

Abstract
In NOD mice and also likely humans, B-lymphocytes play an important role as APC expanding autoreactive T-cell responses ultimately causing type 1 diabetes (T1D). Currently, humans at high future T1D risk can only be identified at late prodromal stages of disease indicated by markers such as insulin autoantibodies (IAA). When commenced in already IAA+ NOD mice, continuous BAFFR-Fc treatment alone or in combination with anti-CD20 (designated combo therapy) inhibited T1D development. Despite eliciting broader B-lymphocyte depletion, continuous combo therapy afforded no greater T1D protection than BAFFR-Fc alone. As previously observed, late disease stage initiated anti-CD20 mono-therapy did not inhibit T1D, and in this study was additionally found to be associated with development of drug blocking antibodies. Promisingly, NOD mice given transient late disease stage BAFFR-Fc mono-therapy were rendered T1D resistant. However, combo treatment abrogated the protective effect of transient BAFFR-Fc mono-therapy. NOD mice receiving transient BAFF blockade were characterized by an enrichment of regulatory B-lymphocytes (Bregs) that inhibit T1D development through IL-10 production, but this population is sensitive to deletion by anti-CD20 treatment. B-lymphocytes from transient BAFFR-Fc treated mice suppressed T-cell proliferation to a greater extent than those from controls. Proportions of B-lymphocytes expressing CD73, an ecto-enzyme operating in a pathway converting pro-inflammatory ATP to immunosuppresive adenosine, were also temporarily increased by transient BAFFR-Fc treatment, but not anti-CD20 therapy. These collective studies indicate transient BAFFR-Fc mediated B-lymphocyte depletion elicits long-term T1D protection by enriching Bregs that are deleted by anti-CD20 co-therapy.
Introduction
β-cell destruction in type 1 diabetes (T1D) is ultimately mediated by T-cells, but based on studies in the NOD mouse model, B-lymphocytes also play a critical role in disease development by serving as a preferential subset of pathogenic APC (1). This appears to result from B-lymphocytes expressing plasma membrane-bound autoreactive Ig molecules that can efficiently capture specific ß-cell antigens for subsequent display to diabetogenic T-cells, expanding such pathogenic effectors more efficiently than other APC subtypes (2–6). In contrast, a subset of B-lymphocytes may normally also play a T1D inhibitory regulatory role that could be defective in NOD mice and human patients (7). These observations place B-lymphocytes at an important nexus between the activation or inhibition of diabetogenic immune responses.
Based on their key pathogenic role, multiple approaches have been tested in NOD mice to develop potential B-lymphocyte-directed T1D interventions. These interventions include depletion of B-lymphocytes with antibodies directed against cell surface CD20 or CD22 molecules (8–11). Blocking B-lymphocyte access to the critical survival factors B-cell activation factor (BAFF) and a proliferation inducing ligand (APRIL) have also been tested as a potential T1D interventions in NOD mice (9, 11–13). BAFF is a B-lymphocyte survival and maturation factor of the TNF superfamily produced by monocytes, macrophages, and some T-cells. Elevated serum BAFF and APRIL levels have been detected in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and Sjögren’s syndrome (SS) (14–16). Partly based on these previous findings, the CD20-specific rituximab antibody was tested in a TrialNet phase II study as a candidate disease intervention agent in patients with recent onset T1D (17). Transient anti-CD20 therapy allowed for preservation of β-cell function one year after initiation of treatment, but such effects were subsequently lost (18). Of note, wide spread deletion of B-lymphocytes by rituximab also failed as a SLE intervention in a clinical trial (19). However, belimumab, an anti-BAFF mAb that only modestly depletes B-lymphocytes, became the first targeted biological treatment for SLE (20). Additionally, belimumab also demonstrated significant efficacy in patients with RA, whereas atacicept (TACI-Fc), another biologic blocking both BAFF and APRIL showed no effect (21, 22). Therefore, previous studies suggest that clinical efficacy of B-lymphocyte-directed therapeutic approaches do not necessarily depend upon how broadly they deplete these cells.
In NOD mice, islet infiltrating B-lymphocytes down-regulate cell surface expression of CD20 (8). We are aware of a report that in humans higher numbers of islet-infiltrating CD20-expressing B-lymphocytes characterize patients most rapidly progressing to overt T1D (23). Methodological differences could account for these differing reports. The human study utilized an immunohistochemistry technique that could detect islet-infiltrating B-lymphocytes potentially expressing intracellular CD20, while our flow cytometric-based protocol only detects this molecule as a cell surface entity. However, if islet infiltrating B-lymphocytes in humans do internalize CD20, this could also limit the effectiveness of rituximab. To be included in T1D prevention trials, humans have to exhibit markers of ongoing islet autoimmunity, such as the presence of circulating islet autoantibodies (24). Thus, another potential clinical translational concern is when used as a mono-therapy, a rituximab-like CD20-specific antibody only inhibited progression to overt T1D in NOD mice when treatment was initiated before, but not after, the appearance of insulin autoantibodies (IAA) (8). These collective issues underscore the need to identify additional modalities that either independently or in synergy with anti-CD20 provide a more efficacious B-lymphocyte-directed T1D intervention approach. Indeed, combining B-lymphocyte-depleting antibodies like anti-CD20 with BAFF blockade have shown to be more efficacious than their use as mono-therapies in murine models of SLE (25). The question of whether combined anti-CD20 and BAFF blockade-mediated B-lymphocyte depletion is superior to their use as mono-therapies for inhibiting T1D development has yet to be addressed. Additionally, a finding that such protection could be achieved when treatment is initiated after the onset of aggressive autoimmunity, marked by the presence IAA, would represent a significant advancement for B-lymphocyte-directed T1D interventions. Hence, we investigated the potential of combination anti-CD20/BAFF blockade therapy as a possible late disease stage B-lymphocyte directed T1D intervention approach in NOD mice.
We also examined whether anti-CD20 or BAFF-blockade treatment, either alone or in combination, inhibit T1D development solely through B-lymphocyte depletion, or whether these interventions also elicit active disease-protective immunoregulatory processes. There have been reports that B-lymphocyte depletion prevents T1D in NOD mice by mechanisms dependent on expansion of regulatory T-cells (Tregs) (8, 13). However, there are other reports that anti-BAFF or anti-CD20-mediated (clone MB20.11, isotype IgG2c) B-lymphocyte depletion exerted T1D-protective effects in NOD mice without a concurrent induction of Tregs (9, 12). IL-10 producing B-lymphocytes (B10 cells) have been reported to ameliorate disease development in mouse models of SLE (26), experimental autoimmune encephalitis (EAE) (27), and RA (28). It has also been reported a subset of B10 cells, decreased in hyperglycemic NOD mice and T1D patients, play an immunoregulatory role in long-term disease protection (7). However, the capacity of B-lymphocyte depletion to modulate levels of B10 cells in the context of T1D is currently unknown. We recently found a sizeable proportion of B-lymphocytes in NOD-Aicdanull mice which are unable to undergo class switch recombination (CSR) and somatic hyper-mutation (SHM) not only fail to support T1D development but also convert to a CD73+ phenotype capable of actively suppressing pathogenic T-cells (29). CD39 and CD73 are ecto-enzymes that respectively convert pro-inflammatory ATP to AMP and then to anti-inflammatory adenosine (30). Thus, we also assessed whether specific B-lymphocyte subsets surviving or rebounding after transient anti-CD20 and/or BAFF-blockade contribute to T1D protection in NOD mice by enrichment towards a regulatory phenotype. An important finding of our collective studies is B-lymphocytes remaining and rebounding after transient BAFFR-Fc mono-therapy are enriched for a T1D protective regulatory population (Bregs), but these are sensitive to deletion by anti-CD20 co-therapy.
Materials and Methods
Mice
NOD/ShiLtDvs (NOD), C57BL/6J (B6), and BALB/cJ (BALB/c) mice are maintained at The Jackson Laboratory under SPF conditions. NOD.Cg-Prkdcscid Emv30b/Dvs (NOD-scid) mice have been previously described (31). NOD mice with CD4 T-cells transgenically expressing the BDC2.5 TCR (NOD-BDC2.5; formal designation NOD.Cg-Tg(TcraBDC2.5,TcrbBDC2.5)1Doi/DoiJ) were acquired from the type 1 diabetes resource (http://type1diabetes.jax.org/) operated at The Jackson Laboratory. NOD.129X1(Cg)-Foxp3tm2Tch/DvsJ (NOD-Foxp3egfp) have also been previously described (32).
Flow Cytometry
Lymphocyte populations in spleen, pancreatic lymph nodes (PLN), bone marrow (BM), and peripheral blood (PBL) were analyzed by flow cytometry. Pancreatic islet-infiltrating leukocytes were isolated as previously described (33). The following fluorochrome-conjugated monoclonal antibodies were used: CD4, CD21, CD62L, CD43, TCRβ (RM4-5, 7G6, MEL-14, S7, H57-597 BD Biosciences), B220, CD8, CD23, CD138, CD73, PD-L2, CD80, IL-10, IgD, CD5, CD11b (RA3-682, 53-6.72, B3B4, 281-2, TY/11.8, TY25, 16-10A1, JES5-16E3, 11-26c.2a, 53-7.3, M1/70 BioLegend), IgM, CD19, CD44 (II/41, ID3, IM7, eBioscience). For intracellular staining of IL-10, 50 ng/ml PMA (Sigma-Aldrich), 500 ng/ml ionomycin (Sigma-Aldrich), and 2 μM monensin (Sigma-Aldrich) were added to the culture for 5h before flow cytometric analysis. Cultured cells were first labeled with a Fixable Viability Dye eFluor 780 (eBiosceince), then surfaced stained for CD19, followed by permeabilization using the Cytofix/Cytoperm kit (BD Bioscience) according to the manufacturer’s protocol. Cells were analyzed on an LSR II flow cytometer (BD Biosciences) using FACSDiva software. The flow cytometric gating strategy used to evaluate lymphocyte subsets is depicted in Supplemental Figure 1. Data were analyzed using FlowJo software.
Immunotherapy
Murine anti-CD20 IgG2c mAb (MB20.11) administered intraperitoneally at 250 μg doses at 2-week intervals, has been described (9) and was provided by MedImmune. BAFFR-Fc, a fusion protein with the extracellular portion of BAFFR fused to the Fc domain of mouse IgG1, was dosed at 200 μg intraperitoneally twice a week and was also obtained from MedImmune (34). An irrelevant control murine IgG1 antibody (ctrl mAb) supplied by MedImmune was dosed at 200 μg intraperitoneally twice a week. Anti-mouse IL-10 (JES5-2A5, BioXcell) was initially dosed at 500 μg intraperitoneally twice a week for 2 weeks followed by a maintenance dose of 200 μg twice a week. Anti-mouse CD25 (PC-61.5.3, BioXcell) administered intraperitoneally at 250 μg doses at 2-week intervals has been described (8).
Assessment of T1D and Insulitis Development
Upon initiating anti-CD20 and/or BAFFR-Fc treatment at 10 weeks of age, NOD female mice were monitored for glycosuria with Ames Diastix (Bayer Diagnostics Division, Elkhart, IN). Mice with two consecutive values of ≥0.25% (corresponds to a blood glucose of ≥300 mg/dl) were scored as diabetic. Mice remaining normoglycemic post-treatment were sacrificed and pancreata were analyzed and assigned an insulitis score of 0–4 by a blinded observer as previously described (33).
Sort-Purification of B-lymphocytes
B-lymphocytes from NOD mice treated with BAFFR-Fc or ctrl mAb antibody from 10 to 14 weeks of age were first enriched from pooled spleens by negative-depletion of CD11b+, CD11c+, CD3ε+ and TER-119+ cells using Biotin Binder Dynabeads (Invitrogen) according to the manufacturer’s protocol. Enriched CD19+ B220+ B-lymphocytes from each treatment group were then sorted to a 99% purity using FACSAriaII SORP sorter (BD Biosciences).
Measurement of IL-10 production
Sort-purified splenic B-lymphocytes isolated from NOD mice treated with BAFFR-Fc or ctrl mAb were stimulated with 10 μg/ml LPS (Sigma-Aldrich) or 10 μg/ml goat anti-mouse IgG + IgM (H+L) (Jackson ImmunoResearch) plus 1 μg/ml anti-CD40 (HM40-3, BD Biosciences) for 3 days. In another experiment, TACIhigh or TACIlow B-lymphocytes isolated from NOD mice were sorted and stimulated with 10 μg/ml LPS (Sigma-Aldrich) for 3 days. IL-10 concentration in culture supernatants was determined using the Mouse IL-10 ELISA MAX kit (Biolegend) according to the manufacturer’s recommended protocol.
Antigen Presentation and Suppression Assay
To assess B-lymphocyte mediated antigen presentation, CD4+ T-cells purified from spleens of 8–10 week old NOD BDC2.5 TCR transgenic mice by negative depletion of CD11b+, CD11c+, TER-119+, B220+, and CD8+ cells using streptavidin microbeads (Miltenyi) were labeled with 2 μM Cell Proliferation Dye eFluor450 (CPD) (eBiosciences) according to the manufacturer’s protocol. Labeled CD4+ T-cells were cultured in the presence of 1 μM BDC2.5 mimotope (AHHPIWARMDA, Mimotopes) with or without sorted B-lymphocytes for 3 days. T-cell proliferation was assessed by flow cytometric analyses of CPD dilution as a measure of antigen presentation. In another experiment, microbead purified CD11c+ DCs (Miltenyi) were used as an APC source. For B-lymphocyte mediated suppression, CD4+ CD25− T-cells purified from spleens of 8–10 week old NOD mice were labeled with 2 μM CPD. Sorted B-lymphocytes and labeled T-cells stimulated with plate-bound 5 μg/mL anti-CD3ε (BD Biosciences) were co-cultured with 0 or 10μg/ml LPS (Sigma-Aldrich) for 3 days. T-cell proliferation (CPD dilution) was assessed by flow cytometry and data analyzed using FlowJo software. Percent suppression was calculated relative to the mean Proliferation Index of anti-CD3 stimulated T-cells co-cultured with B-lymphocytes in medium without LPS.
Adoptive Transfer
Splenic T-cells were purified by negative depletion of CD11b+, CD11c+, TER-119+ and B220+ cells using biotin-conjugated antibodies and streptavidin MicroBeads (Miltenyi) according to manufacturer’s protocol. Indicated NOD-scid recipients were injected i.v. with 5×106 purified T-cells from normoglycemic untreated NOD donors or NOD mice treated with BAFFR-Fc or combo therapy. In another experiment, indicated NOD-scid recipients were injected i.v. with 5×106 T-cells from normoglycemic untreated NOD donors and then intraperitoneally injected with either ctrl mAb or BAFFR-Fc.
IAA assay
Murine IAA were detected using the previously described radioimmunoassay methodology (35).
Serum anti-MB20.11 assays
Serum was collected from mice given anti-CD20 (MB20.11) for the indicated period. Antibodies against anti-CD20 IgG2c (MB20.11), or an irrelevant mouse IgG2c (Southern Biotech) were detected by ELISA.
HEL/CFA Immunization and ELISA
10 week-old NOD mice were immunized i.p. with 50 μg of hen egg lysozyme (HEL) or dPBS emulsified in complete Freund’s adjuvant (CFA). Starting 13 days post-immunization, NOD mice received ctrl mAb antibody or BAFFR-Fc for 4 weeks. Then all mice were immunized with 50 μg HEL emulsified in CFA. This resulted in four separate groups of control or BAFFR-Fc treated NOD mice that had been primed either one or twice with HEL. Serum was collected 13 days following what was either a primary or secondary HEL immunization and assayed for Ag-specific Abs by ELISA. Briefly, serum Abs (Diluted at 1:50) captured on HEL-coated plates were detected with alkaline phosphatase (AP)-conjugated anti-mouse kappa (Jackson ImmunoResearch Laboratories) and subsequent incubation with p-nitrophenyl phosphate (Sigma-Aldrich). The optical density was measured at 405nm using a SpectraMax 190 microplate reader (Molecular Devices).
Statistical analysis
Unless otherwise indicated, all bar and scatter plots are shown as mean±SEM. All graphs and statistics were generated using Prism 6 (GraphPad). Specific statistical analyses are listed within the respective figure legends, and p-values are incorporated within the referenced panels.
Results
Addition of anti-CD20 does not improve the T1D protection by continuous BAFFR-Fc mono-therapy
Initially, we tested if treatment with BAFF blockade, either by itself or combined with anti-CD20 (MB20.11), could provide an effective late prodromal disease stage T1D intervention in NOD mice. Our previous anti-CD20 (18B12) therapy failed within this treatment window (8). Beginning at 10-weeks-of-age, female NOD mice received continuous treatments with BAFFR-Fc alone or in combination (combo) with anti-CD20. Controls consisted of NOD females receiving an irrelevant ctrl mAb. Sera were obtained from all mice just prior to the initiation of treatment regimens. This allowed us to retrospectively stratify the effectiveness of B-lymphocyte depletion in preventing T1D development based on IAA status. Regardless of IAA status at the start of the study, compared to controls, continuous treatment with BAFFR-Fc alone or in combination with anti-CD20 significantly inhibited T1D development (Figure 1A, B). However, there was no significant difference in the extent T1D was inhibited by BAFFR-Fc mono- compared to combo-therapy (Figure 1A, B). The frequency of BAFFR-Fc and combo-treated NOD mice that were IAA positive at the end of the incidence study was no different than when assessed at pre-treatment (data not shown). Meanwhile, continuous anti-CD20 mono-therapy initiated at 10 weeks of age failed to elicit T1D protection (Figure 1A). Consistent with previous studies (8), anti-CD20 was ineffective in inhibiting T1D development when initiated in already IAA+ NOD mice (Figure 1B). However, the BAFFR-Fc mono- or combo-therapy treated NOD mice that remained free of overt T1D at the end of the incidence study (35 weeks of age) still had high levels of insulitis (Figure 1C). In a separate kinetic study, insulitis was similar in NOD mice receiving the ctrl mAb antibody or any of the three B-lymphocyte depletion regimens from 10–14 or 10–20 weeks of age (Figure 1D). Collectively, these data indicate that despite insulitis, continuous BAFFR-Fc mono- and combo-therapy initiated at a late prodromal stage of disease development inhibits T1D onset in NOD mice whereas anti-CD20 mono-therapy was ineffective.
Figure 1. BAFFR-Fc provides late-stage T1D protection and the addition of anti-CD20 does not offer additional benefits.
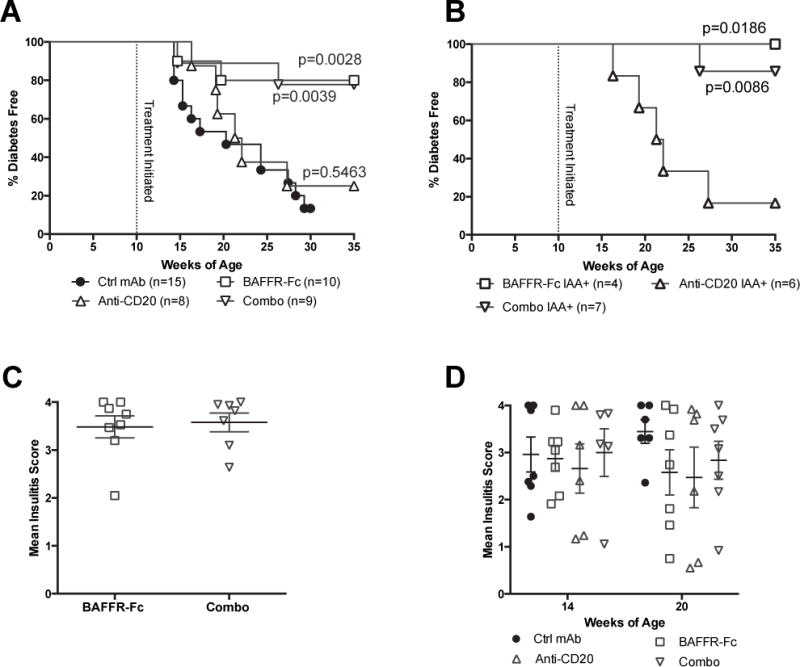
A) Serum was collected from 10-week-old NOD female mice that then immediately began to receive the indicated treatment, (200μg doses of BAFFR-Fc or ctrl mAb twice weekly, 250μg anti-CD20 biweekly, or combo) and followed for diabetes incidence. Statistical comparisons were to ctrl mAb treated NOD mice (Mantel-Cox analysis). B) Stratified diabetes incidence of mice that are retrospectively tested to be IAA+ at the initiation of treatment with BAFFR-Fc, anti-CD20, or combo. Statistical comparisons were to anti-CD20 treated NOD mice (Mantel-Cox analysis). C) Insulitis scores are shown for nondiabetic NOD mice given BAFFR-Fc or combo at the end of incidence study in panel A. D) Insulitis scores are shown for nondiabetic NOD mice on treatment for 4 (14-week-old) or 10 weeks (20-week-old) with ctrl mAb, BAFFR-Fc, anti-CD20, or combo. p-values were calculated using Mann-Whitney analyses. Each symbol represents an individual mouse; small horizontal lines indicate the Mean±SEM.
BAFFR-Fc/anti-CD20 mono- or combo-therapies differentially deplete B-lymphocytes
To better understand the contributions of various B-lymphocytes subsets to T1D development, we quantified the populations remaining present after four weeks of the differing depletion regimens. Compared to controls, total B-lymphocytes were significantly depleted within the spleens and PLNs of all treatment groups, with the greatest reduction observed in the combo therapy cohort (Figure 2A). The number of islets that can be isolated from any given mouse varies. Thus, it is not possible to calculate total yields of islet infiltrating B-lymphocytes. However, proportions of B-lymphocytes among islet infiltrating CD45+ leukocytes were also decreased in all treatment groups (Figure 2A). Anti-CD20 mono-therapy resulted in greater depletion of total B-lymphocytes in spleens and PLNs than that elicited by BAFF blockade alone (Figure 2A). It was previously found B-lymphocytes entering the islets of NOD mice down-regulate cell surface CD20 expression (8). Thus, the proportional decrease of B-lymphocytes among islet infiltrating leukocytes in NOD mice receiving anti-CD20 mono-therapy is likely due to their elimination prior to entering this site. The decrease in total splenic B-lymphocytes in the BAFFR-Fc and combo-therapy groups corresponded to a loss in both frequency and absolute number of transitional stage-2 (T2), mature follicular (FO) and marginal zone (MZ) subsets (Figure 2B). In contrast, the frequency and absolute numbers of transitional stage-1 (T1) cells were respectively increased and marginally decreased with the BAFFR-Fc monotherapy. This observation is consistent with previous findings that BAFF is indispensible for B-lymphocyte survival beyond the T1 stage (36). In previous studies with anti-CD20 (18B12), we found treatment only depleted FO B-lymphocytes. Interestingly, while treatment with the current anti-CD20 (MB20.11) resulted in significant depletion of both MZ and FO subsets, it more efficiently depleted the former population (Figure 2B). Of note, anti-CD20 mono-therapy was more efficient than BAFF blockade in depleting MZ but not FO B-lymphocytes, suggesting the former population is not a necessary contributor to T1D development in NOD mice (Figure 2B). Immature B-lymphocyte populations in the bone marrow have either absent or low CD20 expression and their development is BAFF independent (37). Thus, it was not surprising that none of those subsets were depleted by any of the deployed B-lymphocyte depletion regimens (Supplemental Figure 2). By contrast, mature B-lymphocytes that have re-circulated to the bone marrow were significantly depleted in all treatment groups, with combo-therapy displaying the most efficient reduction, followed by anti-CD20 mono-therapy (Supplemental Figure 2).
Figure 2. The combination of BAFFR-Fc and anti-CD20 synergize in depleting B lymphocytes in the spleen and PLNs.
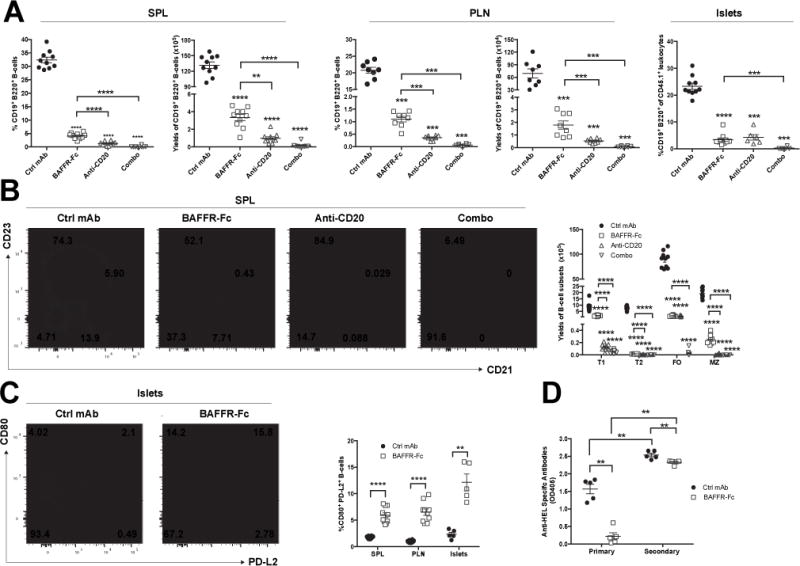
A) Frequency and absolute numbers of B220+ CD19+ B-lymphocytes amongst viable leukocytes in the spleens (left panel) and PLNs (middle panel) of NOD mice treated with ctrl mAb, BAFFR-Fc, anti-CD20, or combo from 10 to 14 weeks of age. Right panel depicts proportions of B-lymphocytes among CD45+ islet associated leukocytes from NOD mice treated with ctrl mAb, BAFFR-Fc, anti-CD20, or combo from 10 to 14 weeks of age. The total number of islets that could be isolated from each mouse varied. Thus, islet associated B-lymphocytes could only be assessed on a proportional basis. Data pooled from two independent experiments. B) Representative flow cytometric plots showing splenic T1 (CD21− CD23−), T2 (CD21hi CD23+), FO (CD21+ CD23+) and MZ (CD21hi CD23−) B-lymphocyte subsets among live CD19+ B220+ cells in NOD mice treated with ctrl mAb, BAFFR-Fc, anti-CD20, or combo regimens from 10 to 14 weeks of age (left panel). Absolute T1, T2, FO, MZ B-lymphocyte numbers calculated from values in the left panel (right panel). (n=10 per group). Data pooled from two independent experiments. C) Left panel-representative flow cytometric plots showing islet infiltrating B-lymphocytes co-expressing CD80 and PD-L2 among live CD19+ B220+ cells in NOD mice treated with ctrl mAb or BAFFR-Fc from 10 to 14 weeks of age. Right-panel-percent CD80+ PD-L2+ amongst splenic, PLN, or islet infiltrating B-lymphocytes in NOD mice treated with ctrl mAb or BAFFR-Fc from 10 to 14 weeks of age. (n=10/group for spleen/PLN); (n=5 mice/group for islets). Data pooled from two independent experiments for spleen and PLN. D) Primary or secondary HEL specific antibodies in NOD mice given ctrl mAb or BAFFR-Fc for 4 weeks starting at 10 weeks of age. (n=5 per group). p-values were calculated using Mann-Whitney analyses, each symbol represents an individual mouse; small horizontal lines indicate the Mean±SEM. Asterisks on top of each B-lymphocyte depleted groups indicate comparison to ctrl mAb (A-C). All other comparisons are drawn as lines between the indicated two groups. (**p<0.01, ***p<0.001, ****p<0.0001).
Despite the drastically decreased number of total B-lymphocytes in each treatment group, the CD80+ PD-L2+ subset, reported to be a memory population (38, 39), was proportionally increased in the islets, PLN, and spleens of BAFFR-Fc-treated mice (Figure 2C). Finally, we tested memory humoral responses in mice given BAFFR-Fc to a prototypic exogenous antigen HEL. As expected, NOD mice in which B-lymphocytes had been depleted by BAFFR-Fc treatment prior to initial priming with HEL only mounted a weak primary antibody response against this antigen (Figure 2D). However, NOD mice initially primed with HEL, followed by subsequent BAFFR-Fc mediated B-lymphocyte depletion, mounted a secondary humoral response to this antigen only slightly less than that observed in controls (Figure 2D) in which the slightly lower antibody titer reflects a decrease in IgM but not class-switched IgG isotypes (data not shown). Hence, functional memory B-lymphocytes are retained in BAFFR-Fc mono-therapy treated NOD mice. A clinically translatable significance of this finding is that any previous vaccination effects may not be lost as a consequence of subsequent BAFFR-Fc mono-therapy. Together, these data indicate while most broadly reduced by both the anti-CD20 and combo regimens, the pattern of B-lymphocyte depletion elicited by BAFFR-Fc mono-therapy is sufficient to provide a late disease stage T1D protective effect in NOD mice.
B-lymphocyte rebound in long-term anti-CD20 treated NOD mice due to the generation of idiotype specific blocking antibodies
In contrast to a previous report that treatment with the MB20.11 CD20 specific antibody significantly delayed, but did not entirely prevent T1D development when initiated in 15-week-old female NOD mice (9), we did not see such an effect in our studies with a 10 week start. Any ability of anti-CD20 mono-therapy to exert T1D protective effects in NOD mice quickly waned by 20 weeks of age (Figure 1A). This loss of protection was associated with B-lymphocytes rebounding in a proportion of diabetic NOD mice receiving 10 weeks of anti-CD20 mono-therapy (data not shown). Therefore, we assessed the effectiveness of anti-CD20 mediated B-lymphocyte depletion in separate cohorts of NOD mice after 10 weeks of treatment. Surprisingly the degree of B-lymphocyte depletion was heterogeneous. Remaining proportions of splenic B-lymphocytes ranged from 0.11% to near control-like 36% in NOD mice treated with anti-CD20 from 10–20 weeks of age (Figure 3A). The reduced ability of long term anti-CD20 mono-therapy to consistently maintain B-lymphocyte depletion in NOD mice led us to explore the possibility this strain produces serum antibodies directed against the utilized IgG2c MB20.11 monoclonal antibody. The presence of such antibodies was indeed detected in anti-CD20 treated mice (Figure 3B). The anti-CD20 binding antibodies were primarily of the class switched IgG isotype (Figure 3C). In contrast, NOD mice receiving long-term combo therapy did not generate high levels of MB20.11 binding antibodies (Figure 3C). This could be explained by the fact the B-lymphocytes that can potentially produce MB20.11 binding antibodies were depleted with the addition of BAFF blockade in the combo therapy group. Furthermore, the degree of B-lymphocyte rebound correlated with the titer of MB20.11 antibodies in sera (Figure 3D). This latter result indicated MB20.11 binding antibodies generated in NOD mice neutralized the B-lymphocyte depletion activity of the reagent.
Figure 3. Differential responses to anti-CD20 (MB20.11) mediated B-lymphocyte depletion due to induction of idiotypic or isotypic directed antibodies.
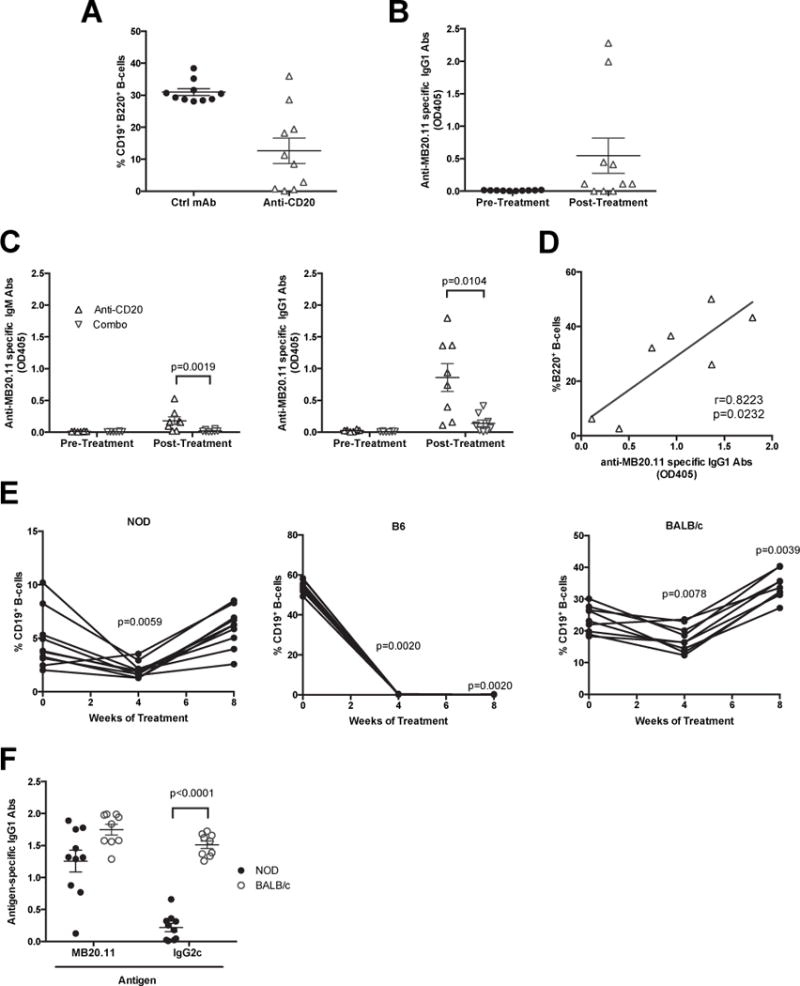
A) Percentages of splenic CD19+ B220+ B-lymphocytes from NOD mice treated with anti-CD20 (MB20.11) or ctrl mAb from 10–20 weeks of age (n=10 per group). B) Sera from anti-CD20 treated NOD mice in Figure 3A were tested for the presence of anti-MB20.11 binding antibodies before and after treatment (n=10 per group). C) Sera from anti-CD20 or combo-treated NOD mice in Figure 1A were compared for the presence and isotypes (IgM or IgG1) of anti-MB20.11 binding antibodies before and after treatment (n=8 per group). D) Correlation of splenic B-lymphocytes rebounding and titer of MB20.11 binding antibodies in anti-CD20 treated NOD mice. p-value was calculated using Pearson correlation. E) Anti-CD20 mediated B-lymphocyte depletion was measured by flow cytometric analyses of CD19+ cells from peripheral blood in NOD (n=10), C57BL/6 (n=10), BALB/c (n=9) mice at 4-week-interval starting at 10 weeks of age. **p< 0.01, p-values were calculated using Wilcoxon test by comparing to pre-treatment. F) Titer of IgG1 antibodies binding anti-CD20 (MB20.11) and an irrelevant IgG2c in NOD (n=10) and BALB/c (n=9) mice after 8 weeks of anti-CD20 treatment. p-value was calculated using Mann-Whitney analysis. Each symbol represents an individual mouse; small horizontal lines indicate the Mean±SEM.
We next tested whether the anti-MB20.11 response is a strain-specific characteristic of autoimmune prone NOD mice. The ability of MB20.11 mono-therapy to maintain a B-lymphocyte depleted state in NOD mice was compared to the non-autoimmune-prone B6 and BALB/c strains. Anti-CD20 completely depleted B-lymphocytes among peripheral blood leukocytes (PBL) at all time points analyzed in B6 mice (Figure 3E). Interestingly, after 8 weeks of treatment, the level of rebounding B-lymphocytes was greater in BALB/c than NOD mice receiving anti-CD20 mono-therapy (Figure 3E). Similar to the NOD strain, BALB/c mice generated MB20.11-binding antibodies (Figure 3F). Of note, mouse strains such as BALB/c and DBA/2 lack the gene encoding IgG2c, an IgG subclass expressed in strains like NOD, B6, and C57BL/10, and instead express the IgG2a isotype (40). Thus, we tested if the MB20.11 binding antibodies generated in BALB/c mice represented an anti-isotypic response. We found that following MB20.11 mono-therapy the levels of antibodies capable of recognizing an irrelevant IgG2c were significantly higher in BALB/c than NOD mice (Figure 3F). Therefore, the anti-MB20.11 response seen in NOD versus BALB/c mice is respectively idiotypic and isotypic directed.
Transient BAFFR-Fc -mediated B-lymphocyte depletion elicits long-term T1D protection that is abrogated by the addition of anti-CD20
To ease patient burden, a B-lymphocyte-directed intervention that is transient in nature, but capable of exerting long-term T1D protection would be desirable. Such a transient therapy would also avoid a sustained state of generalized immunosuppression. Thus, we compared T1D development in female NOD mice injected from 10–14 weeks of age with BAFFR-Fc alone or in combination with anti-CD20, to those treated with ctrl mAb. Transient BAFFR-Fc mono-therapy elicited a strong long-term T1D-protective effect compared to controls. Surprisingly, transient combo therapy failed to inhibit T1D development (Figure 4A). Similar to long-term treatment, the non-diabetic NOD mice given transient BAFFR-Fc at the end of the incidence study (30 weeks) were also characterized by significant levels of insulitis (3.02± 0.41). Several studies reported that B-lymphocytes re-emerging after transient depletion in NOD mice display an immunosuppressive phenotype (10, 11). Thus, we hypothesized the B-lymphocytes that survive or rebound in NOD mice transiently treated with BAFFR-Fc alone might become enriched for a Breg-like population, but this protective population(s) is sensitive to deletion by co-infused anti-CD20. To date, the ability of B-lymphocytes to exert regulatory functions suppressing diabetogenic T-cells in NOD mice is reported to primarily occur through IL-10 production (7). Indeed, a significantly higher proportion B-lymphocytes capable of producing IL-10 upon LPS stimulation (B10 cells) were recovered from mice transiently treated with BAFFR-Fc than ctrl mAb (Figure 4B). Due to their extremely low numbers it was not possible to quantify B10 cells from NOD mice transiently receiving combo therapy. However, transient anti-CD20 mono-therapy depleted such population (Figure 4B). Similarly, secreted levels of IL-10 were also greater in BAFFR-Fc treated mice than in controls upon either LPS or anti-IgM plus anti-CD40 stimulation (Figure 4C). We then asked whether the increased IL-10 producing and memory B-lymphocytes (Figure 2C) present after BAFFR-Fc treatment are distinct or overlapping populations. Similar to what has been reported in humans (41), B10 cells are enriched in the memory subsets and were not changed by BAFFR-Fc mono-therapy (Figure 4D). However, the overall BAFFR-Fc mono-therapy induced increase in B10 cells primarily occurred in the non-memory subset (Figure 4D).
Figure 4. Short-term BAFFR-Fc mediated B-lymphocyte depletion elicits prolonged T1D protection and remaining B-lymphocytes are regulatory in nature.
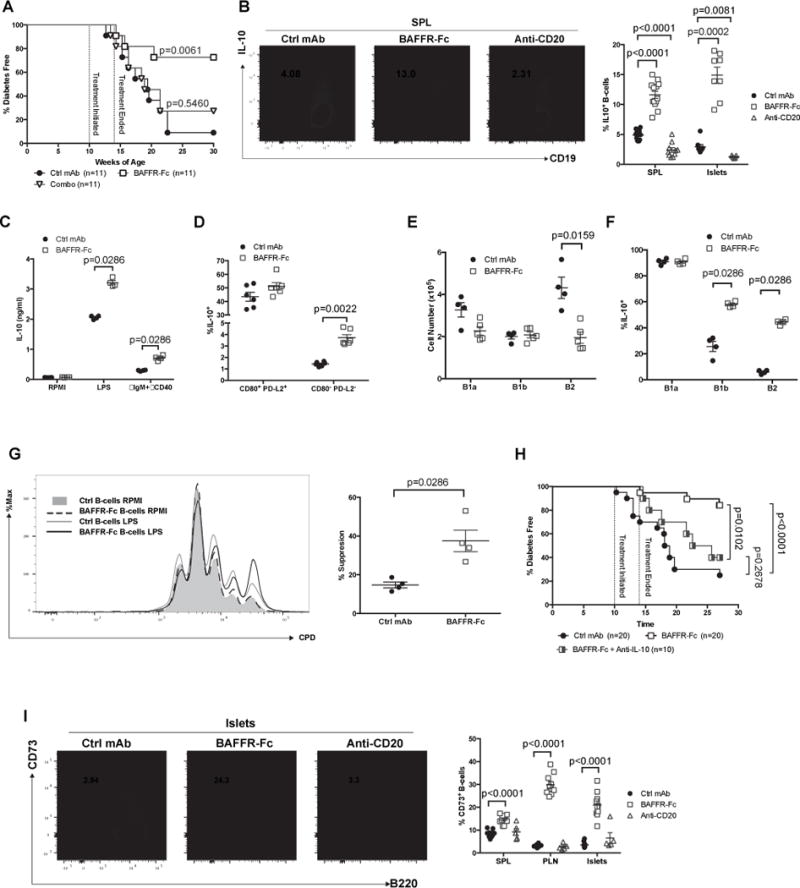
A) NOD female mice treated with BAFFR-Fc alone or combination with anti-CD20 between 10–14 weeks of age were assessed for diabetes development. Diabetes development in each of these experimental groups was statistically compared to NOD females treated with ctrl mAb from 10–14 weeks of age (Mantel-Cox analysis). B) Representative flow cytometric plot showing frequency of B10 cells among CD19+ cells in spleens of NOD mice treated with ctrl mAb, BAFFR-Fc, or anti-CD20 from 10–14 weeks of age (left panel). Summary of the percentage of B10 cells in the spleens and islets (right panel). Data pooled from three and two independent experiments for B10 cells in the spleens and islets respectively. C) 1.0×105 sort-purified splenic B-lymphocytes from ctrl mAb or BAFFR-Fc-treated NOD mice were cultured for 3 days with or without LPS or anti-IgM plus anti-CD40 and culture supernatant was subsequently measured by ELISA for IL-10. Data pooled from two independent experiments. D) Frequency of B10 cells amongst CD80+ PD-L2+ or CD80− PD-L2− B-lymphocytes in spleens of NOD mice treated with ctrl mAb or BAFFR-Fc from 10–14 weeks of age. E-F) Number of B1a, B1b, B2 B-lymphocytes in the peritoneal cavity (E) and percentage of B10 cells within the B1a, B1b, B2 B-lymphocytes subsets (F) in NOD female mice treated with ctrl mAb or BAFFR-Fc from 10–14 weeks of age. G) 1.0×105 CD25− CD4+ T-cells from 10 week-old NOD mice were labeled with Cell Proliferation Dye eFluor450 (CPD) and then stimulated with 5 μg/mL plate bound anti-CD3ε and co-cultured for 3 days with 1.0×105 sorted splenic B-lymphocytes from ctrl mAb or BAFFR-Fc-treated NOD mice with or without 10 μg/mL LPS stimulation (left panel). Quantification of percent T-cell proliferation suppression (right panel) (n=4 per group). Data are representative of two experiments. H) NOD female mice treated with BAFFR-Fc or ctrl mAb between 10–14 weeks of age were assessed for diabetes development. Upon cessation of transient BAFFR-Fc monotherapy a separate cohort of NOD females subsequently received twice-weekly injections of anti-IL-10 while being monitored for diabetes development. Pair-wise comparisons of diabetes development between each group were calculated using Mantel-Cox analysis. I) Representative flow cytometric plots showing islet infiltrating B-lymphocytes expressing CD73 among live CD19+ B220+ cells from NOD mice treated with ctrl mAb, BAFFR-Fc, or anti-CD20 from 10–14 weeks of age (left panel). Percent CD73+ B-lymphocytes from spleens, PLNs, or islets of NOD mice treated with ctrl mAb, BAFFR-Fc, or anti-CD20 from 10–14 weeks of age (right panel). Data pooled from two independent experiments. All p-values were calculated using Mann-Whitney analyses. Each symbol represents an individual mouse except for BAFFR-Fc group in C and D where each symbol represents sorted B-lymphocytes pooled from 2 or 3 mice; small horizontal lines indicate the Mean±SEM.
Peritoneal cavity B cells have been reported to possess regulatory functions through IL-10 production (42). Thus, we also assessed the effects of BAFF blockade on peritoneal B cells numbers and their ability to produce IL-10. We found the number of B1a and B1b cells are not affected by BAFF blockade, whereas the B2 subset was preferentially depleted (Figure 4E). The proportions of B10 cells were found most frequently in the B1a cell subsets and were not changed by BAFF blockade (Figure 4F). However, BAFFR-Fc mono-therapy elicited a significant increase in B10 cells amongst the peritoneal B1b and B2 subsets (Figure 4F). We also found IL-10 was produced at significantly higher levels by TACIhi than TACIlow splenic B-lymphocytes from untreated NOD mice (Supplemental Figure 3A). The TACIhi B-lymphocytes can in theory rely on APRIL for their survival upon BAFF-starvation, and activation of this alternative survival axis might promote IL-10 production (43).
The ability of B-lymphocytes surviving BAFFR-Fc treatment to exert regulatory functions in vitro was then assessed. We compared the ability of unstimulated B-lymphocytes or subsequent to LPS exposure, a condition that can induce IL-10 production (44), to possibly suppress CD4+ CD25− T-cell proliferation. On a per cell basis, LPS stimulated B-lymphocytes from mice that had undergone transient BAFF blockade suppressed T-cell proliferation to a significantly greater extent than those from controls (Figure 4G). Therefore, we tested whether T1D protection observed in NOD mice transiently treated with BAFFR-Fc was dependent on IL-10 production. The administration of an IL-10 neutralizing antibody (JES5-2A5) in NOD mice following transient BAFFR-Fc treatment abrogated the T1D protective effects of this reagent (Figure 4H). We recently identified a subset of CD73+ B-lymphocytes capable of suppressing the activity of NOD diabetogenic T-cells (29). Thus, we characterized the extent B-lymphocytes expressed CD73 following the various depletion regimens. The proportion of CD73+ B-lymphocytes was increased in the spleens, PLNs, and islets of BAFFR-Fc treated mice, while this potentially suppressive population was no longer enriched in the anti-CD20 treated cohort (Figure 4I). TACI expression was significantly higher on CD73 expressing than negative B-lymphocytes (Supplemental Figure 3B). These collective data indicate transient BAFFR-Fc treatment of NOD mice engenders an enrichment of Bregs that can provide for long term T1D protection through IL-10 production, and perhaps CD73 expression, but such disease protective cells are sensitive to depletion by co-treatment with anti-CD20.
B-lymphocytes remaining after BAFFR-Fc treatment have a diminished capacity to present autoantigens to diabetogenic T-cells
Autoreactive B-lymphocytes primarily contribute to T1D in NOD mice and presumably humans by being the subset of APC that most efficiently expands autoreactive CD4+ and CD8+ T-cells (2–6). It was not technically feasible to isolate sufficient numbers of B-lymphocytes from combo-treated mice to assess their APC-capacity. However, we did assess the APC-capacity of B-lymphocytes remaining in NOD mice after transient BAFFR-Fc mono-therapy. These B-lymphocytes had significantly reduced MHC class I and II levels (Figure 5A) and increased CD80 and CD86 expression (Figure 5B). B-lymphocytes sorted from either ctrl mAb or BAFFR-Fc treated mice were tested for ability to serve as APCs supporting proliferation of purified CD4+ autoreactive BDC2.5 TCR transgenic T-cells in the presence of an autoantigenic mimotope peptide. B-lymphocytes from BAFFR-Fc mono-therapy treated mice exhibited a significantly lower ability than those from controls to support proliferation of autoantigenic-peptide-stimulated BDC2.5 CD4+ T-cells (Figure 5C). Similar results were obtained with 1:1 and 2:1 B- to CD4+ T-lymphocyte ratios (Figure 5C). This diminished APC capacity may at least in part be due to reduced cell surface expression of MHC molecules. The decreased proliferation of BDC2.5 CD4+ T-cells in vitro is unlikely due to active suppression by the two aforementioned B10 and CD73+ B-lymphocyte populations as adenosine is rapidly degraded in serum containing culture medium (45) and without LPS stimulation, B-lymphocytes from BAFFR-Fc treated NOD mice failed to produce IL-10 (Figure 4C). Collectively, these data suggest that in addition to becoming enriched for a regulatory phenotype, B-lymphocytes surviving BAFFR-Fc mono-therapy in NOD mice may also contribute to T1D resistance through a diminished capacity to serve as pathogenic APC. Because regulatory B-lymphocytes have been reported to modulate T-cell proliferation by dampening antigen presenting capacity of DCs in a mouse model of Multiple Sclerosis (46), we also assessed the effects of BAFFR-Fc on the ability of DCs to present mimotope peptide. However, we did not find any changes of MHC class II, CD80, CD86, PD-L1, PD –L2 surface expression on splenic DCs (data not shown). When purified DCs from ctrl mAb or BAFFR-Fc treated NOD mice were co-cultured with BDC2.5 CD4+ T-cells in the presence of mimotope peptide, similar levels of cell division were observed (data not shown). Thus, BAFFR-Fc directly diminished the ability of B-lymphocytes to present antigen without altering the ability of DCs to do so.
Figure 5. The B-lymphocytes remaining after BAFF blockade treatment have diminished capacity to present autoantigens to diabetogenic T cells.
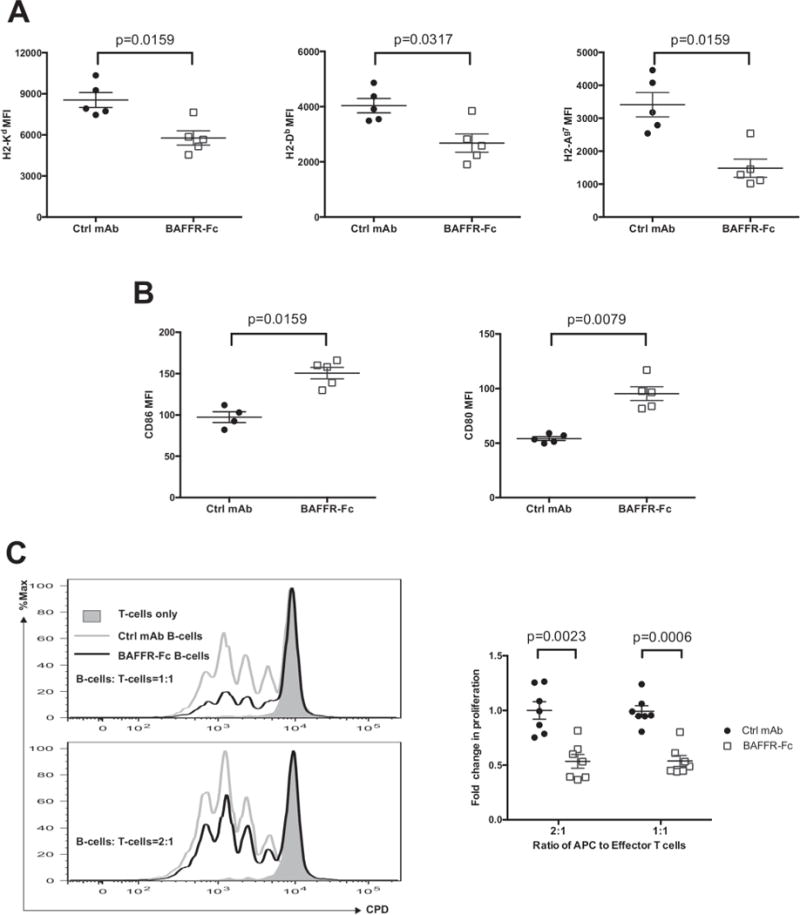
A, B) Assessment by mean fluorescence intensity (MFI) of specific antibody staining of MHC-I (H2-Kd and H2-Db, left and middle panel) or MHC-II (H2-Ag7, right panel) (A) or CD80 and CD86 (B) expression by viable splenic B-lymphocytes from NOD mice treated with ctrl mAb or BAFFR-Fc from 10 to 14 weeks of age (n=5 per group). Data are representative of two experiments. C) 5.0×104 CD4+ T cells purified from BDC2.5 TCR transgenic mice were labeled with Cell Proliferation Dye eFluor450 (CPD) and co-cultured for 3 days with or without 5.0×104 (top) or 1.0×105 (bottom) purified B-lymphocytes from pooled spleens of NOD mice treated with ctrl mAb or BAFFR-Fc from 10 to 14 weeks of age in the presence of 1μM BDC2.5 mimitope peptide (left panels). Summary of the quantification of division index (right panel). Data pooled from two independent experiments. Results are presented relative to the mean of the division index in presence of B-lymphocytes purified from ctrl mAb treated mice. All p-values were calculated using Mann-Whitney analyses. Each symbol represents an individual mouse except for BAFFR-Fc group in C where each symbol represents response in the presence of sorted B-lymphocytes pooled from 2 or 3 mice; small horizontal lines indicate the Mean±SEM.
Rebounding B-lymphocytes after cessation of transient BAFFR-Fc treatment are characterized by increased IL-10-production
We reasoned the B-lymphocytes that survive and/or rebound in NOD mice after transient BAFFR-Fc or combo-treatment may numerically or functionally differ from those in controls. Hence, we analyzed B-lymphocytes subset recovery at the end of the incidence study (30 weeks of age, 16 weeks after cessation of treatment). Following cessation of transient BAFFR-Fc mono-therapy, splenic B-lymphocytes re-populated to the same level as in ctrl mAb-treated mice (Figure 6A). Paradoxically, both the percentage and yields of splenic B-lymphocytes were increased to a significantly greater extent following cessation of combo treatment than in controls (Figure 6A). Following cessation of transient BAFFR-Fc mono-therapy, all examined B-lymphocyte subsets returned to similar levels as in controls (Figure 6B). Interestingly, FO, but not MZ B-lymphocytes preferentially expanded in mice that received transient combo therapy compared to controls (Figure 6B). Our past studies indicate B-lymphocytes entering the pancreatic islets of NOD mice have an FO-like phenotype (47). These rebounding, potentially diabetogenic, FO B-lymphocytes may explain the difference in T1D protection in NOD mice that underwent sustained versus transient combo treatment. Additionally, the potentially regulatory CD73+ B-lymphocytes that were originally proportionally increased in NOD mice immediately after cessation of transient BAFFR-Fc mono-therapy (see Figure 4F) returned to splenic control-like levels 16 weeks later (Figure 6C). Proportions of splenic CD73+ B-lymphocytes were lower than in controls at 16 weeks following cessation of transient combo therapy (Figure 6C). However, as was the case immediately upon cessation of transient BAFFR-Fc mono-therapy (Figure 4B), 16 weeks later the proportion of B10 cells among repopulating B-lymphocytes remained elevated within NOD mice that had received this treatment (Figure 6D). Thus, long after treatment cessation, rebounding B-lymphocytes in NOD mice that had received BAFFR-Fc mono-therapy retain a higher proportion of IL-10 producing, but not CD73+ B-lymphocytes.
Figure 6. Rebounding B-lymphocytes after cessation of transient BAFFR-Fc mono-therapy are characterized by increased IL-10 production.
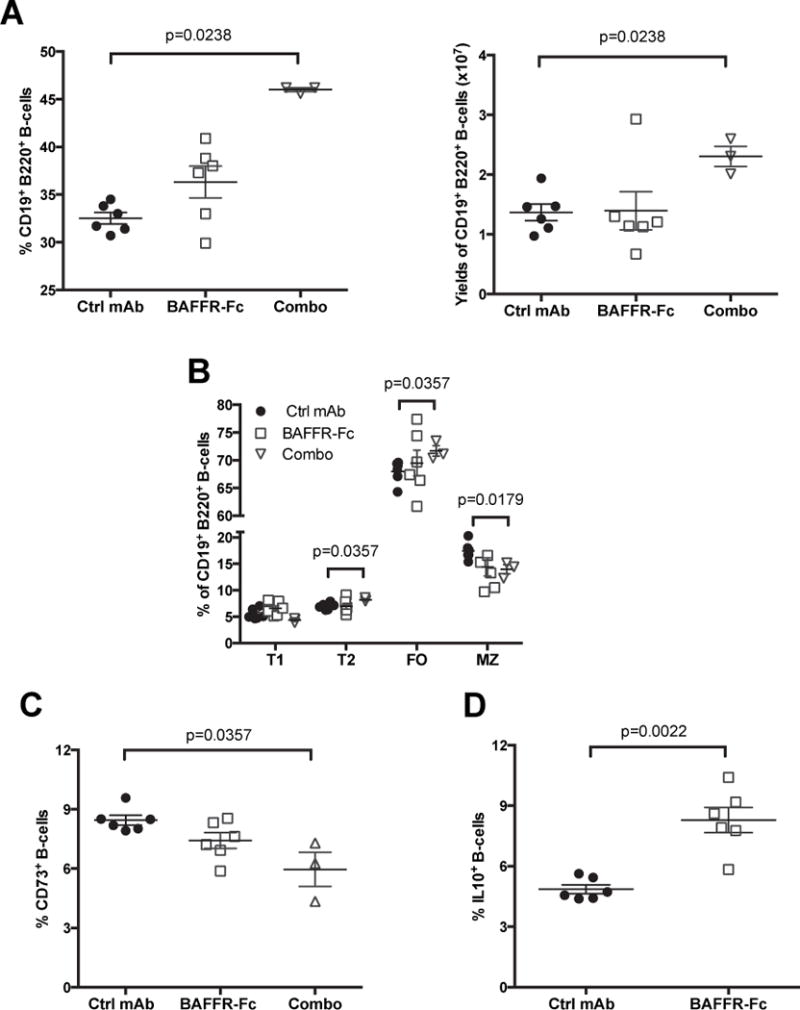
A, B) Splenocytes from non-diabetic NOD mice that had been treated from 10–14 weeks of age with BAFFR-Fc alone or in combination with anti-CD20 (combo) or ctrl mAb antibody were assessed at the 30 weeks of age end of T1D incidence study time point for proportions (A, left panel) and absolute numbers (A, right panel) of B-lymphocytes and their subsets (B). C, D) Proportions of CD73+ and IL-10+ cells among the same splenic B-lymphocytes. All p-values were calculated using Mann-Whitney analyses, each symbol represents an individual mouse; small horizontal lines indicate the Mean±SEM.
BAFFR-Fc treatment does not expand Tregs or directly decrease activity of diabetogenic T-cells in NOD mice
We assessed if B-lymphocyte depletion by BAFFR-Fc treatment altered T-cell numbers, phenotypes, or diabetogenic potential in NOD mice. Consistent with observations in NOD.BAFF−/− mice (48), total numbers of both CD4+ and CD8+ T-cells were reduced within spleens and PLNs following transient BAFFR-Fc or combo therapy treatment from 10–14 weeks of age (Figure 7A). Proportions of islet-infiltrating total T-cells were increased in mice given BAFFR-Fc or combo treatment likely due to the diminished levels of B-lymphocytes at this site (Figure 7A). However, the ratios of CD4+ to CD8+ T-cells were not altered (data not shown). The decrease in splenic CD4+ and CD8+ T-cells corresponds to a decrease in Ki67+ cells likely due to loss of B-lymphocyte support (Figure 7B). Despite numerical changes in the CD4 and CD8 compartments, the frequency of diabetogenic BDC2.5 CD4+ and NY8.3 CD8+ T-cells, relative proportions of activated, and naïve CD4+ and CD8+ T cells did not differ from controls in spleens or PLNs from NOD mice receiving BAFFR-Fc and combo therapy (data not shown). Interestingly, despite no change in the PLN, the proportions of splenic Foxp3-eGFP+ Tregs were decreased in combo, but not BAFFR-Fc mono-therapy treated mice (Figure 7C). We then directly assessed whether Treg induction was a mechanism of BAFF-blockade-mediated T1D protection. Subsequent anti-CD25 (PC61 clone) co-infusion to deplete CD25+ Foxp3+ Tregs did not abrogate the T1D protective effect elicited in NOD mice by prior transient BAFF-blockade (Figure 7D). Previous studies suggest there could be direct signaling of BAFF on T-cells (49, 50). Thus, we tested whether BAFFR-Fc therapy directly affected the diabetogenicity of NOD T-cells. Purified T-cells from 10-week-old NOD mice were transferred into lymphocyte-deficient NOD-scid recipients that then received either BAFFR-Fc or ctrl mAb treatment. NOD-scid recipients treated with BAFF blockade or ctrl mAb developed T1D at a similar rate (Figure 7E). This indicated the diabetogenic activity of NOD T-cells is not directly suppressed by BAFF blockade.
Figure 7. Despite numerical changes the diabetogenic potential of T-cells is not altered after B-lymphocyte depletion.
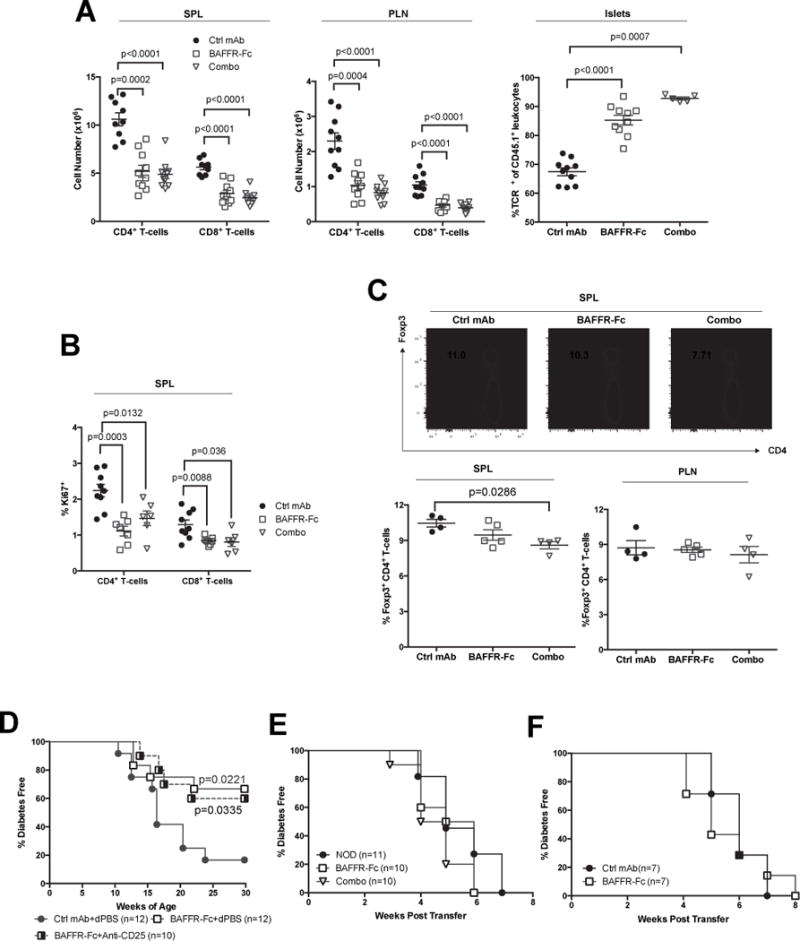
A) Number of CD4 (TCRβ+ CD4+) and CD8 (TCRβ+ CD8+) T-cells in the spleens (left panel), PLNs (middle panel), and proportions of islet infiltrating TCRβ+ T-cell among CD45+ leukocytes (right panel) in NOD female mice treated with ctrl mAb, BAFFR-Fc, or combo from 10–14 weeks of age. Data are pooled from two independent experiments. B) Proportions of CD4 and CD8 splenic T-cells expressing Ki67 in A. C) Representative flow cytometric plots showing splenic CD4+ T-cells expressing FoxP3 driven eGFP in NOD-Foxp3egfp mice treated with ctrl mAb, BAFFR-Fc, or combo from 10–14 weeks of age (top panel). Summary of the percentage of Foxp3-eGFP+ amongst CD4+ T-cells in the spleen and PLN (bottom panels). D) NOD female mice treated with BAFFR-Fc or ctrl mAb from 10–14 weeks of age then started to receive anti-CD25 or dPBS every two weeks and also monitored for T1D. E) 5×106 purified splenic T-cells from 10-week-old female NOD mice were transferred into female NOD-scid recipients, which then began weekly treatment with ctrl mAb or BAFFR-Fc and monitored for T1D. F) 5×106 purified splenic T-cells from BAFFR-Fc or combo treated at the end of T1D incidence study (35 weeks) and as a control from 15-week-old pre-hyperglycemic NOD mice were transferred into female NOD-scid recipients that were then monitored for T1D. Incidence study p-values were calculated using Mantel-Cox analysis; Mann-Whitney analysis was performed for all scattered plots. Each symbol represents an individual mouse; small horizontal lines indicate the Mean±SEM.
Finally, we tested whether continuous BAFFR-Fc or combo-therapy changed the intrinsic diabetogenicity of T-cells in long-term T1D protected mice. Purified splenic T-cells from continuous BAFFR-Fc or combo-treated NOD mice transferred T1D to NOD-scid recipients with equal efficiency as those from 15-week-old pre-hyperglycemic controls (Figure 7F). These collective results indicate depletion of B-lymphocytes by BAFFR-Fc or combo-therapy does not alter diabetogenic T-cell activity. Instead pathogenic T-cells are functionally suppressed by Bregs in NOD mice given BAFF blockade. An additional, non-mutually exclusive mechanism is BAFF blockade likely deprives diabetogenic T-cells of the APC activity they normally require to be expanded and activated to pathogenic levels. The normal pathogenic dependence of diabetogenic T-cells on antigen presenting B-lymphocytes in NOD mice is likely bypassed by an ability of such autoreactive effectors to instead undergo pathogenic homeostatic expansion when transferred into the lymphopenic environment of NOD-scid recipients. Support for this latter conclusion is provided by the finding that the proportion of T-cells showing an activated phenotype was much higher following their engraftment into NOD-scid recipients than was observed in the primary treated donor mice (Supplemental Figure 4). Together, these data indicate BAFF blockade does not have a direct effect on diabetogenic T-cells.
Discussion
While several anti-CD20 mediated B-lymphocyte depleting antibodies prevent (9), and, in some cases, reportedly reverse T1D in NOD mice (10, 11), a clinical study of rituximab use in recent disease onset patients showed only transient efficacy (17). Our previous study found that intra-islet B-lymphocytes in NOD mice down-regulate surface expression of CD20 (8), and if this also occurs in humans, it could possibly explain the limited effectiveness of rituximab as a clinical T1D intervention. Approaches targeting a broader range of B-lymphocytes using combined depleting reagents may improve clinical outcomes. However, we have shown here that in certain scenarios, the combination of therapeutic agents, in this case transient co-treatment with anti-CD20 and BAFFR-Fc, may be more detrimental than using the latter reagent alone in the clinical setting.
BAFFR-Fc treatment induced the depletion of mature recirculating B-lymphocyte in the bone marrow, as well as splenic FO, MZ, T1, and T2 subsets. Pre- and immature B-lymphocytes in the bone marrow are not depleted, consistent with the finding BAFF signaling is dispensable for early B lymphopoiesis (51). BAFF deprivation compensated for the heterogeneity of CD20 expression on B-lymphocytes, resulting in enhanced depletion by combo therapy. The diminished levels of islet infiltrating B-lymphocytes in anti-CD20 treated mice likely reflects their elimination prior to entering this site. Combo therapy induced a further depletion indicating B-lymphocytes surviving after BAFFR-Fc or anti-CD20 treatment are non-overlapping populations. However, the similar degree of disease resistance offered by continuous BAFFR-Fc alone and combo-therapy argues T1D protection does not require the broader B-lymphocyte depletion elicited by the latter approach. In addition, secondary humoral responses against an exogenous antigen were nearly intact in NOD mice given BAFFR-Fc mono-therapy. This observation is consistent with the survival of BAFF-independent memory B-lymphocytes (52). Our finding would also indicate the memory B-lymphocytes that survive after BAFF blockade are likely T1D irrelevant populations or alternatively enriched for B10 cells. These data are in line with the observation that autoreactive B-lymphocytes have increased dependence on BAFF for their survival compared to non-autoreactive populations (53). Therefore, while transient BAFFR-Fc mono-therapy offers long-term T1D protection, it should have minimal impacts on pre-existing memory B-lymphocytes and serum titers arising from childhood vaccines.
A previous study found transient treatment with the MB20.11 CD20 specific antibody elicited T1D protection when initiated in 5-week-old, but not 15-week-old NOD mice (9). Similarly, we found that when initiated in 10-week-old NOD mice, anti-CD20 mono-therapy failed to prevent T1D development. As noted above an accumulation of intra-islet B-lymphocytes that have become CD20 negative may at least partially explain why antibodies directed against this molecule do not elicit a late disease stage T1D prevention effect in NOD mice. However, previous studies did not assess the possibility of anti-idiotypic antibody responses against the MB20.11 anti-CD20 reagent. Our current study indicates an anti-idiotypic antibody response in an autoimmune setting may partially explain the failure of anti-CD20 to exert a late disease stage T1D intervention effect. 5-week-old NOD mice likely have a potentially more limited BCR repertoire than that present >10-weeks of age, allowing MB20.11 treatment to be a more effective early disease stage T1D intervention due to a lessened anti-idiotype response. Another possible explanation is that in our studies, repeated MB20.11 injections result in a feedback loop leading to ever-rising anti-idiotypic response not occurring with transient treatment. In addition, the continuous exposure to MB20.11 allows the generation in NOD mice of isotype switched, and therefore likely high affinity, neutralizing antibodies. The presence of isotype switched neutralizing antibodies also indicates cognate T-cell involvement in MB20.11 clearance. Although there was no reported anti-idioytpe response against rituximab in the T1D clinical trial, in a separate trial using anti-CD20 as a possible SLE intervention anti-chimeric responses account for the poor B-lymphocyte depletion observed in some patients (54). Collectively, these data suggest in efforts to develop T1D immunotherapies, assessment and minimization of drug directed antibody formation is required to improve possible intervention approaches.
In contrast to the NOD.Igμ−/− stock that is insulitis free (1), when B-lymphocyte depletion was initiated in standard NOD mice at a late prodromal stage of T1D development, insulitis levels were not diminished. This holds true even with the combination of BAFFR-Fc and anti-CD20. These data suggest B-lymphocyte depletion is less likely to inhibit further insulitis progression when β-cell autoimmunity is already established. However, the fact NOD mice receiving continuous or transient BAFFR-Fc remained normoglycemic suggests the infiltrating lymphocytes are maintained in an “innocuous” rather than “destructive” state. Therefore, we explored possible regulatory mechanisms that could restrain the pathogenicity of islet infiltrating lymphocytes. We did not find an increased percentage of Tregs after B-lymphocyte depletion, as has been reported in other studies (8, 13). Furthermore, Treg depletion by anti-CD25 does not abrogate T1D protection seen in NOD mice given transient BAFFR-Fc mono-therapy, suggesting other modes of regulation may be involved. Mounting evidence suggests some B-lymphocytes can negatively regulate immune responses and inflammation (55). The published regulatory functions of B-lymphocytes have been primarily through production of IL-10 (7). Our findings that B10 cells are enriched after transient BAFFR-Fc treatment would indicate either this population is less susceptible to BAFF starvation or alternatively, APRIL signaling stimulates IL-10 production. Anti-IL-10 also abrogated the T1D protective effect of transient BAFF blockade. These data indicate T1D protection observed in NOD mice given transient BAFFR-Fc treatment was primarily dependent on IL-10 production. However, this finding does not exclude the possibility of additional IL-10 independent means for B-lymphocytes to exert T1D protective regulatory functions in NOD mice and possibly humans (56). T1D resistance elicited in NOD mice by transient BAFFR-Fc treatment also coincided with an initial expansion of a CD73+ B-lymphocytes. We recently found B-lymphocytes in NOD-Aicdanull mice convert to a CD73+ phenotype capable of suppressing diabetogenic T cell responses through production of immunosuppressive adenosine (29). However, following cessation of treatment, proportions of CD73+ B-lymphocytes do not remain expanded in NOD mice in which long term T1D resistance was established by transient BAFFR-Fc mono-therapy. Hence, while the CD73+ B-lymphocytes that are initially expanded in NOD mice as a consequence of transient BAFFR-Fc mono-therapy might be able to exert T1D protective regulatory activity, they may only do so in a short-term fashion. Instead, our collective data indicate long term T1D resistance established in NOD mice that had undergone transient BAFFR-Fc monotherapy results from an expanded and maintained population(s) of IL-10 producing B-lymphocytes. Meanwhile, the lack of T1D protection in transient combo-treated mice is likely due to the deletion of the regulatory populations (possibly including CD73+ B-lymphocytes and B10 cells) mediated by the co-infused anti-CD20. It remains to be determined to what extent IL-10 producing and the CD73+ B-lymphocytes may be distinct or overlapping subsets of regulatory cells. Accumulating evidence suggests Bregs are a heterogeneous population and it is likely B-lymphocytes acquire different regulatory functions under distinct environment cues. In any case, our studies reveal that T1D protection after transient BAFFR-Fc mediated B-lymphocyte depletion is likely through intertwined mechanisms leading to an expansion of Bregs. Our results also indicate that the potential for B-lymphocyte targeting approaches to ultimately serve as a clinically applicable T1D intervention strategy is more dependent on the pattern rather than the overall level of cellular depletion they mediate.
Supplementary Material
Acknowledgments
We thank staff within The Jackson Laboratory’s Flow Cytometry service and Research Animal Facility for technical support.
Footnotes
Source of Support:
DVS is supported by NIH grants DK-46266, DK-95735, and OD-020351. JJR1 is supported by NIH Fellowship 1F32DK111078. A part of this work was also supported by NCI grant P30CA034196. MAA is supported by NIH grant P01-AI42288.
Contributions: QW designed and conducted experimentation, interpreted data, and wrote the manuscript; JJR1 contributed to discussion of data interpretation and writing of the manuscript; JJR2, SW, and MAA contributed to experimentation; RE helped to interpret data; DVS contributed to study conception, supervised experimental effort, and writing of the manuscript.
Abbreviations: BALB/cJ (BALB/c); Bone Marrow (BM); C57BL/6J (B6); combination treatment with BAFFR-Fc and anti-CD20 (Combo); IL-10 producing B-lymphocytes (B10 cells); insulin autoantibodies (IAA); irrelevant control murine IgG1 antibody (ctrl mAb); Mean fluorescence intensity (MFI); NOD/ShiLtDvs (NOD); NOD.129X1(Cg)-Foxp3tm2Tch/DvsJ (NOD-Foxp3egfp); NOD.Cg-Prkdcscid Emv30b/Dvs (NOD-scid); NOD.Cg-Tg(TcraBDC2.5,TcrbBDC2.5)1Doi/DoiJ CD4 T-cell clones (BDC2.5); pancreatic lymph nodes (PLN); regulatory B-lymphocytes (Bregs); type 1 diabetes (T1D).
References
- 1.Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med. 1996;184:2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silveira PA, Johnson E, Chapman HD, Bui T, Tisch RM, Serreze DV. The preferential ability of B lymphocytes to act as diabetogenic APC in NOD mice depends on expression of self-antigen-specific immunoglobulin receptors. Eur J Immunol. 2002;32:3657–3666. doi: 10.1002/1521-4141(200212)32:12<3657::AID-IMMU3657>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 3.Noorchashm H, Lieu YK, Noorchashm N, Rostami SY, Greeley SA, Schlachterman A, Song HK, Noto LE, Jevnikar AM, Barker CF, Naji A. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol. 1999;163:743–750. [PubMed] [Google Scholar]
- 4.Hulbert C, Riseili B, Rojas M, Thomas JW. B cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J Immunol. 2001;167:5535–5538. doi: 10.4049/jimmunol.167.10.5535. [DOI] [PubMed] [Google Scholar]
- 5.Falcone M, Lee J, Patstone G, Yeung B, Sarvetnick N. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. J Immunol. 1998;161:1163–1168. [PubMed] [Google Scholar]
- 6.Marino E, Batten M, Groom J, Walters S, Liuwantara D, Mackay F, Grey ST. Marginal-zone B-cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T-cells. Diabetes. 2008;57:395–404. doi: 10.2337/db07-0589. [DOI] [PubMed] [Google Scholar]
- 7.Kleffel S, Vergani A, Tezza S, Ben Nasr M, Niewczas MA, Wong S, Bassi R, D’Addio F, Schatton T, Abdi R, Atkinson M, Sayegh MH, Wen L, Wasserfall CH, O’Connor KC, Fiorina P. Interleukin-10+ regulatory B cells arise within antigen-experienced CD40+ B cells to maintain tolerance to islet autoantigens. Diabetes. 2015;64:158–171. doi: 10.2337/db13-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serreze DV, Chapman HD, Niens M, Dunn R, Kehry MR, Driver JP, Haller M, Wasserfall C, Atkinson MA. Loss of intra-islet CD20 expression may complicate efficacy of B-cell-directed type 1 diabetes therapies. Diabetes. 2011;60:2914–2921. doi: 10.2337/db11-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiu Y, Wong CP, Bouaziz JD, Hamaguchi Y, Wang Y, Pop SM, Tisch RM, Tedder TF. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol. 2008;180:2863–2875. doi: 10.4049/jimmunol.180.5.2863. [DOI] [PubMed] [Google Scholar]
- 10.Fiorina P, Vergani A, Dada S, Jurewicz M, Wong M, Law K, Wu E, Tian Z, Abdi R, Guleria I, Rodig S, Dunussi-Joannopoulos K, Bluestone J, Sayegh MH. Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes. 2008;57:3013–3024. doi: 10.2337/db08-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu CY, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, Shlomchik MJ, Wen L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zekavat G, Rostami SY, Badkerhanian A, Parsons RF, Koeberlein B, Yu M, Ward CD, Migone TS, Yu L, Eisenbarth GS, Cancro MP, Naji A, Noorchashm H. In vivo BLyS/BAFF neutralization ameliorates islet-directed autoimmunity in nonobese diabetic mice. J Immunol. 2008;181:8133–8144. doi: 10.4049/jimmunol.181.11.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marino E, Villanueva J, Walters S, Liuwantara D, Mackay F, Grey ST. CD4(+)CD25(+) T-cells control autoimmunity in the absence of B-cells. Diabetes. 2009;58:1568–1577. doi: 10.2337/db08-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001;44:1313–1319. doi: 10.1002/1529-0131(200106)44:6<1313::AID-ART223>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 15.Stohl W. B lymphocyte stimulator protein levels in systemic lupus erythematosus and other diseases. Curr Rheumatol Rep. 2002;4:345–350. doi: 10.1007/s11926-002-0044-7. [DOI] [PubMed] [Google Scholar]
- 16.Tan SM, Xu D, Roschke V, Perry JW, Arkfeld DG, Ehresmann GR, Migone TS, Hilbert DM, Stohl W. Local production of B lymphocyte stimulator protein and APRIL in arthritic joints of patients with inflammatory arthritis. Arthritis Rheum. 2003;48:982–992. doi: 10.1002/art.10860. [DOI] [PubMed] [Google Scholar]
- 17.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, Raskin P, Rodriguez H, Schatz DA, Wherrett D, Wilson DM, Lachin JM, Skyler JS, C. D. S. G. Type 1 Diabetes TrialNet Anti Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pescovitz MD, Greenbaum CJ, Bundy B, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, Moran A, Raskin P, Rodriguez H, Schatz DA, Wherrett DK, Wilson DM, Krischer JP, Skyler JS, C. D. S. G. Type 1 Diabetes TrialNet Anti B-lymphocyte depletion with rituximab and beta-cell function: two-year results. Diabetes Care. 2014;37:453–459. doi: 10.2337/dc13-0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh HJ, Zhang D, Brunetta PG. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–233. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, Leon MG, Tanasescu C, Nasonov E, Lan JL, Pineda L, Zhong ZJ, Freimuth W, Petri MA, B.-S. Group Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 21.Stohl W, Merrill JT, McKay JD, Lisse JR, Zhong ZJ, Freimuth WW, Genovese MC. Efficacy and safety of belimumab in patients with rheumatoid arthritis: a phase II, randomized, double-blind, placebo-controlled, dose-ranging Study. J Rheumatol. 2013;40:579–589. doi: 10.3899/jrheum.120886. [DOI] [PubMed] [Google Scholar]
- 22.Genovese MC, Kinnman N, de La Bourdonnaye G, Pena Rossi C, Tak PP. Atacicept in patients with rheumatoid arthritis and an inadequate response to tumor necrosis factor antagonist therapy: results of a phase II, randomized, placebo-controlled, dose-finding trial. Arthritis Rheum. 2011;63:1793–1803. doi: 10.1002/art.30373. [DOI] [PubMed] [Google Scholar]
- 23.Leete P, Willcox A, Krogvold L, Dahl-Jorgensen K, Foulis AK, Richardson SJ, Morgan NG. Differential Insulitic Profiles Determine the Extent of beta-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes. 2016;65:1362–1369. doi: 10.2337/db15-1615. [DOI] [PubMed] [Google Scholar]
- 24.Bonifacio E, Ziegler AG. Advances in the prediction and natural history of type 1 diabetes. Endocrinol Metab Clin North Am. 2010;39:513–525. doi: 10.1016/j.ecl.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Lin W, Seshasayee D, Lee WP, Caplazi P, McVay S, Suto E, Nguyen A, Lin Z, Sun Y, DeForge L, Balazs M, Martin F, Zarrin AA. Dual B cell immunotherapy is superior to individual anti-CD20 depletion or BAFF blockade in murine models of spontaneous or accelerated lupus. Arthritis Rheumatol. 2015;67:215–224. doi: 10.1002/art.38907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, Sato S, Tedder TF, Fujimoto M. Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol. 2010;184:4801–4809. doi: 10.4049/jimmunol.0902385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003;197:489–501. doi: 10.1084/jem.20021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ratiu JJ, Racine JJ, Hasham MG, Wang Q, Branca JA, Chapman HD, Zhu J, Donghia N, Philip V, Schott WH, Wasserfall C, Atkinson MA, Mills KD, Leeth CM, Serreze DV. Genetic and Small Molecule Disruption of the AID/RAD51 Axis Similarly Protects Nonobese Diabetic Mice from Type 1 Diabetes through Expansion of Regulatory B Lymphocytes. J Immunol. 2017 doi: 10.4049/jimmunol.1700024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serreze DV, Leiter EH, Hanson MS, Christianson SW, Shultz LD, Hesselton RM, Greiner DL. Emv30null NOD-scid mice. An improved host for adoptive transfer of autoimmune diabetes and growth of human lymphohematopoietic cells. Diabetes. 1995;44:1392–1398. doi: 10.2337/diab.44.12.1392. [DOI] [PubMed] [Google Scholar]
- 32.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, Chatila TA. Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol. 2007;178:2961–2972. doi: 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- 33.Johnson EA, Silveira P, Chapman HD, Leiter EH, Serreze DV. Inhibition of autoimmune diabetes in nonobese diabetic mice by transgenic restoration of H2-E MHC class II expression: additive, but unequal, involvement of multiple APC subtypes. J Immunol. 2001;167:2404–2410. doi: 10.4049/jimmunol.167.4.2404. [DOI] [PubMed] [Google Scholar]
- 34.Morissette MC, Gao Y, Shen P, Thayaparan D, Berube JC, Pare PD, Brandsma CA, Hao K, Bosse Y, Ettinger R, Herbst R, Humbles AA, Kolbeck R, Zhong N, Chen R, Stampfli MR. Role of BAFF in pulmonary autoantibody responses induced by chronic cigarette smoke exposure in mice. Physiol Rep. 2016;4 doi: 10.14814/phy2.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonifacio E, Atkinson M, Eisenbarth G, Serreze D, Kay TW, Lee-Chan E, Singh B. International Workshop on Lessons From Animal Models for Human Type 1 Diabetes: identification of insulin but not glutamic acid decarboxylase or IA-2 as specific autoantigens of humoral autoimmunity in nonobese diabetic mice. Diabetes. 2001;50:2451–2458. doi: 10.2337/diabetes.50.11.2451. [DOI] [PubMed] [Google Scholar]
- 36.Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- 37.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. 2009;9:491–502. doi: 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 38.Zuccarino-Catania GV, Sadanand S, Weisel FJ, Tomayko MM, Meng H, Kleinstein SH, Good-Jacobson KL, Shlomchik MJ. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat Immunol. 2014;15:631–637. doi: 10.1038/ni.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomayko MM, Steinel NC, Anderson SM, Shlomchik MJ. Cutting edge: Hierarchy of maturity of murine memory B cell subsets. J Immunol. 2010;185:7146–7150. doi: 10.4049/jimmunol.1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Goldschmidt T, Salter H. Possible allelic structure of IgG2a and IgG2c in mice. Mol Immunol. 2012;50:169–171. doi: 10.1016/j.molimm.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 41.Khoder A, Sarvaria A, Alsuliman A, Chew C, Sekine T, Cooper N, Mielke S, de Lavallade H, Muftuoglu M, Fernandez Curbelo I, Liu E, Muraro PA, Alousi A, Stringaris K, Parmar S, Shah N, Shaim H, Yvon E, Molldrem J, Rouce R, Champlin R, McNiece I, Mauri C, Shpall EJ, Rezvani K. Regulatory B cells are enriched within the IgM memory and transitional subsets in healthy donors but are deficient in chronic GVHD. Blood. 2014;124:2034–2045. doi: 10.1182/blood-2014-04-571125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maseda D, Candando KM, Smith SH, Kalampokis I, Weaver CT, Plevy SE, Poe JC, Tedder TF. Peritoneal cavity regulatory B cells (B10 cells) modulate IFN-gamma+CD4+ T cell numbers during colitis development in mice. J Immunol. 2013;191:2780–2795. doi: 10.4049/jimmunol.1300649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hua C, Audo R, Yeremenko N, Baeten D, Hahne M, Combe B, Morel J, Daien C. A proliferation inducing ligand (APRIL) promotes IL-10 production and regulatory functions of human B cells. J Autoimmun. 2016;73:64–72. doi: 10.1016/j.jaut.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 44.Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF. The development and function of regulatory B cells expressing IL-10 (B10 cells) requires antigen receptor diversity and TLR signals. J Immunol. 2009;182:7459–7472. doi: 10.4049/jimmunol.0900270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780–6786. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 46.Matsushita T, Horikawa M, Iwata Y, Tedder TF. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J Immunol. 2010;185:2240–2252. doi: 10.4049/jimmunol.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leeth CM, Racine J, Chapman HD, Arpa B, Carrillo J, Carrascal J, Wang Q, Ratiu J, Egia-Mendikute L, Rosell-Mases E, Stratmann T, Verdaguer J, Serreze DV. B-lymphocytes expressing an immunoglobulin specificity recognizing the pancreatic ss-cell autoantigen peripherin are potent contributors to type 1 diabetes development in NOD mice. Diabetes. 2016 doi: 10.2337/db15-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marino E, Walters SN, Villanueva JE, Richards JL, Mackay CR, Grey ST. BAFF regulates activation of self-reactive T cells through B-cell dependent mechanisms and mediates protection in NOD mice. Eur J Immunol. 2014;44:983–993. doi: 10.1002/eji.201344186. [DOI] [PubMed] [Google Scholar]
- 49.Ye Q, Wang L, Wells AD, Tao R, Han R, Davidson A, Scott ML, Hancock WW. BAFF binding to T cell-expressed BAFF-R costimulates T cell proliferation and alloresponses. Eur J Immunol. 2004;34:2750–2759. doi: 10.1002/eji.200425198. [DOI] [PubMed] [Google Scholar]
- 50.Huard B, Schneider P, Mauri D, Tschopp J, French LE. T cell costimulation by the TNF ligand BAFF. J Immunol. 2001;167:6225–6231. doi: 10.4049/jimmunol.167.11.6225. [DOI] [PubMed] [Google Scholar]
- 51.Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173:2245–2252. doi: 10.4049/jimmunol.173.4.2245. [DOI] [PubMed] [Google Scholar]
- 52.Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, Noelle RJ. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. 2008;180:3655–3659. doi: 10.4049/jimmunol.180.6.3655. [DOI] [PubMed] [Google Scholar]
- 53.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, Cyster JG. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 54.Albert D, Dunham J, Khan S, Stansberry J, Kolasinski S, Tsai D, Pullman-Mooar S, Barnack F, Striebich C, Looney RJ, Prak ET, Kimberly R, Zhang Y, Eisenberg R. Variability in the biological response to anti-CD20 B cell depletion in systemic lupus erythaematosus. Ann Rheum Dis. 2008;67:1724–1731. doi: 10.1136/ard.2007.083162. [DOI] [PubMed] [Google Scholar]
- 55.Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42:607–612. doi: 10.1016/j.immuni.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 56.Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol. 2015;27:479–486. doi: 10.1093/intimm/dxv038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.