

Abstract
Mass spectrometry (MS) has become an increasingly important technique to analyze proteins. In popular bottom-up MS-based proteomics, reduction and alkylation are routine steps to facilitate peptide identification. However, the reaction incompletion and side reactions may occur, which compromise the experimental results. In this work, we systematically evaluated the reduction step with the commonly used reagents, i.e., dithiothreitol, 2-mercaptoethanol, tris(2-carboxyethyl)phosphine, or tris(3-hydroxypropyl)phosphine, and alkylation with iodoacetamide, acrylamide, N-ethylmaleimide, or 4-vinylpyridine. By using digested peptides from a yeast whole-cell lysate, the number of proteins and peptides identified were very similar using four different reducing reagents. The results from four alkylating reagents, however, were dramatically different with iodoacetamide giving the highest number of peptides with alkylated cysteine and the lowest number of peptides with incomplete cysteine alkylation and side reactions. Alkylation conditions with iodoacetamide were further optimized. To identify more peptides with cysteine, Thiopropyl-Sepharose 6B resins were used to enrich them, and the optimal conditions were employed for the reduction and alkylation. The enrichment resulted in over three times more cysteine-containing peptides than without enrichment. Systematic evaluation of the reduction and alkylation with different reagents can aid in a better design of bottom-up proteomic experiments.
Introduction
Mass spectrometry (MS)-based proteomics is currently very powerful for protein identification and quantification.1–5 There are three major approaches, i.e. top-down, middle-down, and bottom-up proteomics.6–11 For bottom-up proteomics, proteins are digested enzymatically and/or chemically into peptides. The resulting peptides are separated by high-performance liquid chromatography (HPLC), and the eluted peptides are subsequently subjected to mass spectrometric (MS) analysis.1, 4, 12 Despite the fact that some protein structure information is compromised, bottom-up proteomics currently is still the most popular method because it has been proven to be extremely powerful to identify and quantify proteins,13–21 including the study of protein post-translational modifications (PTMs),22–33 and investigation of protein interactions with other proteins and small molecules.34–37
Disulfide bonds between sulfhydryl groups of cysteine side chains often regulate protein folding and final functional structures.38–40 A typical workflow for bottom-up proteomics includes the reduction of disulfide bonds and the alkylation of sulfhydryl groups.7, 41, 42 Without the reduction and alkylation, peptides involved in the disulfide bonds would be difficult to identify during database searching.43 Currently, the reduction and alkylation are routine steps for bottom-up proteomics.
Several reducing and alkylating reagents have been commonly used in this field. The following compounds have frequently been reported to serve as reducing reagents: dithiothreitol (DTT), 2-mercaptoethanol (2-ME), tris(2-carboxyethyl)phosphine (TCEP) and tris(3-hydroxypropyl)phosphine (THPP). Sulfur-containing reagents reduce disulfide bonds through the thiol-disulfide exchange (Eq. 1),44 while those containing phosphorous form phosphine oxide after disulfide reduction (Eq. 2).45
| (1) |
| (2) |
Regarding the alkylation, reagents typically alkylate nucleophiles through bimolecular nucleophilic substitution (S2N) for haloalkanes or Michael addition for maleimide.46 The following compounds have normally been employed for alkylation: iodoacetamide (IAA), acrylamide (AA), N-ethylmaleimide (N-EM), and 4-vinylpyridine (4-VP).41, 47–50
Although the reduction and alkylation are critical to bottom-up proteomics, this process may pose some problems, including the incompleteness of the reactions and side reactions. Besides the sulfhydryl group of cysteine, the alkylation may also happen at other chemical groups, including the amino group of the peptide N-terminus, and those on the side chains of lysine, aspartic acid, glutamic acid, methionine, and histidine.51, 52 Recently, Muller and Winter have reported the systematic evaluation of protein reduction and alkylation, which has also included side reactions on the side chains of tyrosine, serine, and threonine.53 Therefore, it is critical to push the desired reaction toward completion and minimize the side reactions. Compared to that paper,53 the current work has several differences: (1) The experimental designs are different. Here we compared only one parameter with all other parameters fixed for each individual experiment, while in that paper, different combinations of reducing and alkylating reagents were compared. (2) The reducing and alkylating reagents compared are different. For the alkylating reagents, we compared IAA, AA, N-EM, and 4-VP while they evaluated IAA, AA, iodoacetic acid (IAC), and chloroacetamide (CAA). For the reducing reagents, besides DTT, TCEP, and 2-ME in that paper, we also included THPP. (3) Here we systematically investigated the effect of the alkylation reaction conditions (concentration, temperature, and reaction time) on the cysteine alkylation completion and side reactions on other amino acids and the peptide N- and C-termini. (4) We further identified cysteine-containing peptides in yeast cells using the best reducing and alkylating reagents and optimal reaction conditions after the enrichment with Thiopropyl-Sepharose 6B resins.
In this work, we systematically evaluated the reduction and alkylation using the most commonly used reagents, investigated side reactions during the alkylation step, and further optimized the experimental conditions to maximize peptide identifications. The reduction of the disulfide bonds with commonly used DTT, 2-ME, TCEP, or THPP was systematically compared. After the reduction, the alkylation of sulfhydryl groups with the popular reagents of iodoacetamide, acrylamide, N-EM, or 4-VP was also evaluated, and the completion of cysteine alkylation and undesired side reactions were systematically investigated. Alkylating conditions were further optimized for iodoacetamide, and the optimal conditions were then used for the analyses of the enriched cysteine-containing peptides from yeast.
Materials and methods
Yeast cell culture, lysis, and digestion
BY4742 MAT alpha yeast (Saccharomyces cerevisiae) was cultured in Difco™ YPD broth (BD) overnight at 31 °C with shaking until the optical density was ~1.0 as measured by UV-Vis spectrometry at 600 nm. Cells were harvested by centrifugation at 800 g, 4 °C for 5 minutes, and washed with water. Lysis buffer containing 0.5% sodium deoxycholate (SDC, Sigma-Aldrich), 8 M urea (Sigma-Aldrich), 1 pill/10 mL cOmplete ULTRA Tablets protease inhibitor cocktail (Roche), and 75 mM sodium chloride (Sigma-Aldrich) in 50 mM, pH=8.2 N-(2-hydroxyethyl)piperazine-N’-2-ethanesulfonic acid (HEPES, Biobuffer) buffer, along with 0.5 mm zirconia/silica beads (BioSpec), were added to the XXTuff 2 mL microvials (BioSpec). The cells were lysed using a Mini BeadBeater (BioSpec) at the maximum speed for three cycles, 30 seconds each with a resting period of 2 minutes on ice between cycles. The lysate was then collected.
Chloroform/methanol precipitation of proteins was performed by adding methanol (EMD Millipore), chloroform (EMD Millipore), and water to the lysate in the ratio of 4:1:3, respectively. The protein pellet was collected and air-dried. Digestion buffer containing 5% acetonitrile (ACN, Sigma-Aldrich), 1.6 M urea, and 50 mM, pH=8.2 HEPES was added to the dried pellet so that the concentration of proteins is ~1 mg/mL. Protein digestion was performed with lysyl endopeptidase (Lys-C, Wako) at 31 °C overnight and subsequently with sequencing grade modified trypsin (Promega) at 37 °C for 4 hours (enzyme:substrate ratio of 1:100 for both enzymes). Trifluoroacetic acid (TFA, Sigma-Aldrich) was added to a final pH of ~2 to quench the digestion. Peptides were desalted using a Sep-Pak Vac tC18 cartridge (Waters). Desalted peptides were aliquoted, dried using a vacuum concentrator, and frozen at −80 °C until used.
Comparison of reducing reagents
Dried peptides were reconstituted in 50 mM, pH=8.2 HEPES buffer to the concentration of 1 µg/µL. Four reducing reagents, DTT (Sigma-Aldrich), 2-ME (Sigma-Aldrich), TCEP (Calbiochem), and THPP (Santa Cruz Biotechnology), were compared at the same concentration of 5 mM. The reduction took place at 56 °C for 25 minutes. Then the mixtures were cooled down to room temperature, and iodoacetamide was added to the final concentration of 14 mM to alkylate the peptides. The alkylation was performed in the dark at room temperature for 30 minutes, followed by addition of the same amount of each reducing reagent to quench the alkylation. The mixtures were left in the dark for another 15 minutes. After the comparison, DTT was chosen as the reducing reagent. Triplicate runs were performed in each experiment. For parallel experiments, except the reducing reagents, the amount of starting materials and any other steps were kept the same.
Comparison of alkylating reagents
Similar to the reduction experiment in the previous section, dried peptides were dissolved in 50 mM, pH=8.2 HEPES buffer to the final concentration of 1 µg/µL. After the reduction with DTT, the alkylation with iodoacetamide, acrylamide, N-EM, or 4-VP (all from Sigma-Aldrich) was performed and compared at the same concentration of 14 mM in the dark at room temperature for 30 minutes. Then DTT was added to the final concentration of 5 mM to quench the alkylation. Based on the current experimental results, iodoacetamide was selected as the alkylating reagent for further experiments. The following parameters were optimized: concentration (1, 2, 4, 8, 14, and 20 mM), temperature (room temperature, 40, 70, and 85 °C), and reaction time (10, 20, and 30 minutes). Similarly, triplicate runs were performed in each experiment.
Enrichment of peptides with cysteine from yeast whole-cell lysate
The protocol for the enrichment of peptides containing cysteine using Thiopropyl-Sepharose 6B resin was modified from the method described by Guo et al.54 In summary, 300 µg of dried peptides from a yeast whole-cell lysate in 100 µL 50 mM, pH=8.2 HEPES buffer was reduced with 5 mM DTT for 25 minutes at 56 °C. The mixture was cooled down and incubated with rehydrated resin. Unbound or non-specifically bound peptides were washed according to the original protocol. Enriched peptides were eluted by incubation with DTT, and alkylated with 14 mM iodoacetamide at room temperature in the dark for 30 minutes. The samples were quenched with 5 mM DTT for another 15 minutes in the dark at room temperature prior to drying in the vacuum concentrator.
LC-MS/MS analysis
Eluted peptides were purified, and dried peptides were subsequently dissolved in a solution with 5% ACN and 4% formic acid (FA) prior to LC-MS/MS analysis. Peptides were separated by a Dionex UltiMate 3000 UHPLC system (Thermo Scientific), which is coupled to an LTQ Orbitrap Elite Hybrid Mass Spectrometer (Thermo Scientific) with Xcalibur software (version 3.0.63). A total of ~1 µg of peptides was loaded onto a C18-packed microcapillary column (Magic C18AQ, 3 μm, 200 Å, 100 μm × 16 cm, Michrom Bioresources) by a Dionex WPS-3000TPL RS autosampler (Thermostatted Pulled Loop Rapid Separation Nano/Capillary Autosampler). Peptides were separated by reversed-phase chromatography using an UltiMate 3000 binary pump with a 90-minute gradient of 4–30% ACN containing 0.125% FA. MS/MS analysis was performed with a data-dependent Top20 method. For each cycle, a full MS scan (resolution: 60,000) in the Orbitrap with 1 million automatic gain control (AGC) target was followed by up to 20 MS/MS in the LTQ for the most intense ions.55, 56 Selected ions were excluded from further sequencing for 90 seconds. Ions with single or unassigned charge were not sequenced. Maximum ion accumulation times were 1,000 ms for each full MS scan and 50 ms for MS/MS scans.
Database Search
Raw files from MS/MS analysis were converted to mzXML files and searched by SEQUEST algorithm (version 28).57 The following parameters were used: 10 ppm precursor mass tolerance, 0.5 Da fragment ion mass tolerance, fully tryptic digestion, up to two missed cleavages, methionine oxidation (+15.9949 Da), alkylation at cysteine, histidine, lysine, aspartic acid, glutamic acid, tyrosine, the peptide N- and C-termini (+57.0215 Da for iodoacetamide, +71.0371 Da for acrylamide, +126.0555 Da for N-EM, and +105.0579 Da for 4-VP). The target-decoy method was employed to estimate the false discovery rate (FDR).58, 59 Linear discriminant analysis (LDA) was used to filter and control peptide identifications by parameters such as XCorr, , and precursor mass error.60 The minimum-length peptides contain at least six amino acid residues. Peptide spectral matches were filtered to less than 1% FDR.
Results and discussion
Comparison of reducing reagents
In this study, Baker’s yeast (Saccharomyces cerevisiae) was chosen as a model, and the peptides used were from its whole-cell lysate. The typical bottom-up approach includes the reduction of disulfide bonds, followed by the alkylation of sulfhydryl groups prior to chromatographic separation and MS analysis. For the comparison of the reduction, the amount of the starting peptides and all other steps were the same except using different reducing reagents, and the same is for the alkylation comparison (Fig. 1 A and B).
Fig. 1.
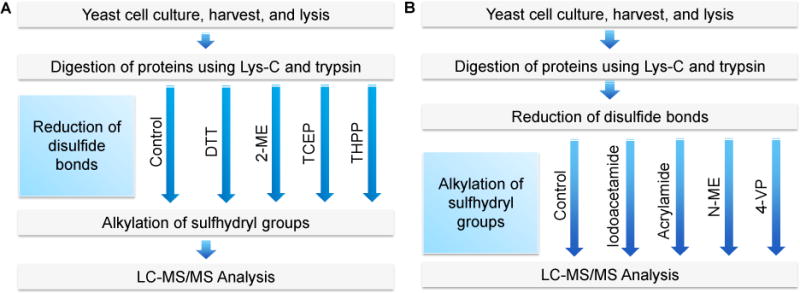
Experimental Procedure. (A) Comparison of the reduction with DTT, 2-ME, TCEP, or THPP. (B) Comparison of the alkylation with iodoacetamide, acrylamide, N-EM, or 4-VP. The reduction and alkylation were performed at the peptide level.
In the first experiment, four commonly used reducing reagents were compared, i.e. DTT, 2-ME, TCEP, and THPP. Each of them was used to reduce peptides at the same concentration of 5 mM at 56 °C for 25 minutes. A control group without the reduction and alkylation was included (Fig. 1). Each experiment was run in triplicate, and the number of identified peptides and proteins are the average ones with the standard deviation shown in Fig. 2. There were no obvious differences among four reducing reagents, which clearly demonstrated that all these reducing reagents are similarly effective to reduce disulfide bonds. Interestingly, without the reduction and alkylation, we obtained very similar results. Theoretically, the reduction and alkylation increase the identification of cysteine-containing peptides, albeit only a small portion of peptides contain cysteine. In addition, some peptides may be lost due to the side reactions of the alkylation, as discussed below.
Fig. 2.
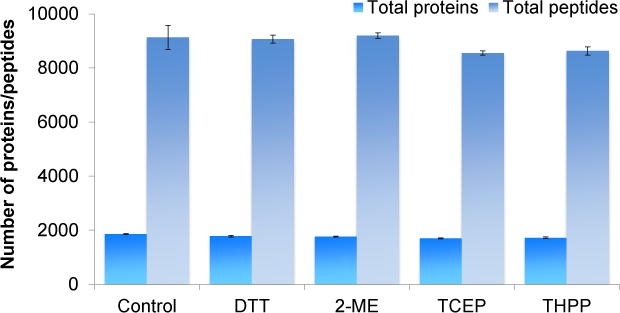
Comparison of the reducing reagents. The number of proteins and total peptides identified from each reducing reagent were compared.
Each reducing reagent has its advantages and disadvantages. TCEP is not stable in phosphate buffer under physiological pH,45 but it is more stable than DTT when there is no metal chelator. However, a metal chelator, such as ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), can increase the stability of DTT but decrease TCEP stability.61 Both DTT and 2-ME have shown to decrease their stability at higher pH and temperature.62 THPP is more stable than both TCEP and DTT at room temperature and pH=8.63 The current results demonstrated that all reducing reagents had similar performance. DTT was chosen for further experiments because of its popularity.
It should be noted that the number of proteins or peptides may be more fairly compared within the same experiment, as there are some variations among different batch of samples, including variations from the LC column and MS conditions.
Side reactions during the alkylation reaction
After the reduction, the alkylation follows to stabilize free sulfhydryl groups. Several alkylating reagents have been commonly used, including iodoacetamide, acrylamide, N-EM, and 4-VP. Scheme 1A shows the desired alkylation at the side chain of cysteine by iodoacetamide. Three other possible side reactions at the peptide N-terminus and the side chains of lysine and aspartic acid are displayed in Scheme 1B. Side reactions may occur during the alkylation reactions with different reagents. There have been reports regarding the alkylation at the side chains of other amino acids besides the desired alkylation of cysteine, including aspartic acid, glutamic acid, lysine, histidine, tyrosine, serine, and threonine.51–53, 64 Even di- and tri-alkylation on the side chains of some amino acid residues of peptides may further occur,65 but it is less common. In this study, we mainly focused on mono-alkylation at cysteine, histidine, lysine, aspartic acid, glutamic acid, tyrosine, and the peptide N- and C-termini.
Scheme 1.
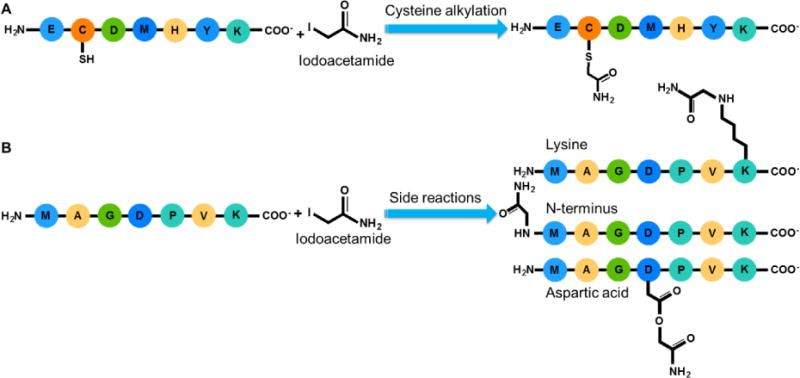
Possible alkylation reactions with iodoacetamide. Ideal alkylation is on the sulfhydryl group of cysteine (A). Side reactions may occur, and the alkylation reactions at the peptide N-terminus and the side chains of lysine and aspartic acid are shown here as examples (B).
Examples of MS/MS spectra of peptides with side reactions, i.e. carbamidomethylated N-terminus and lysine, are in Fig. 3A and 3B, respectively. Peptide G]LVSDPAGSDALNVLK (“]” denotes the alkylation of the amino group of the peptide N-terminus) is highly confidently identified with XCorr of 4.30 and a mass accuracy of −0.48 ppm (Fig. 3A). This peptide is from protein YML126C, which is a hydroxymethylglutaryl-CoA synthase protein. The peptide does not contain any cysteine, and the peptide N-terminus was undesirably alkylated.
Fig. 3.
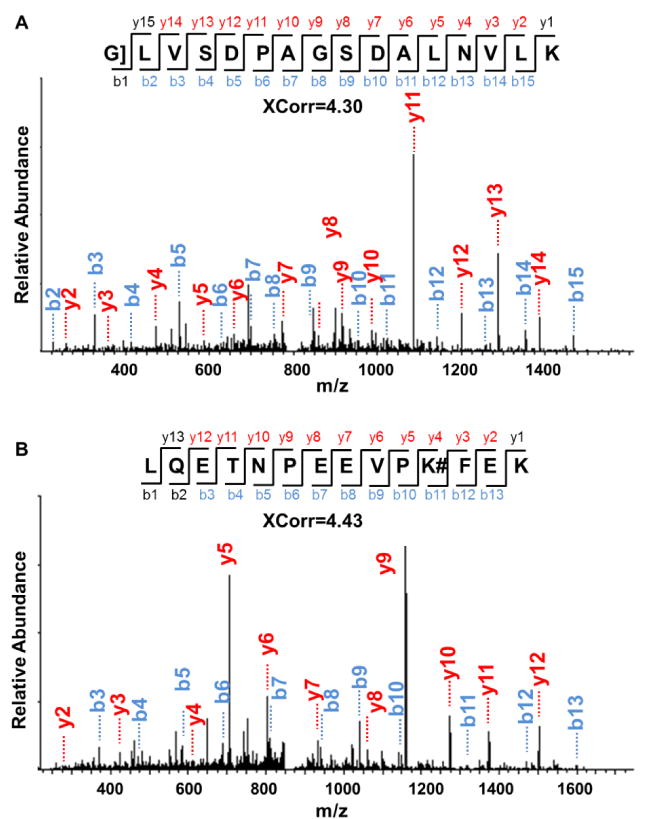
Examples of MS/MS spectra from peptides with (A) the alkylated N-terminus, as shown by the “]” sign and (B) alkylated lysine, as shown by the “#” sign.
Another example is peptide LQETNPEEVPK#FEK (“#” refers to the alkylation at the side chains of other amino acids except cysteine) (Fig. 3B). Similarly, this peptide from protein YKL056C, a translationally-controlled tumor protein homolog, was also confidently identified with XCorr of 4.43 and a mass accuracy of −0.21 ppm. There is no cysteine in the identified peptide, and undesired alkylation occurred at the side chain of lysine.
Comparison of alkylation with different reagents
Compared to the reduction, the alkylation of free sulfhydryl groups of cysteine is more complex. Besides the potential incompletion of the reaction, side reactions from reactive alkylating reagents can also occur. Here we compared four commonly used alkylating reagents: iodoacetamide, acrylamide, N-EM, and 4-VP. Peptide samples were reduced with 5 mM DTT for 25 minutes at 56 °C before alkylated with each reagent at the same concentration of 14 mM for 30 minutes in the dark at room temperature. A control group without the reduction or alkylation was included as well.
The number of proteins detected was similar, i.e. 1,700–1,800 proteins in each experiment, except for the results with N-EM (1,447 ± 153 proteins) (Fig. 4A). Similarly, the number of total peptides was about 8,200 except for the samples treated with N-EM (6,672 ± 589 peptides) (Fig. 4B), as further discussed below.
Fig. 4.
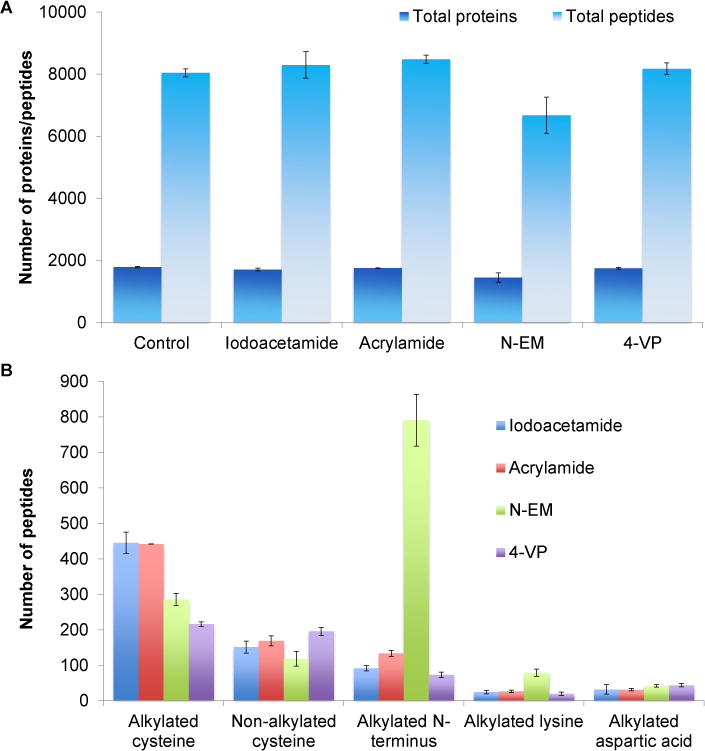
Comparison of the alkylating reagents. (A) Effects of different alkylating reagents on the identification of proteins and peptides. (B) Comparison of identified peptides with alkylated cysteine, free cysteine (due to incomplete reaction), or the side reactions on the peptide N-terminus and the side chains of lysine and aspartic acid.
Side reactions of the alkylation from different reagents
With reactive alkylating reagents, side reactions frequently happen, especially in complex biological samples. These four alkylating reagents resulted in different degrees of alkylation at other functional groups, especially the amino group at the peptide N-terminus and the side chain of lysine. Among the four alkylating reagents, N-EM also resulted in the greatest number of peptides with the alkylated N-termini (791 ± 73), which is even greater than the alkylation on cysteine. There are 133 ± 9 peptides with the alkylated N-termini from acrylamide, 92 ± 8 peptides from iodoacetamide, and 73 ± 8 peptides from 4-VP (Fig. 4B).
For the side reaction on the side chain of lysine, the alkylation with N-EM resulted in the highest number of peptides with alkylated lysine among four alkylating reagents. These results are highly consistent with previous data, i.e. the least number of total peptides was identified using N-EM as the alkylating reagent because more peptides were lost due to the side reactions than those rescued from the cysteine alkylation. Compared to 4-VP, N-EM was more effective to alkylate cysteine, but the side reactions were much severer.
Side reactions at other amino acid residues were also studied, as shown in Fig. S1. Based on the current results, the extent of side reactions was the N-terminus > glutamic acid > the C-terminus ≈ lysine > aspartic acid > tyrosine > histidine. As mentioned previously, the alkylation using iodoacetamide could occur through bimolecular nucleophilic substitution, where the nucleophilic sulfhydryl group attacks the C2 of iodoacetamide with iodine leaving the molecule at the same time.66 The electron-rich groups of the side chains of other amino acids also carry out the nucleophilic substitution reactions, as shown in this work.
Among the parallel experiments, 4-VP resulted in the least side reactions, but the completion rate of the cysteine alkylation was the lowest. These results suggest that 4-VP is relatively less reactive, and it is not a good choice as the alkylating reagent. N-EM has the highest level of side reactions, especially with the amino group at the peptide N-terminus and the side chain of lysine. After evaluating all these results with different alkylating reagents, iodoacetamide is still the best choice because it provided the highest completion rate of the cysteine alkylation and relatively lower side reactions. Therefore, iodoacetamide was chosen for further experiments. Acrylamide could also be used as an alternative since similar results were obtained.
Optimization of the alkylation with iodoacetamide
As shown above, even for the best alkylating reagent, the reaction completion and side reactions still pose problems. In order to push the reaction towards completion and to minimize side reactions, we further optimized several reaction conditions for iodoacetamide, including concentration, temperature, and reaction time. Peptides were first reduced with 5 mM DTT in 50 mM, pH=8.2 HEPES buffer, and then the samples were alkylated with iodoacetamide under different conditions.
The iodoacetamide concentrations of 1, 2, 4, 8, 14, or 20 mM were examined. Overall, the number of proteins and peptides identified were similar in all cases (Fig. 5A). However, more peptides with alkylated cysteine were identified as the concentration increased. The number of peptides with alkylated cysteine was the highest at the concentration of 14 mM (446 ± 13 peptides) and started to level off at 20 mM, while at the lowest iodoacetamide concentration tested here, 1 mM, 217 ± 10 peptides were detected (Fig. 5B). However, at 14 mM iodoacetamide, 144 ±11 peptides were still not alkylated. Alkylation at the N-terminus is still the major side reaction, and the number of peptides with side reactions slightly increased as the concentration went up (Fig. 5B and Fig. S2A).
Fig. 5.
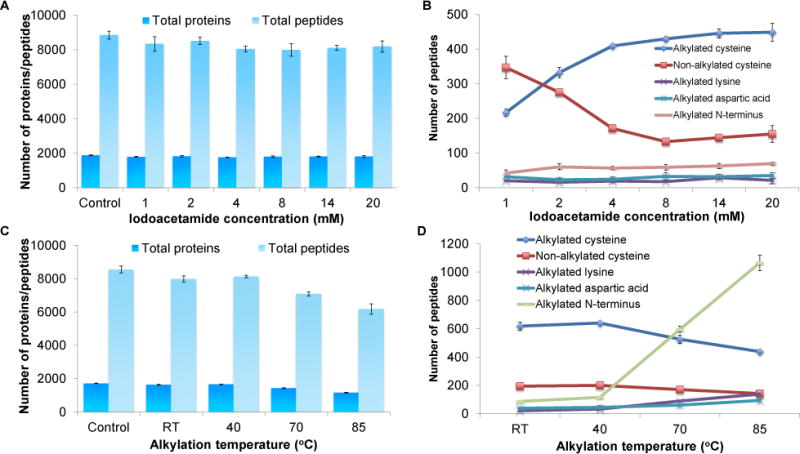
Optimization of alkylation conditions. (A) Effects of the iodoacetamide concentration on the identification of proteins and peptides. (B) The number of identified peptides with alkylated cysteine, free cysteine (due to incomplete reaction), or the side reactions on the peptide N-terminus and the side chains of lysine and aspartic acid as a function of the iodoacetamide concentration. (C) Effects of the alkylation temperature on the identification of proteins and peptides. (D) The number of identified peptides with alkylated cysteine, free cysteine, or the side reactions on the peptide N-terminus and the side chains of lysine and aspartic acid at different alkylation temperatures.
The temperature effect on the alkylation was also investigated, and the following temperatures were tested: room temperature, 40 °C, 70 °C, or 85 °C. As the temperature became higher, the number of detected proteins and peptides slightly decreased. The highest number of proteins identified were at room temperature (1,631 ± 33 proteins) and 40°C (1,655 ± 9) and the number of peptides were 7,982 ± 183 and 8,132 ±72, respectively. At 85°C, only 1,157 ± 32 proteins and 6,178 ± 315 peptides were identified (Fig. 5C). Temperature affected the alkylation at the peptide N-terminus the most, as shown in Fig. 5D. The number of peptides with the alkylated N-terminus dramatically increased from 87 ± 4 at room temperature to 1,065 ± 55 at 85°C (Fig. 5D). Alkylation at the side chains of lysine, glutamic acid, aspartic acid, and histidine also increased slightly with the temperature (Fig. S2B). This resulted in lower number of peptides with alkylated cysteine as the temperature elevated.
The effect of the alkylation reaction time on the identification of peptides and proteins was tested as well. Peptide samples were incubated with iodoacetamide for 10, 20, or 30 minutes, followed by quenching with DTT as mentioned above. Alkylation time did not markedly affect the degrees of the cysteine alkylation with iodoacetamide because a similar number of peptides with alkylated cysteine were identified. Similarly, degrees of side reactions barely changed for the different reaction durations tested here (Fig. S3A–C).
Enrichment of peptides with cysteine
Next, we sought to identify peptides with cysteine from the yeast whole-cell lysate. To maximize the number of cysteine-containing peptides, enrichment of peptides containing cysteine from the lysate is critical. In addition, we need to effectively reduce disulfide bonds and alkylate sulfhydryl groups to boost the identification of these peptides. The enrichment was performed by incubating peptides with Sepharose resin containing sulfhydryl groups.
Scheme 2 shows the enrichment and alkylation of peptides containing cysteine. A commercially available Thiopropyl-Sepharose 6B was employed for the enrichment. The resin contains 2-thiopyridyl disulfide groups attached to Sepharose through an ester linkage. The lyophilized resin was first rehydrated in water and subsequently incubated with peptides that were treated with DTT to reduce disulfide bonds. Several washing steps were performed to remove non-specifically bound peptides. Enriched peptides were eluted with 20 mM DTT solution in 25 mM NH4HCO3 buffer before alkylating with 14 mM iodoacetamide. During the incubation, active sulfhydryl groups on the resin should form disulfide bonds with cysteine on peptides. This resulted in effective and selective alkylation of peptides containing cysteine. A control group without the enrichment nor the reduction/alkylation and a group without the enrichment, but with the reduction/alkylation were also compared.
Scheme 2.
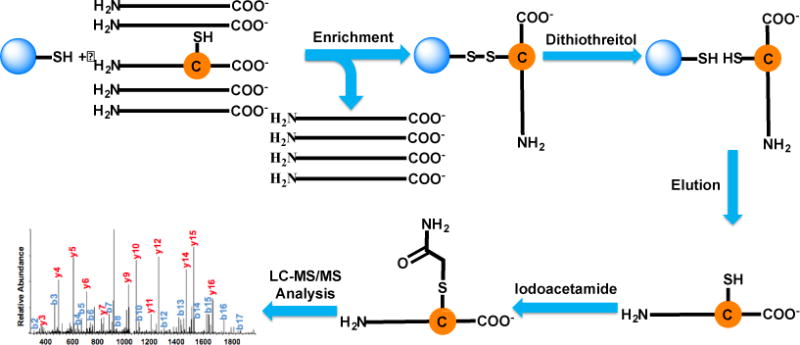
Enrichment of peptides containing cysteine using Thiopropyl Sepharose 6B resin. Peptides from the yeast whole-cell lysate are incubated with the resin. Peptides containing cysteine are bound to the resin through disulfide bonds while unbound peptides are removed. Enriched peptides are cleaved with DTT, and then alkylated with iodoacetamide prior to LC-MS/MS analysis.
The overlap of cysteine-containing peptides identified from the three experiments is in Fig. 6, and only peptides with unique sequence were compared. Without the reduction and alkylation, 288 unique peptides, corresponding to 216 proteins, containing cysteine were identified (Table S1, Fig. 6 and S4A). With the reduction and alkylation, but not the enrichment, 831 unique peptides with alkylated and free cysteine, corresponding to 504 proteins, were detected (Table S2). However, 2,730 unique peptides with alkylated and free cysteine and 1,398 proteins were identified with the enrichment (Table S3, and Fig. 6). Cysteine-containing peptides identified from the enrichment were about nine times of those without the enrichment and reduction/alkylation. The cysteine-containing peptides identified with the enrichment covers 81.9% of those detected from the experiment without the enrichment, but with the reduction/alkylation. The enrichment dramatically enhanced the identification of peptides and proteins containing cysteine.
Fig. 6.
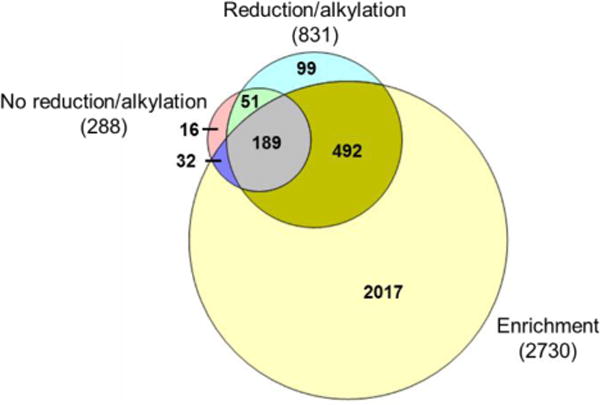
Comparison of peptides containing cysteine identified in yeast cells with or without the enrichment. Overlap of unique peptides containing cysteine (both alkylated and non-alkylated) identified from the experiments without enrichment nor reduction/alkylation (No reduction/alkylation), the experiments with only reduction/alkylation (Reduction/alkylation), and the experiments with both the enrichment and reduction/alkylation (Enrichment).
Proteins with cysteine from the enrichment experiment were subjected to clustering analysis using The Database for Annotation, Visualization and Integrated Discovery 6.8 (DAVID).67 Based on the biological process, proteins with the organic acid metabolic process were highly enriched with a very low P-value of 2.7×10−32 (Fig. S4B). Other enriched processes include the organic acid biosynthesis process (P=9.4×10−17), nuclear transport (P=7.2×10−8), carbohydrate biosynthesis process (P=8.6×10−5), and cellular response to oxidative stress (P=1.3×10−5), which correspond excellently with the well-known function of cysteine that plays a critical role in oxidation-reduction processes.
Conclusions
Bottom-up proteomics has proven to be very powerful for protein analysis. The reduction and alkylation are routine steps during the sample preparation prior to MS analysis. In this work, we systematically evaluated the commonly used reducing and alkylating reagents. Four most commonly used reducing reagents examined had very comparable performances while the results from four alkylating reagents were notably different. In addition to the completion rate differences among alkylation reagents, side reactions also varied at several side chains of the amino acid residues, including histidine, lysine, aspartic acid, glutamic acid, tyrosine, and the peptide N- and C-termini. The extents of the alkylation reaction with iodoacetamide on cysteine and undesired side reactions were also affected by its concentration and the alkylation temperature. Based on the current results, either of the tested reducing reagents (DTT, 2-ME, TCEP, or THPP) can be used to perform the reduction reaction. Regarding the alkylation, iodoacetamide provided us the best results considering the reaction completion rate and side reactions. The optimal alkylation conditions included 14 mM iodoacetamide, room temperature, and the reaction time of 30 minutes. The similar results were obtained using acrylamide, which may be used as an alternative alkylation reagent. The optimal alkylation conditions were employed to maximize the coverage of cysteine-containing peptides identified from the yeast whole-cell lysate. The enrichment markedly enhanced the identification of cysteine-containing peptides. The current results provide valuable information for choosing right reagents and optimal experimental conditions to maximize the identification of peptides, especially cysteine-containing peptides.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM118803, and the National Science Foundation (CAREER Award, CHE-1454501).
Footnotes
Electronic Supplementary Information (ESI) available: Four supplementary figures and three supplementary tables.
During the preparation of this manuscript, one paper about the evaluation of the reduction and alkylation was published online (T. Muller and D. Winter, Mol. & Cell. Proteomics, 2017, DOI: 10.1074/mcp.M116.064048).
The raw files are publicly accessible at http://www.peptideatlas.org/PASS/PASS01072 (Username: PASS01072; Password: JJ7355doh)
References
- 1.Aebersold R, Mann M. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin MA. Mol Cell Proteomics. 2004;3:1–9. doi: 10.1074/mcp.R300012-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Cottrell JS. J Proteomics. 2011;74:1842–1851. doi: 10.1016/j.jprot.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Washburn MP, Wolters D, Yates JR. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 5.Keshishian H, Addona T, Burgess M, Kuhn E, Carr SA. Mol Cell Proteomics. 2007;6:2212–2229. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moradian A, Kalli A, Sweredoski MJ, Hess S. Proteomics. 2014;14:489–497. doi: 10.1002/pmic.201300256. [DOI] [PubMed] [Google Scholar]
- 7.Resing KA, Ahn NG. FEBS Lett. 2005;579:885–889. doi: 10.1016/j.febslet.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Catherman AD, Skinner OS, Kelleher NL. Biochem Biophys Res Commun. 2014;445:683–693. doi: 10.1016/j.bbrc.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heck AJR. Nat Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 10.Li HL, Sheng YW, McGee W, Cammarata M, Holden D, Loo JA. Anal Chem. 2017;89:2731–2738. doi: 10.1021/acs.analchem.6b02377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai WX, Tucholski T, Chen BF, Alpert AJ, McIlwain S, Kohmoto T, Jin S, Ge Y. Anal Chem. 2017;89:5467–5475. doi: 10.1021/acs.analchem.7b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mann M. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 13.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 14.Wu RH, Haas W, Dephoure N, Huttlin EL, Zhai B, Sowa ME, Gygi SP. Nat Methods. 2011;8:677–683. doi: 10.1038/nmeth.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aebersold R, Goodlett DR. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 16.Sun LL, Hebert AS, Yan XJ, Zhao YM, Westphall MS, Rush MJP, Zhu GJ, Champion MM, Coon JJ, Dovichi NJ. Angew Chem-Int Edit. 2014;53:13931–13933. doi: 10.1002/anie.201409075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen WX, Smeekens JM, Wu RH. Chem Sci. 2016;7:1393–1400. doi: 10.1039/c5sc03826j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smeekens JM, Xiao HP, Wu RH. J Proteome Res. 2017;16:1039–1049. doi: 10.1021/acs.jproteome.6b00953. [DOI] [PubMed] [Google Scholar]
- 19.Feist PE, Sidoli S, Liu X, Schroll MM, Rahmy S, Fujiwara R, Garcia BA, Hummon AB. Anal Chem. 2017;89:2773–2781. doi: 10.1021/acs.analchem.6b03602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang XR, Cimermancic P, Yu C, Schweitzer A, Chopra N, Engel JL, Greenberg C, Huszagh AS, Beck F, Sakata E, Yang YY, Novitsky EJ, Leitner A, Nanni P, Kahraman A, Guo X, Dixon JE, Rychnovsky SD, Aebersold R, Baumeister W, Sali A, Huang L. Mol Cell Proteomics. 2017;16:840–854. doi: 10.1074/mcp.M116.065326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miah S, Banks CAS, Adams MK, Florens L, Lukong KE, Washburn MP. Mol Biosyst. 2017;13:42–55. doi: 10.1039/c6mb00639f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su D, Gaffrey MJ, Guo J, Hatchell KE, Chu RK, Clauss TR, Aldrich JT, Wu S, Purvine S, Camp DG, Smith RD, Thrall BD, Qian WJ. Free Radic Biol Med. 2014;67:460–470. doi: 10.1016/j.freeradbiomed.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang XS, Yuan ZF, Fan J, Karch KR, Ball LE, Denu JM, Garcia BA. Mol Cell Proteomics. 2016;15:2462–2475. doi: 10.1074/mcp.O115.049627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLachlin DT, Chait BT. Curr Opin Chem Biol. 2001;5:591–602. doi: 10.1016/s1367-5931(00)00250-7. [DOI] [PubMed] [Google Scholar]
- 25.Zhu ZK, Desaire H. Annual Review of Analytical Chemistry, Vol 8. In: Cooks RG, Pemberton JE, editors. Annual Reviews. Vol. 8. Palo Alto: 2015. pp. 463–483. [DOI] [PubMed] [Google Scholar]
- 26.Thaysen-Andersen M, Packer NH, Schulz BL. Mol Cell Proteomics. 2016;15:1773–1790. doi: 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng JN, Xiao HP, Wu RH. Angew Chem-Int Edit. 2017;56:7107–7111. doi: 10.1002/anie.201702191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen WX, Smeekens JM, Wu RH. Chem Sci. 2015;6:4681–4689. doi: 10.1039/c5sc01124h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao HP, Wu RH. Anal Chem. 2017;89:3656–3663. doi: 10.1021/acs.analchem.6b05064. [DOI] [PubMed] [Google Scholar]
- 30.Xiao HP, Wu RH. Chem Sci. 2017;8:268–277. doi: 10.1039/c6sc01814a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi TJ, Gao YQ, Gaffrey MJ, Nicora CD, Fillmore TL, Chrisler WB, Gritsenko MA, Wu CC, He JT, Bloodsworth KJ, Zhao R, Camp DG, Liu T, Rodland KD, Smith RD, Wiley HS, Qian WJ. Anal Chem. 2015;87:1103–1110. doi: 10.1021/ac503797x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu LQ, Robinson RAS. Analyst. 2016;141:3904–3915. doi: 10.1039/c6an00417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng JM, Gu W, Zhao YM. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smits AH, Vermeulen M. Trends Biotechnol. 2016;34:825–834. doi: 10.1016/j.tibtech.2016.02.014. [DOI] [PubMed] [Google Scholar]
- 35.Abu-Farha M, Elisma F, Figeys D. Adv Biochem Eng Biotechnol. 2008;110:67–80. doi: 10.1007/10_2007_091. [DOI] [PubMed] [Google Scholar]
- 36.Vasilescu J, Guo X, Kast J. Proteomics. 2004;4:3845–3854. doi: 10.1002/pmic.200400856. [DOI] [PubMed] [Google Scholar]
- 37.Xiao YS, Guo L, Wang YS. Anal Chem. 2013;85:7478–7486. doi: 10.1021/ac401415z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bulaj G. Biotechnol Adv. 2005;23:87–92. doi: 10.1016/j.biotechadv.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Woycechowsky KJ, Raines RT. Curr Opin Chem Biol. 2000;4:533–539. doi: 10.1016/s1367-5931(00)00128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feige MJ, Hendershot LM. Curr Opin Cell Biol. 2011;23:167–175. doi: 10.1016/j.ceb.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gundry RL, White MY, Murray CI, Kane LA, Fu Q, Stanley BA, Van Eyk JE. Curr Protoc Mol Biol. 2009 doi: 10.1002/0471142727.mb1025s88. Chapter 10, Unit10 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Fonslow BR, Shan B, Baek MC, Yates JR., 3rd Chem Rev. 2013;113:2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sechi S, Chait BT. Anal Chem. 1998;70:5150–5158. doi: 10.1021/ac9806005. [DOI] [PubMed] [Google Scholar]
- 44.Singh R, Lamoureux GV, Lees WJ, Whitesides GM. Method Enzymol. 1995;251:167–173. doi: 10.1016/0076-6879(95)51119-9. [DOI] [PubMed] [Google Scholar]
- 45.Burns JA, Butler JC, Moran J, Whitesides GM. J Org Chem. 1991;56:2648–2650. [Google Scholar]
- 46.Paulech J, Solis N, Cordwell SJ. Bba-Proteins Proteom. 2013;1834:372–379. doi: 10.1016/j.bbapap.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 47.Hill BG, Reily C, Oh JY, Johnson MS, Landar A. Free Radic Biol Med. 2009;47:675–683. doi: 10.1016/j.freeradbiomed.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Righetti PG. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;841:14–22. doi: 10.1016/j.jchromb.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 49.Crankshaw MW, Grant GA. Curr Protoc Protein Sci. 2001 doi: 10.1002/0471140864.ps1501s03. Chapter 15, Unit15 11. [DOI] [PubMed] [Google Scholar]
- 50.Zoeller RT, Fletcher DL, Butnariu O, Lowry CA, Moore FL. J Histochem Cytochem. 1997;45:1035–1041. doi: 10.1177/002215549704500712. [DOI] [PubMed] [Google Scholar]
- 51.Boja ES, Fales HM. Anal Chem. 2001;73:3576–3582. doi: 10.1021/ac0103423. [DOI] [PubMed] [Google Scholar]
- 52.Guo MZ, Weng GF, Yin DY, Hu XX, Han J, Du Y, Liu YQ, Tang DQ, Pan YJ. Rsc Adv. 2015;5:103662–103668. [Google Scholar]
- 53.Muller T, Winter D. Mol Cell Proteomics. 2017 doi: 10.1074/mcp.M116.064048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo J, Gaffrey MJ, Su D, Liu T, Camp DG, 2nd, Smith RD, Qian WJ. Nat Protoc. 2014;9:64–75. doi: 10.1038/nprot.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen WX, Smeekens JM, Wu RH. Mol Cell Proteomics. 2014;13:1563–1572. doi: 10.1074/mcp.M113.036251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao HP, Tang GX, Wu RH. Anal Chem. 2016;88:3324–3332. doi: 10.1021/acs.analchem.5b04871. [DOI] [PubMed] [Google Scholar]
- 57.Eng JK, McCormack AL, Yates JR. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 58.Peng JM, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 59.Elias JE, Gygi SP. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 60.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Getz EB, Xiao M, Chakrabarty T, Cooke R, Selvin PR. Anal Biochem. 1999;273:73–80. doi: 10.1006/abio.1999.4203. [DOI] [PubMed] [Google Scholar]
- 62.Stevens R, Stevens L, Price NC. Biochem Educ. 1983;11:70–70. [Google Scholar]
- 63.McNulty J, Krishnamoorthy V, Amoroso D, Moser M. Bioorg Med Chem Lett. 2015;25:4114–4117. doi: 10.1016/j.bmcl.2015.08.027. [DOI] [PubMed] [Google Scholar]
- 64.Fruchter RG, Crestfield AM. J Biol Chem. 1967;242:5807–5812. [PubMed] [Google Scholar]
- 65.Yang Z, Attygalle AB. J Mass Spectrom. 2007;42:233–243. doi: 10.1002/jms.1157. [DOI] [PubMed] [Google Scholar]
- 66.Paulsen CE, Carroll KS. Chem Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang DW, Sherman BT, Lempicki RA. Nature Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.