. Br J exp Pathol. 1929;10:226–236. [Google Scholar]
- 29.Frieden T. CDC reports Department of Health and Human services. Centers for Disease Control and Prevention; Washington, DC, USA: 2013. Antibiotic Resistance Threats in the United States. [Google Scholar]
- 30.Rossolini GM, Arena F, Pecile P, Pollini S. Update on the antibiotic resistance crisis. Curr Opin Pharmacol. 2014;18:56–60. doi: 10.1016/j.coph.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature. 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- 32.Ferreira VP, Pangburn MK, Cortes C. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol. 2010;47:2187–2197. doi: 10.1016/j.molimm.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong SM, Shaughnessy J, Ram S, Akerley BJ. Defining the Binding Region in Factor H to Develop a Therapeutic Factor H-Fc Fusion Protein against Non-Typeable Haemophilus influenzae. Front Cell Infect Microbiol. 2016;6:40. doi: 10.3389/fcimb.2016.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaughnessy J, Vu DM, Punjabi R, Serra-Pladevall J, DeOliveira RB, Granoff DM, Ram S. Fusion protein comprising factor H domains 6 and 7 and human IgG1 Fc as an antibacterial immunotherapeutic. Clin Vaccine Immunol. 2014;21:1452–1459. doi: 10.1128/CVI.00444-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaughnessy J, Gulati S, Agarwal S, Unemo M, Ohnishi M, Su XH, Monks BG, Visintin A, Madico G, Lewis LA, Golenbock DT, Reed GW, Rice PA, Ram S. A Novel Factor H-Fc Chimeric Immunotherapeutic Molecule against Neisseria gonorrhoeae. J Immunol. 2016;196:1732–1740. doi: 10.4049/jimmunol.1500292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collin M, Olsen A. Generation of a mature streptococcal cysteine proteinase is dependent on cell wall-anchored M1 protein. Mol Microbiol. 2000;36:1306–1318. doi: 10.1046/j.1365-2958.2000.01942.x. [DOI] [PubMed] [Google Scholar]
- 37.Berge A, Kihlberg BM, Sjoholm AG, Bjorck L. Streptococcal protein H forms soluble complement-activating complexes with IgG, but inhibits complement activation by IgG-coated targets. J Biol Chem. 1997;272:20774–20781. doi: 10.1074/jbc.272.33.20774. [DOI] [PubMed] [Google Scholar]
- 38.Kihlberg BM, Cooney J, Caparon MG, Olsen A, Bjorck L. Biological properties of a Streptococcus pyogenes mutant generated by Tn916 insertion in mga. Microb Pathog. 1995;19:299–315. doi: 10.1016/s0882-4010(96)80003-9. [DOI] [PubMed] [Google Scholar]
- 39.Akesson P, Schmidt KH, Cooney J, Bjorck L. M1 protein and protein H: IgGFc- and albumin-binding streptococcal surface proteins encoded by adjacent genes. Biochem J. 1994;300(Pt 3):877–886. doi: 10.1042/bj3000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sim E, Palmer MS, Puklavec M, Sim RB. Monoclonal antibodies against the complement control protein factor H (beta 1 H) Biosci Rep. 1983;3:1119–1131. doi: 10.1007/BF01120205. [DOI] [PubMed] [Google Scholar]
- 41.Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther. 2008;7:2517–2527. doi: 10.1158/1535-7163.MCT-08-0201. [DOI] [PubMed] [Google Scholar]
- 42.Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, Tzekou A, DeAngelis RA, Resuello RR, Lupu F, Barlow PN, Lambris JD. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. 2013;218:496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cattaneo M, Cerletti C, Harrison P, Hayward CP, Kenny D, Nugent D, Nurden P, Rao AK, Schmaier AH, Watson SP, Lussana F, Pugliano MT, Michelson AD. Recommendations for the Standardization of Light Transmission Aggregometry: A Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J Thromb Haemost. 2013 doi: 10.1111/jth.12231. [DOI] [PubMed] [Google Scholar]
- 44.Reuter M, Caswell CC, Lukomski S, Zipfel PF. Binding of the human complement regulators CFHR1 and factor H by streptococcal collagen-like protein 1 (Scl1) via their conserved C termini allows control of the complement cascade at multiple levels. J Biol Chem. 2010;285:38473–38485. doi: 10.1074/jbc.M110.143727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blackmore TK, V, Fischetti A, Sadlon TA, Ward HM, Gordon DL. M protein of the group A Streptococcus binds to the seventh short consensus repeat of human complement factor H. Infect Immun. 1998;66:1427–1431. doi: 10.1128/iai.66.4.1427-1431.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferreira VP, Herbert AP, Cortes C, McKee KA, Blaum BS, Esswein ST, Uhrin D, Barlow PN, Pangburn MK, Kavanagh D. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol. 2009;182:7009–7018. doi: 10.4049/jimmunol.0804031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frank MM, Joiner K, Hammer C. The function of antibody and complement in the lysis of bacteria. Rev Infect Dis. 1987;9(Suppl 5):S537–545. doi: 10.1093/clinids/9.supplement_5.s537. [DOI] [PubMed] [Google Scholar]
- 48.Joiner KA, Brown EJ, Frank MM. Complement and bacteria: chemistry and biology in host defense. Annual review of immunology. 1984;2:461–491. doi: 10.1146/annurev.iy.02.040184.002333. [DOI] [PubMed] [Google Scholar]
- 49.Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci U S A. 1976;73:3268–3272. doi: 10.1073/pnas.73.9.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whaley K, Ruddy S. Modulation of C3b hemolytic activity by a plasma protein distinct from C3b inactivator. Science. 1976;193:1011–1013. doi: 10.1126/science.948757. [DOI] [PubMed] [Google Scholar]
- 51.Fujita T, Gigli I, Nussenzweig V. Human C4-binding protein. II. Role in proteolysis of C4b by C3b-inactivator. J Exp Med. 1978;148:1044–1051. doi: 10.1084/jem.148.4.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gigli I, Fujita T, Nussenzweig V. Modulation of the classical pathway C3 convertase by plasma proteins C4 binding protein and C3b inactivator. Proc Natl Acad Sci U S A. 1979;76:6596–6600. doi: 10.1073/pnas.76.12.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haapasalo K, Vuopio J, Syrjanen J, Suvilehto J, Massinen S, Karppelin M, Jarvela I, Meri S, Kere J, Jokiranta TS. Acquisition of complement factor H is important for pathogenesis of Streptococcus pyogenes infections: evidence from bacterial in vitro survival and human genetic association. J Immunol. 2012;188:426–435. doi: 10.4049/jimmunol.1102545. [DOI] [PubMed] [Google Scholar]
- 54.Haapasalo K, Jarva H, Siljander T, Tewodros W, Vuopio-Varkila J, Jokiranta TS. Complement factor H allotype 402H is associated with increased C3b opsonization and phagocytosis of Streptococcus pyogenes. Mol Microbiol. 2008;70:583–594. doi: 10.1111/j.1365-2958.2008.06347.x. [DOI] [PubMed] [Google Scholar]
- 55.Kihlberg BM, Collin M, Olsen A, Bjorck L. Protein H, an antiphagocytic surface protein in Streptococcus pyogenes. Infect Immun. 1999;67:1708–1714. doi: 10.1128/iai.67.4.1708-1714.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gustafsson MC, Lannergard J, Nilsson OR, Kristensen BM, Olsen JE, Harris CL, Ufret-Vincenty RL, Stalhammar-Carlemalm M, Lindahl G. Factor H binds to the hypervariable region of many Streptococcus pyogenes M proteins but does not promote phagocytosis resistance or acute virulence. PLoS Pathog. 2013;9:e1003323. doi: 10.1371/journal.ppat.1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akesson P, Cooney J, Kishimoto F, Bjorck L. Protein H–a novel IgG binding bacterial protein. Mol Immunol. 1990;27:523–531. doi: 10.1016/0161-5890(90)90071-7. [DOI] [PubMed] [Google Scholar]
- 58.Berends ET, Dekkers JF, Nijland R, Kuipers A, Soppe JA, van Strijp JA, Rooijakkers SH. Distinct localization of the complement C5b-9 complex on Gram-positive bacteria. Cellular microbiology. 2013;15:1955–1968. doi: 10.1111/cmi.12170. [DOI] [PubMed] [Google Scholar]
- 59.Suresh R, Chandrasekaran P, Sutterwala FS, Mosser DM. Complement-mediated ‘bystander’ damage initiates host NLRP3 inflammasome activation. Journal of cell science. 2016;129:1928–1939. doi: 10.1242/jcs.179291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bindon CI, Hale G, Bruggemann M, Waldmann H. Human monoclonal IgG isotypes differ in complement activating function at the level of C4 as well as C1q. J Exp Med. 1988;168:127–142. doi: 10.1084/jem.168.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nilsson M, Weineisen M, Andersson T, Truedsson L, Sjobring U. Critical role for complement receptor 3 (CD11b/CD18), but not for Fc receptors, in killing of Streptococcus pyogenes by neutrophils in human immune serum. Eur J Immunol. 2005;35:1472–1481. doi: 10.1002/eji.200424850. [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Wang L, Xiang Y, Ricklin D, Lambris JD, Chen G. Using an in vitro xenoantibody-mediated complement-dependent cytotoxicity model to evaluate the complement inhibitory activity of the peptidic C3 inhibitor Cp40. Clin Immunol. 2016;162:37–44. doi: 10.1016/j.clim.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krych-Goldberg M, Atkinson JP. Structure-function relationships of complement receptor type 1. Immunol Rev. 2001;180:112–122. doi: 10.1034/j.1600-065x.2001.1800110.x. [DOI] [PubMed] [Google Scholar]
- 65.Tacconelli E, Magrini N. GLOBAL PRIORITY LIST OF ANTIBIOTIC-RESISTANT BACTERIA TO GUIDE RESEARCH, DISCOVERY, AND DEVELOPMENT OF NEW ANTIBIOTICS. World Health Organization; 2017. [Google Scholar]