

Abstract
An accurate and reliable UPLC-MS/MS method is reported for quantification of endogenous Prostaglandin E2 (PGE2) in rat colon mucosa and polyps. This method adopted the “surrogate analyte plus authentic bio-matrix” approach, using two different stable isotopic labeled analogs — PGE2-d9 as the surrogate analyte and PGE2-d4 as the internal standard. Quantitative standard curve was constructed with the surrogate analyte in colon mucosa homogenate; and the method was also successfully validated with the authentic bio-matrix. Concentrations of endogenous PGE2 in both normal and inflammatory tissue homogenates were back-calculated based on the regression equation. Because there is no any endogenous interference on the surrogate analyte determination, the specificity is particularly good. By using authentic bio-matrix for validation, the matrix effect and exaction recovery are identically same for the quantitative standard curve and actual samples – this notably increases the assay accuracy. The result proves that this method is easy, fast, robust and reliable for colon PGE2 determination. This “surrogate analyte” approach was applied to measure the Pirc (an Apc-mutant rat kindred that models human FAP) mucosa and polyps PGE2, one of the strong biomarkers of colorectal cancer. The similar concept could be also applied for other assays of endogenous biomarkers in other tissues.
Keywords: PGE2, COX-2, Colon, UPLC-MS/MS, Pirc rat, Endogenous biomarker, FAP, CRC
1. Introduction
Familial Adenomatous Polyposis (FAP) is a rare inherited disease [1] that results from an autosomal dominant genetic alteration in the adenomatous polyposis coli (APC) gene [2]. Patients with this disease characteristically develop hundreds to thousands of potentially pre-cancerous polyps (adenomas) in the duodenum, colon and rectum beginning in adolescence. Left untreated, patients have almost 100% lifetime risk of developing colorectal cancer (CRC) ultimately. Beyond early detection and surgical removal of the adenomatous precursor lesions, there is currently no a safe and convenience therapeutic approach to FAP [3].
One potential pharmacologic target in impeding growth of adenomatous tissue has been the cyclooxygenase (COX) enzyme. Elevated levels of COX-2 are found in many pre-malignant lesions; in particular, it has been demonstrated that COX-2 expression is low to undetectable in normal colorectal mucosa, whereas COX-2 expression is considerably increased in the majority of colorectal adenomas and adenocarcinomas [4, 5]. PGE2 is one of the most important COX-2 catalyzed metabolites (prostaglandins) driving the tumor growth. The positive feedback loop [6] between PGE2 and COX-2 is major tumorigenesis mechanism. Several lines of evidences [4, 7] have suggested that application of COX-2 inhibitors have therapeutic utility in reducing colorectal neoplasia. Much more powerful evidence from two human clinical trials, APC (Pfizer) and APPROVe (Merck), proved that celecoxib and refecoxib can significantly reduce both the number and size of colon polyps [8–12].
The Apc-mutant (Pirc) rat was developed by targeted ENU induced germline mutagenesis of the inbred F344/NTac rat strain [13, 14]. Pirc rats have a unique biology that mimics many characters of human FAP and colon cancer [15]. Previous studies [13–20] prove the Pirc rat is a reliable preclinical experimental platform for human FAP. For examples, the polyps multiplicity significantly regressed when the Pirc rat was feed with celecoxib for 20–24 weeks [13]. This result is consistent to the human FAP clinical data [9]. Similar studies are also found for another COX-2 inhibitor Sulindac in Pirc rat and human [19, 21, 22].
Although many strong evidences [23–26] prove that PGE2 is a potent factor promoting intestinal polyps growth in APCmin/+ mouse (an Apc-gene knock-out mouse model created 20 years prior to Pirc rat), the relationship between PGE2 and Pirc colon polyps is yet not reported. Knowing such relationship is very important and meaningful, because it would help to predict the COX-2 inhibition of Pirc colon polyps growth.
To solve this problem, we established a new UPLC-MS/MS to accurately quantify colon tissue PGE2. The basic concept is to use a surrogate analyte PGE2-d9 replacing of the native PGE2, so that all the validation can be conducted with the authentic bio-matrices. We also prove the MS response of PGE2 and PGE2-d9 are identically the same, hence the authentic bio-matrix based standard curve constructed with the surrogate analyte is adequately to quantify the tissue PGE2 concentration. This method was well validated with authentic tissue homogenate following FDA Guidance for Bioanalytical Method Validation (short for “FDA guidance” below). It was successfully applied for the quantification of colon tissue PGE2 for both wild type and Pirc rat.
2. Experiment
2.1 Materials and reagents
PGE2 (authentic analyte), PGE2-d4 (internal standard, I.S.) and PGE2-d9 (surrogate analyte) were purchased from Cayman Chemical Inc. (Ann Arbor, MI). Ethyl acetate, K2HPO4 and KH2PO4 were bought from VWR Corporate (Radnor, PA). Ammonium hydroxide and sucrose were supplied by Sigma-Aldrich (St. Louis, MO); and EDTA-Na2·6H2O by J. T. Baker (Phillipsburg, NJ). Acetonitrile (HPLC grade) was obtained from OmniSolv (Billerica, MA). Distilled water, prepared from demineralized water, was used throughout the study.
2.2 Instruments and conditions
The UPLC-MS/MS consists of a Waters ACQUITY UPLC System (a quaternary solvent manager, a column manager, and a sample manager; Waters Corporation, MA. USA) and an API 5500 Q-Trap (AB Sciex, MA. USA) with an ESI interface. Data was processed with Analyst software (version 1.52).
Chromatographic separation was conducted on a UPLC BEH C18 column (2.1×100 mm, 1.7 μm, Waters). A binary gradient consisting of 0.1% ammonium hydroxide solution (A) and acetonitrile (B) eluted in program: 0–6.5 min 13% B; 6.5–7.5 min from 13% to 80% B; 7.5–9.5 min 80% B; 9.5–10 min from 80% to 13% B; 10–15 min 13% B. The flow rate was 0.3 ml/min and the column temperature was 25 °C. Mass spectrometer run in ESI (-) mode with following the parameters: Source Temperature 600 °C; Curtain Gas 30 psi; Gas1 40 psi; and Gas2 30 psi. MRM transitions for PGE2 (authentic analyte, Figure 1A), PGE2-d4 (internal standard, I.S., Figure 1B) and PGE2-d9 (surrogate analyte, Figure 1C) were m/z 351.3->271.2 (Figure 2A), m/z 355.3->275.2 (Figure 2B) and m/z 360.2->280.3 (Figure 2C). The optima DP and CE were −230 V and −22 V for all the compounds.
Figure 1.
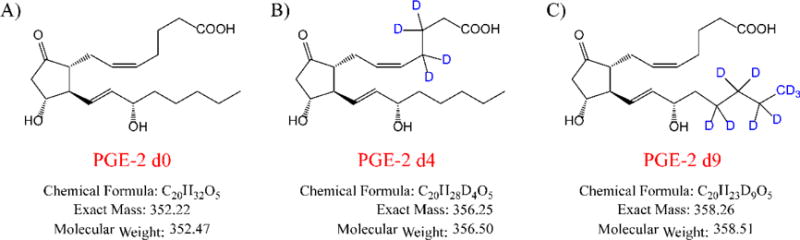
Chemical structures of PGE2 (A), PGE2-d4 (B) and PGE2-d9 (C).
Figure 2.
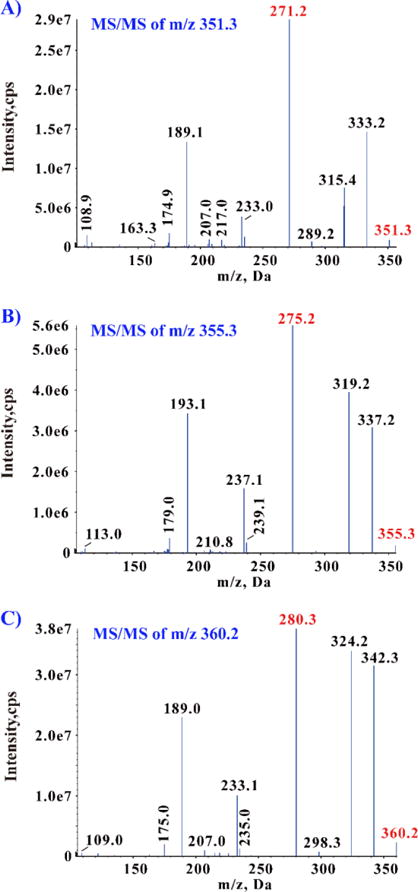
Product ion spectra of PGE2 (A), PGE2-d4 (B) and PGE2-d9 (C).
2.3 Animal treatments and sample collections
Pirc rats (raised on Center for Epigenetics & Disease Prevention, Houston TX) were orally dosed with drug suspensions (dissolved in 0.5% Methyl cellulose and 0.1% Tween-80, 40 mg/Kg, twice daily) for successive 4 days (12-hour dosing interval, and 8 doses in total). The normal control (wild type rat) and disease control (Pirc rat) were dosed with the same amount of blank vehicle solvent. Animals were sacrificed 6 hours after receiving the last dose on Day 4; the colon polyps and mucosa were taken immediately and then kept in the −80 degree freezer until analysis.
2.4 Homogenization buffer preparation
60 ml of 100 mM phosphate buffer solution (pH =7), 51.344 g sucrose, 0.224 g EDTA dihydrate and 540 ml purified water were mixed and sonicated. The total volume was 600 ml, and the pH of the buffer was adjusted to 7.4.
2.5 Preparation of calibration curve and quality control
Stock solutions of 1 mg/ml PGE2, 50 μg/ml PGE2-d4 (internal standard, I.S.) and 20 μg/ml PGE2-d9 were prepared in acetonitrile. A series of PGE2 and PGE2-d9 standard working solutions ranging from 3.908 to 1,000 ng/ml were obtained by further dilution of the stock solution with acetonitrile. Matrix-matched calibration standards of PGE2-d9 at concentration levels of 0.098, 0.195, 0.391, 0.781, 1.563, 3.125, 6.25, 12.50 and 25.00 ng/ml were prepared by spiking an appropriate volume of the working solutions in nine aliquots of 2 ml tissue homogenate, respectively. The quality control (QC) samples were prepared at concentrations of 0.195, 1.563 and 20.00 ng/ml in the same bio-matrix. An internal standard working solution of 5 μg/ml was prepared by diluting the stock solution of PGE2-d4 with acetonitrile. All the solutions were stored in −80 °C.
2.6 Sample preparation
The colon mucosa and polyps were homogenized with 10 ml homogenization buffer after their weights were measured. 2 ml fraction of the homogenization samples and 20 μl PGE2-d4 (5 μg/ml internal standard stock solution, and final concentration in 50 ng/ml) were mixed with 8 ml ethyl acetate for liquid-liquid extraction (LLE). The total 10 ml mixture was vortexed and centrifuged; then the upper supernatant was collected and transferred to another tube for nitrogen air dry. The collective dried residual was dissolved with 100 μl reconstitution solvent (acetonitrile: 0.1% ammonium hydroxide = 2:8, v/v), then vortexed and centrifuged (15,000 rpm). 5 μl of the supernatant was injected to UPLC-MS/MS for analysis.
2.7 Determination of relative response factor (RRF)
RRF was measured by UPLC-MS/MS using a series of neat solutions containing both authentic and surrogate analytes. The calibration curve was prepared the same as the matrix-matched calibration standards described on section 2.5, except using 2 ml of neat solution (acetonitrile: water = 2:8) to replace the tissue homogenate. The nominal concentrations of PGE2 and PGE2-d9 are exactly the same in each point of this neat solution based calibration curve. The peak area ratios of authentic/surrogate analyte to internal standard were plotted against the nominal concentration, and standard curves were constructed using linear regression analysis with a 1/x2 weighting factor (Equation 1). The actual PGE2 and PGE2-d9 of each nominal concentration is back calculated using their individual fitting standard curves (Equation 2), and then the individual RRF is the ratio of actual PGE2 to actual PGE2-d9 in the same nominal concentration (Equation 3). Finally, the average RRF is the calculated by averaging all the 9 individual RRFs.
| (Equation 1) |
Where, a is the slope; b is the intercept;
| (Equation 2) |
| (Equation 3) |
2.8 Method validation
The selectivity was assessed by comparing the chromatograms of 8 different blank mucosa and polyps homogenates from wild type and Pirc rats. No authentic analyte, surrogate analyte or internal standard was added into the blanks.
Calibration curves were constructed by analyzing spiked calibration surrogate analyte into the wild type mucosa homogenate. Samples were quantified using the peak area ratios of PGE2-d9 to PGE2-d4 (the I.S.). The peak area ratios were plotted against nominal concentration of PGE2-d9, and standard curves were constructed using linear regression analysis with a 1/x2 weighting factor. The LLOQ was defined as the lowest concentration on the calibration curve; and its precision and accuracy should be within ± 20%.
To evaluate the precision and accuracy of the method, QC samples at three concentration levels (0.195, 1.563 and 20.00 ng/ml) were analyzed in five replicates on three different analysis batches. Assay precision is defined as the relative standard deviation (SD) from the mean (M), calculated by the equation RSD% = SD/M× 100%. Accuracy is defined as the relative deviation in the calculated value (E) of a standard from that of its true value (T) expressed as a percentage RE % (RE % = (E − T)/T × 100%). The accuracy, intra- and inter-day precisions are all required not to exceed 15%.
The matrix effect and recovery were determined by comparing responses from the analysis of five neat standards (set 1), five extracts of each of the different tissue homogenate samples spiked with the analyte and internal standard after extraction (set 2), and five extracts of each of the different tissue homogenate samples spiked with the compounds before extraction (set 3) at 0.195, 1.563 and 20.00 ng/ml of PGE2-d9. The matrix effect (%) was calculated by dividing peak areas of each compound in samples from set 2 by those in samples from set 1. Recovery (%) was calculated by dividing peak areas in samples from set 3 by those in samples from set 2.
The stabilities of PGE2-d9 in tissue homogenate were evaluated by analyzing replicates (n = 3) of samples at the concentrations of 0.195, 1.563 and 20.00 ng/ml. These samples were exposed to different conditions (time and temperature). Results were compared with those obtained from freshly prepared samples. The analytes are considered stable in the biological matrix when 85 – 115% of their initial concentrations are found. The bench-top (short-term) stability was determined after the exposure of the spiked samples at room temperature for 12 h, and the post-preparative stability in the auto-sampler (10 °C) was determined for 18 h. The long-term stability was assessed after storage of the standard spiked bio-samples at −80 °C for 2 week. The freeze-thaw stability was evaluated after three complete freeze-thaw cycles on consecutive days.
2.9 Tissue PGE2 calculation
Tissue homogenate PGE2 concentrations were calculated based on the regression equation of the calibration curve and the peak area ratio of endogenous analyte to internal standard. Because the RRF of PGE2 to PGE2-d9 is almost equal 1 (0.99, see the result and discussion), the tissue PGE2 concentration can be directly obtained from the PGE2-d9 matrix-match standard curve (Equation 2). Finally, tissue PGE2 level was expressed as PGE2 amount per unit tissue weight (Equation 4).
| Equation 4 |
Where Analyte Conc. is the calculated PGE2 concentration by using the PGE2-d9 matrix-matched standard curve; 10 ml is the total homogenate buffer volume for each sample; Tissue weight is measure with highly accurate analytical balance.
3. Result and discussion
3.1 Brief review of current PGE2 assay methods
Current PGE2 assay methods can be classified in to 3 different categories: ELISA binding, GC-MS and LC-MS. The commercial PGE2 ELISA Kits use particular monoclonal anti-bodies binding with PGE2; and the PGE2 concentration is calculated based on the visible light absorbance of the total Schiff Base (Supplementary Figure 2) [27]. However, many other endogenous PGs have the same cross-reaction (Supplementary Figure 3). Lacking of enough specificity, the ELISA methods always overestimate the actual PGE2 in biometrics [28]. The GC-MS can well separate the PGs in sequence, so that the specificity and selectivity are well improved in comparison to ELISA. Incapable to gasify in GC-MS, PGE2 needs chemical derivations in sample preparation prior to assay. This time and labor consuming step undermines the assay precision and accuracy.
Only 18 pieces of papers were found with the key words “PGE2 and LC-MS” in Pubmed.gov from year of 2000. Most of them are designed to measure the PGE2 in cell line [28–37] and plasma/serum [38–41], hence difficult to be applied for tissue PGE2 assays (since the endogenous interference is much more complex in the tissue). M.Y. Golovko’s team members keep improving the LC-MS assay method for brain tissue PGE2, and they have published 3 papers [42–44]. Their studies makes good contributions on sample purifications and chromatographic separations. A UPLC-MS/MS method was reported for determination of tissue PGE2 in their latest paper [42], but what kind of matrix used for standard curve preparation as well as method validation is not clear specified – this has great influence on the assay accuracy. If the validation matrix was buffer or neat solution (like the experiment in which the actual bio-matrix was replaced by artificial cerebrospinal fluid for PGE2 assay [45]), the tissue PGE2 will be highly underestimated. The PGE2 signal response is much higher in neat solution than in tissue homogenate (as we found it the experiment), because the neat solution does not suffer from ion-suppression (matrix effect) or extraction lose (recovery) [46]. On the other hand, if the validation matrix was the authentic tissue homogenate, the assay unavoidably suffers the background interference from endogenous PGE2. According to the FDA guidance, the limit of quantitation (LOQ) of analyte should be necessary to generate a characteristic signal at least 10-fold higher than the background noise, but the inflammation tissue cannot generate PGE2 10 fold higher its background level. In summary, whatever matrices they used for validation, there is always a concern for accuracy.
3.2 Quantification of endogenous compounds
The FDA guidance strongly recommends using the same matrices as the test sample for standard curve preparation, to ensure equal signal responses for equal concentrations of analyte in both standard reference and test sample. For endogenous substances, however, there is no real analyte-free bio-matrices available. Four approaches [46, 47] are commonly adopted to assay endogenous compounds by LC-MS/MS lacking of blank matrices, including background subtraction, standard addition, surrogate matrix, and the surrogate analyte approaches.
Both of “background subtraction” and “standard addition” approaches require that the analyte signal in all the test samples should be much higher than the endogenous background (at least 20 folds); otherwise the haunting error will lead to an inaccurate quantification. Since the inflammation tissue PGE2 only is several folds higher than the normal tissue in this study, these two methods are obviously not suitable for this study. The surrogate matrix method uses neat solutions, artificial matrices or stripped matrices to lower down even diminish the endogenous background. However, it usually does not account for actual extraction recovery and matrix effect that are highly different between authentic bio-matrices and artificial matrices. Finally, the remaining choice is the surrogate analyte approach. The surrogate analyte method was firstly proposed by W. Li and L. Cohen from Pfizer Company on 2003 [48]. Its basic concept is to build the calibration curve using the signal response from a stable labeled surrogate analyte; and calculate the sample concentrations of endogenous analyte based on the regression equation from the surrogate standard. This concept has been successfully applied for determinations of other endogenous biomarkers [47, 49–54].
In our study, the calibration standard was prepared with PGE2-d9 combing with colon mucosa homogenate (the authentic bio-matrix), and no background interference from biological matrices was found for PGE2-d9 (Figure 3 F and M). Because the intrinsic physical and chemical properties of PGE2 and PGE2-d9 are almost identical, their chromatographic retentions, MS ionization efficiencies, extraction recoveries and matrix effects from tissue homogenate are the same. The individual RRF ranges from 0.95 to 1.04 in this assay, and the average RRF is 0.99 (Table-1). It proves the sample PGE2 can be directly calculated by the PGE2-d9 standard curve.
Figure 3.
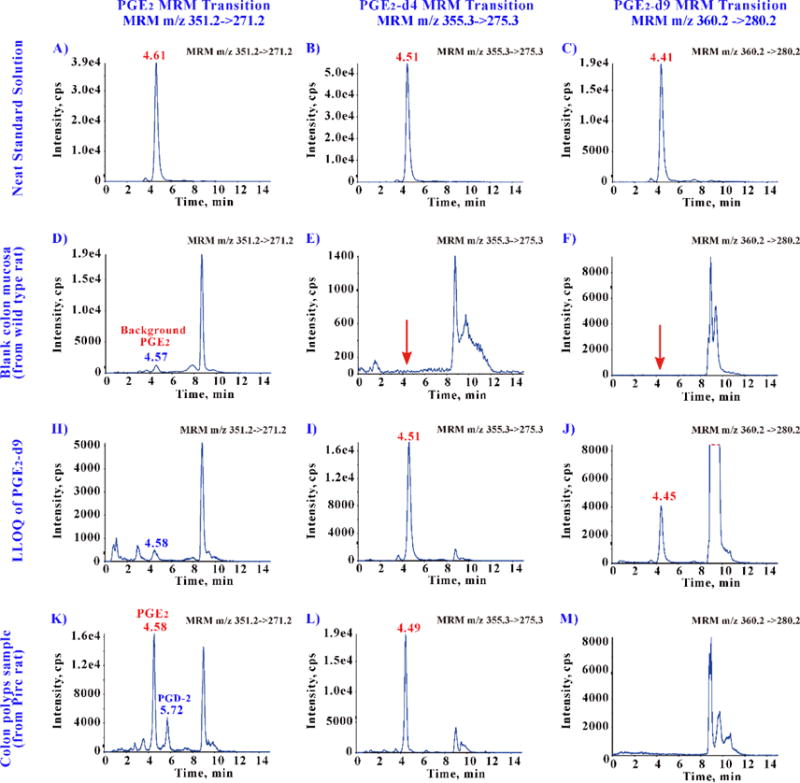
Typical chromatograms for method specificity.
The retention times of PGE2 (A), PGE2-d4 (B) and PGE2-d9 (C) reference standards were almost the same at about 4.5 min. A low level of endogenous PGE2 was detected on wild type colon mucosa (D), but no any background interference is found for PGE2-d4 (E) and PGE2-d9 (F). The LLOQ (as well as quantification standard curve) was constructed with wild type colon mucosa by adding the surrogate analyst PGE2-d9 (J), and then measured by adding the internal standard PGE2-d4 (I). Similar endogenous background was also found for PGE2 (H) in the LLOQ. This method can well separate the PGE2 and PGD2 (confirmed with PGD2 reference standard) in the actual colon polyps samples (K).
Table-1.
Calculation of average relative response factor (RRF) between PGE2 and PGE2-d9. Both standard curves were constructed with mixed water-organic solution (water: acetonitrile, 8:2 v/v).
| Nominal Conc. | Calculated Conc.
|
RRF | |
|---|---|---|---|
| PGE2 | PGE2-d9 | ||
|
| |||
| (ng/ml) | |||
| 0.098 | 0.099 | 0.096 | 1.03 |
| 0.195 | 0.197 | 0.200 | 0.99 |
| 0.391 | 0.374 | 0.394 | 0.95 |
| 0.781 | 0.758 | 0.781 | 0.97 |
| 1.563 | 1.510 | 1.540 | 0.98 |
| 3.125 | 2.970 | 3.140 | 0.95 |
| 6.250 | 6.160 | 6.220 | 0.99 |
| 12.50 | 12.30 | 12.30 | 1.00 |
| 25.00 | 25.80 | 24.90 | 1.04 |
|
| |||
| Average RRF | 0.99 | ||
Three steps to calculate the average RRF.
Obtain the actual PGE2 and PGE2-d9 concentration of each point from their individual standard curves;
Calculate the individual RRF (RRF = PGE2 concentration / PGE2-d9 concentration);
Average the 9 individual RRF and obtain the average RRF of 0.99 (almost equal to 1).
Important note: If RRF is not equal to 1, the concentration calculated by the surrogate standard curve should be calibrated by this factor. (Some factors may result in response difference [47, 48] including, ionization difference, gas-phase deuterium exchange, inaccurate purity characterization, kinetic isotope effect and compression of isotope distribution for stable isotope labeled analyte comparing to the native analyte.)
Notably, some issues [46] should be aware when the “surrogate analyte” approach is used, such as contamination of labeled standards with trace levels of unlabeled compounds, unequal MS responses, commercial availability, stability and degradation to the unlabeled standards over time. However, there are some rules of choosing the stable labeled surrogate which can avoid the unnecessary troubles: 1) The mass difference between any two analytes should be more than 3 Da; 2) the label position should not be the metabolism spot or the active hydrogen (e.g. –OH or –COOH); 3) All the solutions and samples are highly suggested to store in the −80 °C freezer.
3.3 LC method development
A good LC separation guarantees the reliability and robustness of this assay, because the MS analyzer cannot separate PGE2 from many other endogenous prostaglandin isomers that have the same MRM transitions as PGE2. The UPLC C18 BEH column (2.1 mm * 100mm, 1.7μm) was finally used, because it had the best separation performance among all the tested columns. Pure water, water with ammonium acetate and formic acid were initially tested, but they are incapable to achieve a good baseline separation for PGE2 and PGD2 before 20 minutes (which is too long for the assays).
In contrast, the 0.1% NH4OH solution can make an ideal separation easily with much more flexibility and convenience. The separation performance of 0.1% NH4OH for PGE2 and PGD2 was at least 10 folds higher than that of other mobile phase mentioned above. In this reported method, PGE2 and PGD2 are fully separated before 6 min (in a flow rate 0.3 ml/min, Figure 3 K). The mechanism is probably to increase the polarity of PGE2, making its retention behavior much different from other endogenous interferences.
Another advantage of 0.1% NH4OH solution is to enhance the sensitivity. In ESI negative ionization, the analyte loses one proton in order to carry one charge. Containing one carboxyl acid moiety, PGE2 can easily deprotonate with 0.1% NH4OH mobile phase thereby increasing its MS response on negative mode. We found that the PGE2 sensitivity increases 10 folds when the aqueous phase was changed from 0.1% HCOOH to 0.1% NH4OH. (Note: 0.1% HCOOH highly inhibit the ESI (-) signal response.)
One important information is worthy of mention for the NH4OH mobile phase, since it is not a common mobile phase. The safe pH range for most conventional HPLC column is from 2 to 8, and any pH beyond this range may ruin the silicon bedding of the column. The waters BEH material can withstand a pH range from 1–12, therefore it is safe for the 0.1% NH4OH (pH =10).
3.4 Method validation
Figure 3 shows the specificity of this method. The retention times of PGE2, PGE2-d4 and PGE2-d9 were about 4.5–4.6 min (Figure 3 A, B and C; Note: the number of deuterium slightly changes the retention time [48]). A low level of endogenous PGE2 was detected on wild type rat colon mucosa (Figure 3 D), but no any background interference is found for PGE2-d4 (Figure 3E) or PGE2-d9 (Figure 3F). The LLOQ was constructed with wild type colon mucosa by adding the surrogate analyst PGE2-d9 (Figure 3 J), and then measured by adding the internal standard PGE2-d4 (Figure 3 I). Similar endogenous background was also found for PGE2 (Figure 3 H) in the LLOQ. This method can well separate the PGE2 and PGD2 (confirmed with PGD2 reference standard) in the actual colon polyps samples (Figure 3K).
The purity of the stable labeled compounds (surrogate analyte and internal standard) is critically important. The detection limit, linearity and accuracy will be undermined if the isotope labeled substances do not have sufficient purity (> 98%). Therefore, 3–5 ng/ml neat standard solutions were injected. No unlabeled impurity or cross-talk influence was found (Figure 4).
Figure 4.
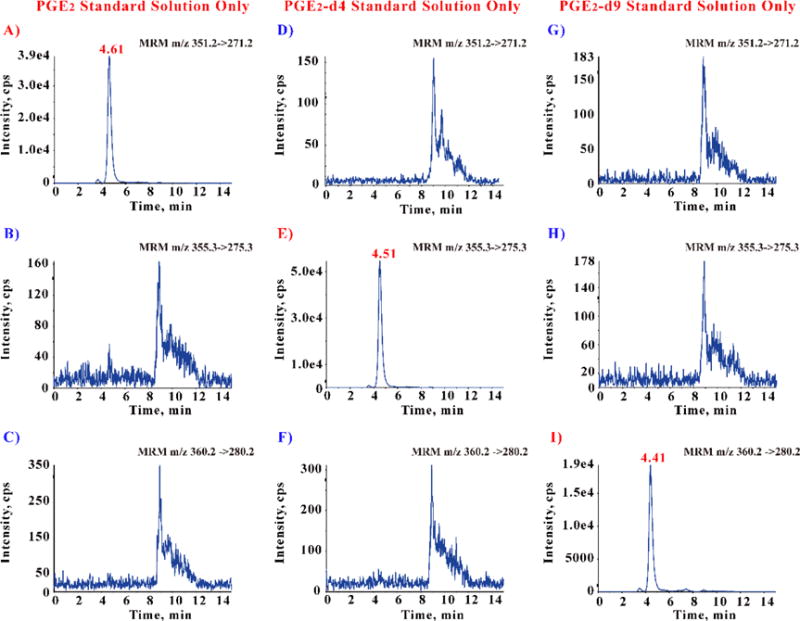
Disproval of contamination and cross-talk among PGE2, PGE2-d4 and PGE2-d9. (Limit of detection is about 0.5 pg/ml)
Three different neat standard solutions (concentration in 3–5 ng/ml, about 10,000 folds above LOD) were injected separately, and the MS analyzer detected three different MRM transitions simultaneously. The PGE2 solution only shows one peak in its owned MRM transition at 4.6 min (A), and no any peak at the same retention time showed in other two MRM transitions that belong to PGE2-d4 (B) and PGE2-d9 (C). Similarly, PGE2-d4 (E) and PGE2-d9 (I) only show one peak in their individual owned MRM transition.
The selected assay range for PGE2-d9 fulfilled the criteria for the LLOQ concentration and the calibration curve. The typical regression equation from calibration curves was:
Where f represents the peak area ratio of surrogate analyte to the I.S.; and C represents the concentration of PGE2-d9. Because the RRF is 0.99 (almost equal to 1, Table-1), the authentic analyte PGE2 can be directly calculated by this PGE2-d9 bio-matrix based standard curve. The LLOQ was 97 pg/ml for PGE2 with good accuracy and precision (90% and 3%, n=5).
Table-2 summarizes the intra- and inter-assay precision and accuracy of the method. Both intra- and inter-assay precisions tested with PGE2-d9 were less than 10%, and the precision of three bathes endogenous PGE2 was 6%. The above results in together demonstrated that the values were within the acceptable range and the method was accurate and precise.
Table-2.
Precision and accuracy of PGE2-d9 in colon tissue homogenate
| Concentration (ng/ml)
|
Precision, RSD%
|
Accuracy, RE% | ||
|---|---|---|---|---|
| Nominal | Actual | Intra-batch | Inter-batch | |
| 0.195 | 0.194 | 2.8 | 7.8 | −0.5 |
| 1.563 | 1.555 | 5.6 | 3.5 | −0.3 |
| 20.00 | 20.81 | 2.6 | 5.8 | 4.0 |
The absolute matrix effects for PGE2-d9 at concentrations of 0.195, 1.560 and 20.00 ng/ml were 96, 94 and 99%, respectively; for the I.S. was 106%. Mean extraction recoveries of PGE2-d9 were 99%, 95% and 99% and that of the internal standard was 97%.
Sample stability evaluations were designed to cover the anticipated conditions of processing of the bio-samples (Table-3). PGE2-d9 was stable during sample storage (at room temperature for 12 h, at −80 °C for 2 week), processing (three freeze–thaw cycles) and post-treatment (in the reconstituted extract at 10 °C for 18 h). All RE values between post-storage and initial QC samples were within ± 15%. Meanwhile, the authentic analyte (background PGE2) was also proven to be stable (Table-3).
Table-3.
Stability of PGE2-d9 in colon tissue homogenate in various storage conditions
| Stability Conditions | **Authentic (Background PGE2) | Analyte | *Surrogate Analyte (Spiked PGE2 d9) | ||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| % of Control | RE% | RSD% | QC samples (ng/ml) | % of Control | RE% | RSD% | |
| QC-L (0.195) |
104.1% | 4.1% | 3.5% | ||||
| Auto-sampler Stability | 101.6% | 1.6% | 2.3% | QC-M (1.560) |
104.5% | 4.5% | 4.6% |
| QC-H (20.00) |
108.0% | 8.0% | 4.1% | ||||
| QC-L (0.195) |
103.1% | 3.1% | 4.8% | ||||
| Bench-top Stability | 98.9% | −1.1% | 2.8% | QC-M (1.560) |
97.2% | −2.8% | 4.4% |
| QC-H (20.00) |
109.2% | 9.1% | 4.3% | ||||
| QC-L (0.195) |
105.1% | 5.1% | 3.4% | ||||
| Freeze-Thaw Stability | 100.1% | 0.1% | 2.8% | QC-M (1.560) |
104.0% | 4.0% | 2.6% |
| QC-H (20.00) |
111.7% | 11.7% | 0.9% | ||||
| QC-L (0.195) |
94.4% | −5.6% | 2.9% | ||||
| Long-term Stability | 98.4% | −1.6% | 1.5% | QC-M (1.560) |
100.0% | 0.0% | 3.4% |
| QC-H (20.00) |
103.9% | 3.9% | 3.6% | ||||
RE: Relative Error; RSD: Relative Standard Deviation
3 replicates for each QC sample with surrogate analyte;
Combining 9 QC samples to calcuate the average value of authentic analyte, because of the same amount of matrix background PGE2.
3.5 Application
PGE2 was unusually high in Pirc colon mucosa (21.0 ± 2.0 pg/mg tissue, Figure 5), in comparison with about 5.6 ± 0.4 pg/mg tissue in the wild type rats colon mucosa. Data suggest COX-2 enzyme upregulation in the Pirc colon tissue. In untreated Pirc, the polyps PGE2 level was two folds higher than that of mucosa (p=0.016) thus confirms the current belief that PGE2 promotes the colorectal cancer growth. In this study, a short-term celecoxib treatment reduced mucosa PGE2 by 57% (p=0.014) and polyps PGE2 by 29% (p=0.12). The result proves celecoxib decreases colon PGE2 and is capable of suppresses the polyps’ growth. The variant in inhibition observed between PGE2 within polyps and colon mucosa might be contributed by the discrepancy in surface area and physiological barrier. Celecoxib can easily hit the COX-2 target in the barrier-free mucosa that has large surface area. Inspiringly, celecoxib reduced the Pirc mucosa PGE2 back to normal (compared to wild type, p=0.788) with a short treatment. We expect that mucosa PGE2 should be maintained in this reduced level with a long-term treatment (24 weeks). The PGE2 reduction mechanism could explain why a 24-weeks celecoxib treatment (similar dose as we used in this study) remarkably decrease the Pirc polyps burden [13]. The only regret in this study is that the polyps PGE2 did not significantly decline as we expected initially. Perhaps the treatment duration is too short (only 4 days). A longer dosing (2–3 months) would have a big chance to hit this expectation.
Figure 5.
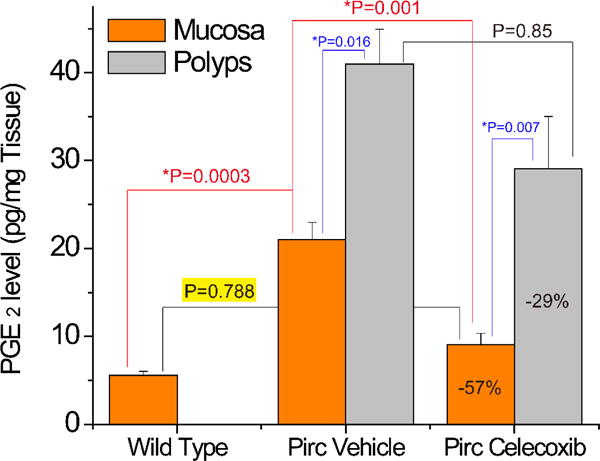
Colon tissue PGE2 levels of wild type and Pirc rats
In conclusion, Pirc rat is a good animal model for human FAP study; and the mucosa PGE2 inhibition is a good indicator for anti-polyps efficacy in short-term study when the Pirc is treated with COX-2 inhibitors.
Supplementary Material
Highlights.
The colon PGE2 of normal (wild type) rat and Pirc rat (an Apc-mutant rat) were firstly accurately determined by specific and sensitive UPLC-MS/MS.
PGE2 was confirmed to promote Pirc rat colon polyps growth.
An unusual mobile phase (0.1% ammonia hydroxide) was found to greatly improve the chromatographic separation of prostaglandins.
Major abbreviations
- PGE2 / PGD2
Prostaglandin E2/D2
- COX-2
Cyclo-oxygenase-2
- Pirc
polyposis in the rat colon
- FAP
familial adenomatous polyposis
- APC gene
Adenomatous Polyposis Coli gene
- CRC
Colorectal cancer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101:385–398. doi: 10.1111/j.1572-0241.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 2.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 3.Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. doi: 10.1186/1750-1172-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- 5.Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, Sahai E, Reis e Sousa C. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell. 2015;162:1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M, Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- 7.FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345:433–442. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- 8.Baron JA, Sandler RS, Bresalier RS, Quan H, Riddell R, Lanas A, Bolognese JA, Oxenius B, Horgan K, Loftus S, Morton DG, A.P.T. Investigators A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131:1674–1682. doi: 10.1053/j.gastro.2006.08.079. [DOI] [PubMed] [Google Scholar]
- 9.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, Hess TM, Woloj GM, Boisserie F, Anderson WF, Viner JL, Bagheri D, Burn J, Chung DC, Dewar T, Foley TR, Hoffman N, Macrae F, Pruitt RE, Saltzman JR, Salzberg B, Sylwestrowicz T, Gordon GB, Hawk ET, A.P.C.S. Investigators Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–884. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 10.RS Bresalier RS, Sandler H, Quan JA, Bolognese B, Oxenius K, Horgan C, Lines R, Riddell D, Morton A, Lanas MA, Konstam JA, Baron I, Adenomatous Polyp Prevention on Vioxx Trial Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 11.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee D, Nissen SE, Topol EJ. Cox-2 inhibitors and cardiovascular risk: we defend our data and suggest caution. Cleve Clin J Med. 2001;68:963–964. doi: 10.3949/ccjm.68.11.963. [DOI] [PubMed] [Google Scholar]
- 13.Amos-Landgraf JM, Kwong LN, Kendziorski CM, Reichelderfer M, Torrealba J, Weichert J, Haag JD, Chen KS, Waller JL, Gould MN, Dove WF. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc Natl Acad Sci U S A. 2007;104:4036–4041. doi: 10.1073/pnas.0611690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Boxtel R, Gould MN, Cuppen E, Smits BM. ENU mutagenesis to generate genetically modified rat models. Methods Mol Biol. 2010;597:151–167. doi: 10.1007/978-1-60327-389-3_11. [DOI] [PubMed] [Google Scholar]
- 15.Irving AA, Yoshimi K, Hart ML, Parker T, Clipson L, Ford MR, Kuramoto T, Dove WF, Amos-Landgraf JM. The utility of Apc-mutant rats in modeling human colon cancer. Dis Model Mech. 2014;7:1215–1225. doi: 10.1242/dmm.016980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwong LN, Dove WF. APC and its modifiers in colon cancer. Adv Exp Med Biol. 2009;656:85–106. doi: 10.1007/978-1-4419-1145-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Femia AP, Becherucci C, Crucitta S, Caderni G. Apc-driven colon carcinogenesis in Pirc rat is strongly reduced by polyethylene glycol. Int J Cancer. 2015;137:2270–2273. doi: 10.1002/ijc.29581. [DOI] [PubMed] [Google Scholar]
- 18.Femia AP, Luceri C, Soares PV, Lodovici M, Caderni G. Multiple mucin depleted foci, high proliferation and low apoptotic response in the onset of colon carcinogenesis of the PIRC rat, mutated in Apc. Int J Cancer. 2015;136:E488–495. doi: 10.1002/ijc.29232. [DOI] [PubMed] [Google Scholar]
- 19.Femia AP, Soares PV, Luceri C, Lodovici M, Giannini A, Caderni G. Sulindac, 3,3′-diindolylmethane and curcumin reduce carcinogenesis in the Pirc rat, an Apc-driven model of colon carcinogenesis. BMC Cancer. 2015;15:611. doi: 10.1186/s12885-015-1627-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris KL, Pulliam SR, Okoro E, Guo Z, Washington MK, Adunyah SE, Amos-Landgraf JM, Ramesh A. Western diet enhances benzo(a)pyrene-induced colon tumorigenesis in a polyposis in rat coli (PIRC) rat model of colon cancer. Oncotarget. 2016;7:28947–28960. doi: 10.18632/oncotarget.7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giardiello FM, Hamilton SR, Krush AJ, Piantadosi S, Hylind LM, Celano P, Booker SV, Robinson CR, Offerhaus GJ. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. 1993;328:1313–1316. doi: 10.1056/NEJM199305063281805. [DOI] [PubMed] [Google Scholar]
- 22.Cruz-Correa M, Hylind LM, Romans KE, Booker SV, Giardiello FM. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology. 2002;122:641–645. doi: 10.1053/gast.2002.31890. [DOI] [PubMed] [Google Scholar]
- 23.Mahmoud NN, Boolbol SK, Dannenberg AJ, Mestre JR, Bilinski RT, Martucci C, Newmark HL, Chadburn A, Bertagnolli MM. The sulfide metabolite of sulindac prevents tumors and restores enterocyte apoptosis in a murine model of familial adenomatous polyposis. Carcinogenesis. 1998;19:87–91. doi: 10.1093/carcin/19.1.87. [DOI] [PubMed] [Google Scholar]
- 24.Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000;60:4705–4708. [PubMed] [Google Scholar]
- 25.Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. 2000;60:5040–5044. [PubMed] [Google Scholar]
- 26.Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(Min/+) mice. Cancer Res. 2002;62:403–408. [PubMed] [Google Scholar]
- 27.Gao Y, Hou R, Liu F, Cai R, Fang L, Peng C, Qi Y. Highly specific and sensitive immunoassay for the measurement of prostaglandin E2 in biological fluids. Bioanalysis. 2015;7:2597–2607. doi: 10.4155/bio.15.130. [DOI] [PubMed] [Google Scholar]
- 28.Gandhi AS, Budac D, Khayrullina T, Staal R, Chandrasena G. Quantitative analysis of lipids: a higher-throughput LC-MS/MS-based method and its comparison to ELISA. Future Sci OA. 2017;3:FSO157. doi: 10.4155/fsoa-2016-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao H, Xiao L, Park G, Wang X, Azim AC, Christman JW, van Breemen RB. An improved LC-MS/MS method for the quantification of prostaglandins E(2) and D(2) production in biological fluids. Anal Biochem. 2008;372:41–51. doi: 10.1016/j.ab.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kempen EC, Yang P, Felix E, Madden T, Newman RA. Simultaneous quantification of arachidonic acid metabolites in cultured tumor cells using high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Anal Biochem. 2001;297:183–190. doi: 10.1006/abio.2001.5325. [DOI] [PubMed] [Google Scholar]
- 31.Martins CA, Weffort-Santos AM, Gasparetto JC, Trindade AC, Otuki MF, Pontarolo R. Malva sylvestris L. extract suppresses desferrioxamine-induced PGE(2) and PGD(2) release in differentiated U937 cells: the development and validation of an LC-MS/MS method for prostaglandin quantification. Biomed Chromatogr. 2014;28:986–993. doi: 10.1002/bmc.3106. [DOI] [PubMed] [Google Scholar]
- 32.Nithipatikom K, Laabs ND, Isbell MA, Campbell WB. Liquid chromatographic-mass spectrometric determination of cyclooxygenase metabolites of arachidonic acid in cultured cells. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;785:135–145. doi: 10.1016/s1570-0232(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 33.Takabatake M, Hishinuma T, Suzuki N, Chiba S, Tsukamoto H, Nakamura H, Saga T, Tomioka Y, Kurose A, Sawai T, Mizugaki M. Simultaneous quantification of prostaglandins in human synovial cell-cultured medium using liquid chromatography/tandem mass spectrometry. Prostaglandins Leukot Essent Fatty Acids. 2002;67:51–56. doi: 10.1054/plef.2002.0381. [DOI] [PubMed] [Google Scholar]
- 34.Shin JS, Peng L, Kang K, Choi Y. Direct analysis of prostaglandin-E2 and -D2 produced in an inflammatory cell reaction and its application for activity screening and potency evaluation using turbulent flow chromatography liquid chromatography-high resolution mass spectrometry. J Chromatogr A. 2016;1463:128–135. doi: 10.1016/j.chroma.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka M, Moran S, Wen J, Affram K, Chen T, Symes AJ, Zhang Y. WWL70 attenuates PGE2 production derived from 2-arachidonoylglycerol in microglia by ABHD6-independent mechanism. J Neuroinflammation. 2017;14:7. doi: 10.1186/s12974-016-0783-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas D, Suo J, Ulshofer T, Jordan H, de Bruin N, Scholich K, Geisslinger G, Ferreiros N. Nano-LC-MS/MS for the quantitation of prostanoids in immune cells. Anal Bioanal Chem. 2014;406:7103–7116. doi: 10.1007/s00216-014-8134-8. [DOI] [PubMed] [Google Scholar]
- 37.Cao H, Yu R, Tao Y, Nikolic D, van Breemen RB. Measurement of cyclooxygenase inhibition using liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2011;54:230–235. doi: 10.1016/j.jpba.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komaba J, Matsuda D, Shibakawa K, Nakade S, Hashimoto Y, Miyata Y, Ogawa M. Development and validation of an on-line two-dimensional reversed-phase liquid chromatography-tandem mass spectrometry method for the simultaneous determination of prostaglandins E(2) and F(2alpha) and 13,14-dihydro-15-keto prostaglandin F(2alpha) levels in human plasma. Biomed Chromatogr. 2009;23:315–323. doi: 10.1002/bmc.1117. [DOI] [PubMed] [Google Scholar]
- 39.Shinde DD, Kim KB, Oh KS, Abdalla N, Liu KH, Bae SK, Shon JH, Kim HS, Kim DH, Shin JG. LC-MS/MS for the simultaneous analysis of arachidonic acid and 32 related metabolites in human plasma: Basal plasma concentrations and aspirin-induced changes of eicosanoids. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;911:113–121. doi: 10.1016/j.jchromb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Chen CY, Lin P, Tsai MH, Lee HL. Targeted lipidomics profiling of acute arsenic exposure in mice serum by on-line solid-phase extraction stable-isotope dilution liquid chromatography-tandem mass spectrometry. Arch Toxicol. 2017 doi: 10.1007/s00204-017-1937-6. [DOI] [PubMed] [Google Scholar]
- 41.Araujo P, Mengesha Z, Lucena E, Grung B. Development and validation of an extraction method for the determination of pro-inflammatory eicosanoids in human plasma using liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2014;1353:57–64. doi: 10.1016/j.chroma.2013.10.082. [DOI] [PubMed] [Google Scholar]
- 42.Brose SA, Baker AG, Golovko MY. A fast one-step extraction and UPLC-MS/MS analysis for E2/D 2 series prostaglandins and isoprostanes. Lipids. 2013;48:411–419. doi: 10.1007/s11745-013-3767-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brose SA, Thuen BT, Golovko MY. LC/MS/MS method for analysis of E(2) series prostaglandins and isoprostanes. J Lipid Res. 2011;52:850–859. doi: 10.1194/jlr.D013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Golovko MY, Murphy EJ. An improved LC-MS/MS procedure for brain prostanoid analysis using brain fixation with head-focused microwave irradiation and liquid-liquid extraction. J Lipid Res. 2008;49:893–902. doi: 10.1194/jlr.D700030-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt R, Coste O, Geisslinger G. LC-MS/MS-analysis of prostaglandin E2 and D2 in microdialysis samples of rats. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;826:188–197. doi: 10.1016/j.jchromb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 46.Thakare R, Chhonker YS, Gautam N, Alamoudi JA, Alnouti Y. Quantitative analysis of endogenous compounds. J Pharm Biomed Anal. 2016;128:426–437. doi: 10.1016/j.jpba.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 47.Jones BR, Schultz GA, Eckstein JA, Ackermann BL. Surrogate matrix and surrogate analyte approaches for definitive quantitation of endogenous biomolecules. Bioanalysis. 2012;4:2343–2356. doi: 10.4155/bio.12.200. [DOI] [PubMed] [Google Scholar]
- 48.Li W, Cohen LH. Quantitation of endogenous analytes in biofluid without a true blank matrix. Anal Chem. 2003;75:5854–5859. doi: 10.1021/ac034505u. [DOI] [PubMed] [Google Scholar]
- 49.Goodenough AK, Onorato JM, Ouyang Z, Chang S, Rodrigues AD, Kasichayanula S, Huang SP, Turley W, Burrell R, Bifano M, Jemal M, LaCreta F, Tymiak A, Wang-Iverson D. Quantification of 4-beta-hydroxycholesterol in human plasma using automated sample preparation and LC-ESI-MS/MS analysis. Chem Res Toxicol. 2011;24:1575–1585. doi: 10.1021/tx2001898. [DOI] [PubMed] [Google Scholar]
- 50.Jemal M, Schuster A, Whigan DB. Liquid chromatography/tandem mass spectrometry methods for quantitation of mevalonic acid in human plasma and urine: method validation, demonstration of using a surrogate analyte, and demonstration of unacceptable matrix effect in spite of use of a stable isotope analog internal standard. Rapid Commun Mass Spectrom. 2003;17:1723–1734. doi: 10.1002/rcm.1112. [DOI] [PubMed] [Google Scholar]
- 51.Kang S, Oh SM, Chung KH, Lee S. A surrogate analyte-based LC-MS/MS method for the determination of gamma-hydroxybutyrate (GHB) in human urine and variation of endogenous urinary concentrations of GHB. J Pharm Biomed Anal. 2014;98:193–200. doi: 10.1016/j.jpba.2014.05.028. [DOI] [PubMed] [Google Scholar]
- 52.Liang HR, Takagaki T, Foltz RL, Bennett P. Quantitative determination of endogenous sorbitol and fructose in human nerve tissues by atmospheric-pressure chemical ionization liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:2284–2294. doi: 10.1002/rcm.2055. [DOI] [PubMed] [Google Scholar]
- 53.Liang HR, Takagaki T, Foltz RL, Bennett P. Quantitative determination of endogenous sorbitol and fructose in human erythrocytes by atmospheric-pressure chemical ionization LC tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;824:36–44. doi: 10.1016/j.jchromb.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 54.Lindsay C, Moutquin J-M, C-Gaudreault R, Forest J-C. Development of an enzyme-linked immunosorbent assay for 2,3-dinor-6-keto-prostagland in F1α in urine using a monoclonal antibody. Clinical Biochemistry. 1995;28:395–400. doi: 10.1016/0009-9120(95)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.