

Abstract
Background
Food allergies are a growing health problem and the development of therapies that prevent disease onset is limited by the lack of adjuvant-free experimental animal models. We compared allergic sensitization in patients with food allergy or Wiskott-Aldrich syndrome (WAS) and defined whether spontaneous disease in Was−/− mice recapitulates the pathology of a conventional disease model and/or human food allergy.
Methods
Comparative ImmunoCAP ISAC microarray was performed in patients with food allergy or WAS. Spontaneous food allergy in Was−/− mice was compared to an adjuvant-based model in wild-type mice (WT-OVA/alum). Intestinal and systemic anaphylaxis was assessed and the role of the high affinity IgE Fc receptor (FcεRI) in allergic sensitization was evaluated using Was−/−Fcer1a−/− mice.
Results
Polysensitization to food was detected in both WAS and food allergic patients which was recapitulated in the Was−/− model. Oral administration of OVA in Was−/− mice induced low titers of OVA-specific IgE compared to the WT-OVA/alum model. Irrespectively, 79% of Was−/− mice developed allergic diarrhea following oral OVA challenge. Systemic anaphylaxis occurred in Was−/− mice (95%) with a mortality rate >50%. Spontaneous sensitization and intestinal allergy occurred independent of FcεRI expression on mast cells and basophils.
Conclusions
Was−/− mice provide a model of food allergy with the advantage of mimicking polysensitization and low food-antigen IgE titers as observed in humans with clinical food allergy. This model will facilitate studies on aberrant immune responses during spontaneous disease development. Our results imply that therapeutic targeting of the IgE/FcεRI activation cascade will not affect sensitization to food.
Keywords: Food allergy; Adjuvant-free polysensitization; FcεRI, high-affinity IgE receptor; Mast cells; Wiskott-Aldrich syndrome protein
Introduction
Immunoglobulin E (IgE)-mediated food allergies are a rapidly growing health problem in industrialized societies. Although the determination of the exact prevalence is hampered by non-overlapping diagnostic definitions, estimates for US adults and children currently estimated to be 5% and 8% respectively [1, 2]. In food allergic patients, exposure to minor quantities of food allergens elicits type I hypersensitivity reactions, which are responsible for symptoms that range in severity from oral tingling and nausea to profuse diarrhea and anaphylactic shock [3–6]. Allergic reactions to food can be fatal and are a common reason for emergency room visits and hospitalization [5, 7–9]. Therefore, considerable research effort is dedicated to understanding the pathophysiology of IgE-mediated food allergies [10, 11].
Allergic sensitization results from perturbations in the pathways that normally maintain a state of immune tolerance to innocuous food antigens [12, 13]. Under physiological circumstances, antigen presentation of food components by dendritic cells results in anergy or deletion of reactive T cells and the induction of FOXP3+ regulatory T cells (Tregs). In food allergic patients, however, exposure to intestinal allergens gives rise to Th2-primed effector T cells [14]. These allergen-specific T cells will respond to subsequent allergen encounters with production of IL-4 and IL-13, which induces class-switching to IgE and IgG4 (in humans) and IgG1 (in mice) in allergen-specific B cells [3, 4]. Food-specific IgE circulates in serum and binds to its high-affinity IgE Fc receptor, FcεRI, which is constitutively expressed on mast cells and basophils [15]. During the allergic effector phase, the innate effector cells respond to antigen-mediated crosslinking of IgE-loaded FcεRI with rapid secretion of pre-formed inflammatory mediators, such as histamine and proteases, that mediate the local and systemic symptoms associated with type I allergic reactions [3, 16].
Epidemiologic studies have identified numerous associations between microbial, environmental, and nutritional factors and the occurrence of food allergy [17–20], but experimental and mechanistic evidence to prove causality remains scarce. This gap in our understanding is, at least in part, attributable to the lack of animal models that mimic allergic sensitization as it occurs in humans. Wild-type mice are highly resistant to the induction of IgE-mediated immune responses to food, attesting to the robustness of the regulatory mechanisms that underlie oral tolerance [21]. Consequently, experimental models of murine food allergy require sensitization to induce systemic production of antigen-specific IgE prior to intestinal allergen challenge [21]. Commonly strategies rely on intraperitoneal sensitization to a model antigen (e.g., ovalbumin (OVA)) emulsified in aluminum hydroxide as a Th2-skewing adjuvant or, alternatively, oral sensitization by co-administration of adjuvants such as cholera toxin or staphylococcal enterotoxin B with the model antigen [22]. These ‘isomorphic’ models have demonstrated great utility in the study of allergic effector responses in vivo. However, such models only recapitulate the symptoms and treatment responses of established food allergy and cannot mimic the early phases of sensitization to food. Study of spontaneous, adjuvant-free induction of IgE responses against food allergens instead requires a ‘homologous’ animal model, which for food allergy has not been developed [21].
Recently, we demonstrated that patients with Wiskott-Aldrich syndrome (WAS) caused by loss-of-function mutations in the Wiskott-Aldrich syndrome protein (WASP) are at increased risk of developing food allergy [23]. Similarly, mice deficient in the murine ortholog (Was−/− mice) develop IgE responses against food allergens in chow [23]. We aimed to characterize Was−/− mice as a potential homologous model of IgE-mediated food allergy. Here we show that Was−/− mice become sensitized to ovalbumin (OVA) upon oral administration in the absence of adjuvant, and that subsequent IgE-mediated mast cell responses and small intestinal inflammation results in systemic and tissue specific food allergy. Importantly, serum sensitization in Was−/− mice mimics polysensitization and the low food-specific IgE titer observed in humans with clinical food allergy which is not observed in common adjuvant-based experimental murine models.
Materials and methods
Patients
Serum from WAS patients was obtained from a biorepository at Boston Children’s Hospital (IRB number P00000529). Samples from age- and gender-matched food allergic and non-allergic control patients were obtained from a longitudinal cohort study on intestinal allergies at Boston Children’s Hospital (IRB number 07-11-0460, [24]). All patients or their legal guardians provided informed consent prior to sample donation.
ImmunoCAP ISAC microarray
Allergen-specific IgE and IgG4 was detected using the ImmunoCAP® ISAC 112 solid phase assay following the manufacturer´s guidelines (Thermo Scientific, Waltham, MA, 81-1012-01). Allergen-specific Igs were quantified as relative ISAC standard units between with Phadia Microarray Image Analysis (MIA®) and Xplain® software by ThermoFisher (Waltham, MA).
Mice
Was−/− and Wasfl/flFoxp3-Cre mice have been described previously [23]. Was−/− and Was+/− on the colitis-resistant BALB/c and C57BL/6 background or WT controls were bred in house under SPF conditions. Hemizygous Was− males are also referred to as Was−/−. Experiments were performed with mixed gender cohorts in littermate-controlled and cohoused settings following the guidelines of the Society of Mucosal Immunology. C57BL/6 Fcer1a−/− mice [25] were crossed with C57BL/6 Was−/− mice to generate cohorts of Was−/−Fcer1a−/− and Was−/−Fcer1a+/− littermates. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC 13-06-2415R).
ELISA experiments
IgE and IgG levels were detected with anti-human IgE (clone: MHE-18, BioLegend, San Diego, CA) and anti-human IgG (Southern Biotech, Birmingham, AL, 2014-01) and horseradish peroxidase-conjugated anti-human IgE (Sigma-Aldrich, Natick, MA A9667) or biotin-conjugated anti-human IgG (eBioscience, San Diego, CA, 13-4998-83). Serial dilutions of human IgE (NBS-C BioScience, Vienna, AT, 0911-0-050) or human IgG (Southern Biotech 0150-01) were used as standards. Quantification of murine total, food-antigen-specific, and OVA-specific IgE and IgG1 has been published [23, 26].
Experimental murine food allergy
Following an established protocol [26, 27], WT BALB/c mice were immunized with three intraperitoneal injections of OVA/alum (OVA (Sigma), 50μg/100μl aluminum hydroxide (alum, Thermo Scientific)) in seven-day intervals (WT-OVA/alum model). Adjuvant-free sensitization to OVA in Was−/− mice was achieved by gavaging OVA (5mg/200μl in PBS) seven times in five day intervals. Sensitized mice were challenged by gavage with 50 mg OVA in 200 μl PBS. Anaphylaxis was measured as drop in core temperature with implantable temperature transponders (IPTT-300, Biomedic Data Systems, Seaford, Del) [28].
In vitro mast cell degranulation assay
Bone marrow cells from WT or Was−/− mice on BALB/c or C57BL/6 background were differentiated in vitro with 10 ngml-1 IL3 and 20 ngml-1 SCF in RPMI 1640 (Gibco, Thermo Fisher, Waltham, MA) supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml-1 streptomycin, 2mM L-glutamine, and 1% 2-Mercaptoethanol (MC medium). Purity of cultures was assessed by FACS (mAb 2B8 for c-kit, mAb MAR-1 for FcεRI; Biolegend). For degranulation assays, cells were loaded with IgE overnight using serum or TNP-specific IgE and challenged with Ova or TNP-OVA at the indicated doses. Degranulation was assessed by staining with an anti-LAMP-1 mAb (1D4B, Biolegend) and viability dye (eBioscience). Cells were acquired on a FACSCanto II flow cytometer (BD Bioscience, San Jose, CA) and analyzed using FlowJo (Treestar, Ashland, OR).
Flow cytometric analysis of basophils
Blood samples were treated with RBC lysis buffer (eBioscience) and mononuclear cells were stained with an Ab cocktail containing: anti-CD16/CD32 (2.4G2, BD Pharmingen, San Jose, CA), anti-CD45 (30-F11), anti-CD49b (DX5), anti-FcεRIa (MAR-1), and anti-IgE (RME-1, all from Biolegend). Basophils were identified as CD45MidCD49b+ and IgE+.
mRNA expression analysis
Intestinal Mcpt1 mRNA levels were determined as described using iQ SYBR Green Supermix (Hercules, CA) [23].
Mast cell histology
MCs were quantified using by chloroacetate esterase staining as previously described [26].
Results
Patients with Wiskott-Aldrich syndrome and children with food allergy show similar patterns of polysensitization
We previously documented the increased prevalence of physician-diagnosed food allergy in WAS patients [23]. To define if sensitization patterns in WAS-associated food allergy compare with those in patients with food allergy, we analyzed serum IgE from WAS patients (14 months to 7 years), patients with food allergy, and non-allergic controls. Both patients with food allergy and those with WAS show increased serum IgE levels when compared to controls without allergies (Fig. S1). While total IgE levels were increased in WAS compared to food allergic patients, no differences were observed for total serum IgG between any of the groups (Fig. S1).
Patients with food allergy are at increased risk of sensitization to other food antigens [29, 30]. To assess whether polysensitization is also a feature of WAS-associated food allergy, we performed an immunochip-based microarray that screens IgE reactivity against 112 molecular allergens, 43 of which are antigens from 17 different foods including peanut, tree nuts, milk, soy, and shrimp (Fig. 1). In 3/5 patients with WAS, IgE with specificity for food was detected. Serum sensitized WAS patients contained IgE with reactivity for more than one allergen in a single food group or IgE with specificity for at least two different foods. Similarly, 4/6 food allergic patients were polysensitized to food (Fig. 1A). To further compare the humoral anti-food response, we screened for the presence of food-specific IgG4 [31, 32]. IgG4 reactivity against food antigens was common even in control subjects and did not distinguish between groups (Fig. 1B). These results demonstrate a comparable degree of polysensitization between patients with food allergy and patients with WAS with a high degree of overlap in sensitization profiles.
Figure 1.
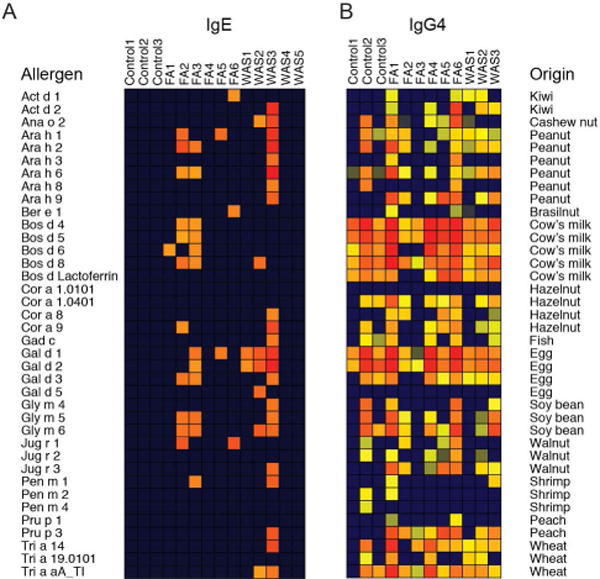
Polysensitization in patients with food allergy and Wiskott-Aldrich syndrome. Antigen specificity profiling for common food allergens for (A) IgE and (B) IgG4 in serum from controls, food allergic (FA), and Wiskott Aldrich syndrome (WAS) patients.
Was−/− mice mount OVA-specific IgE responses following oral antigen exposure without adjuvant
IgE with specificity for multiple components of chow is found in Was−/− mice [23], and mimics the polysensitization pattern seen in food allergic patients. In order to compare the development of antigen-specific food allergy in the spontaneous Was−/− model to a well-established experimental model, we selected ovalbumin (OVA) as a model allergen [33]. For the established allergy model we utilized systemic sensitization via aluminum hydroxide injection in wild type animals (WT-OVA/alum model [27]; Fig. 2A). For adjuvant-free spontaneous allergic sensitization, Was−/− mice received 8 oral gavages of OVA dissolved in PBS (spontaneous Was−/− model; Fig. 2B). The adjuvant-free approach induced OVA-specific IgE selectively in Was−/− serum but not in Was+/− or WT littermates (Fig. 2C). Titers of OVA-specific IgE were significantly lower in Was−/− mice than in the WT-OVA/alum model (Fig. 2C). Since Was−/− mice have higher serum IgE levels than OVA/alum-sensitized WTs (Fig. 2D), the reduction of antigen-specific IgE in Was−/− mice was even more pronounced when displayed as fraction of total IgE (Fig. 2D).
Figure 2.
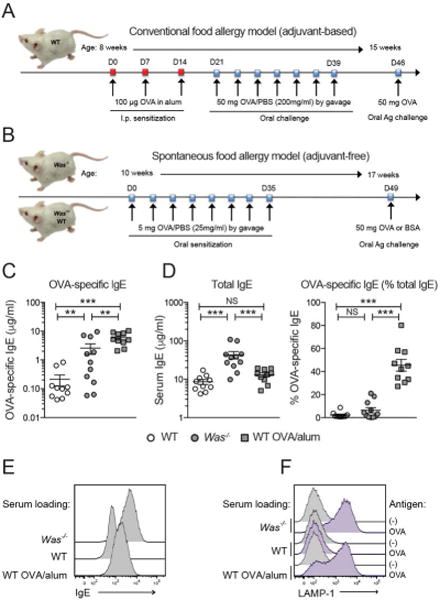
Adjuvant-based sensitization in WT and spontaneous sensitization Was−/− mice. (A-B) Sensitization strategies. (C) OVA-IgE, D, total IgE and %OVA-IgE of total IgE in OVA/alum sensitized WT (squares), OVA-gavaged Was−/− (gray circles), and littermates (open circles). E, IgE on serum-loaded MCs. F, Degranulation after OVA- crosslinking. SEM, *p<0.05; **p<0.01; ***p<0.001; NS: not significant, ANOVA with Tukey’s multiple comparison test.
To assess whether the different fractions of antigen-specific IgE affected the efficiency of antigen-mediated IgE/FcεRI activation, we loaded WT bone-marrow-derived mast cells (BMMCs) with serum from either the WT-OVA/alum model or spontaneous Was−/− model. When BMMCs were loaded with serum that contained equivalent concentrations of OVA-specific IgE, as determined by ELISA, we detected increased surface IgE with Was−/− serum (Fig. 2E). This was not surprising given the higher concentration of serum IgE in Was−/− mice, which increases the surface IgE by stabilizing FcεRI [34]. Even though the amount of OVA-specific IgE was lower in serum from the Was−/− model, the extent of degranulation induced with OVA-induced crosslinking was comparable to MCs loaded with WT-OVA/alum serum (Fig. 2F). When surface loading was corrected for differences in total IgE concentrations, we observed similar degrees of degranulation despite the approximately 20-fold lower concentration of OVA-specific IgE in Was−/− serum (Fig. S2). The finding that the low fraction of OVA-specific IgE in Was−/− serum induced efficient MC degranulation is reminiscent of the clinical observation that severity of food allergic responses cannot always be predicted based on allergen-specific IgE titers ([35] in food allergic patients (see also Fig. S1).
Pronounced allergic diarrhea and intestinal mast cell expansion in Was−/− mice
A valuable homologous model of food allergy needs to recapitulate the IgE effector phase as seen in food allergic patients. Since IgE-mediated anaphylaxis is a phenotype in food allergic individuals, we sought to determine if OVA-specific IgE in Was−/− mice would be associated with anaphylaxis in vivo. When screening both models for signs of allergic diarrhea following a single gavage containing OVA, which has been defined as intestinal anaphylaxis [27], we found that 79% of Was−/− exhibited symptoms of intestinal anaphylaxis compared to 60% of WT-OVA/alum mice (Fig. 3A and S3). No signs of intestinal anaphylaxis were observed in Was+/−, WT controls, or in Was−/− mice challenged with bovine serum albumin (BSA) as a specificity control. In line with previously reported data [22, 27], WT-OVA/alum mice were resistant to oral anaphylaxis defined as a drop in body temperature upon intragastric OVA challenge after sensitization (Fig. S3B). MCPT1 is a mast cell protease that is released from mucosal mast cells upon allergen-dependent crosslinking of IgE and is used as a serum marker for the severity of experimental food allergy [26]. We have shown that serum MCPT1 levels in Was−/− mice are strongly correlated with the degree of intestinal mast cell expansion and food-specific IgE titers [23]. Gastric OVA challenges resulted in increases in MCPT1 in the spontaneous Was−/− model and WT-OVA/alum mice (Fig. 3B). Despite the higher levels of serum MCPT1 in Was−/− mice, a comparable expansion of mucosal mast cells was found in both models (Fig. 3C). These data demonstrate strong MC-dependent innate IgE effector responses in spontaneous Was−/− model even though food-antigen-specific IgE titers are lower than in the WT-OVA/alum model.
Figure 3.
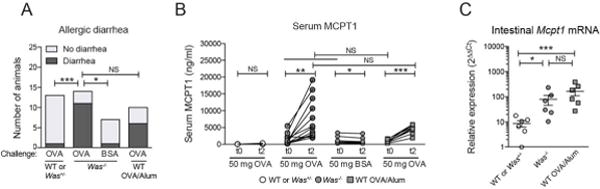
IgE effector responses in Was−/− mice. (A) Allergic diarrhea after challenge with OVA or BSA assessed with Fisher’s exact test. (B) Serum MCPT1 after oral challenge. (C) Mcpt1 mRNA expression. SEM, *p<0.05; **p<0.01; ***p<0.001; NS: not significant as assessed by paired t-test or Wilcoxon matched-pair rank test for intra-individual analyses (B), or ANOVA on log-transformed data with Tukey’s multiple comparison test (C).
Systemic anaphylaxis in Was−/− mice and efficient IgE-mediated degranulation of WASP-deficient mast cells
Lethal anaphylactic shock is the most severe outcome of accidental antigen exposure in food allergic patients [17, 36]. When studying anaphylactic shock to systemic allergen exposure, we observed a rapid temperature drop in sensitized Was−/− mice but not their Was+/− or WT littermates (Fig. 4A and 4B). Systemic anaphylaxis occurred in 19 out of 20 Was−/− animals (95%) with a mortality rate greater than 50% (Fig. 4C). These results show that spontaneous sensitization to food antigens in Was−/− mice in the absence of adjuvant is sufficient to mediate lethal anaphylaxis upon systemic antigen exposure. This susceptibility of Was−/− mice to lethal anaphylaxis was unexpected since decreased mediator release in the absence of WASP and resistance to passive anaphylaxis in Was−/− mice has been reported [37]. To investigate the consequences of WASP deficiency for MC degranulation, we generated BMMCs from WT and Was−/− mice on the BALB/c or C57BL/6 background (Fig. S4). Mast cells from each strain differentiated normally with no significant difference in surface expression of cKit or FcεRI (Fig. S4). A modest decrease in degranulation upon challenge in the low antigen concentration range was observed, but this difference did not achieve statistical significance, nor were differences observed over a broad range of concentrations (Fig. 4D). This set of experiments demonstrated that the spontaneous Was−/− model is a valuable asset for studying anaphylactic shock in hosts with low allergen-specific IgE titers.
Figure 4.
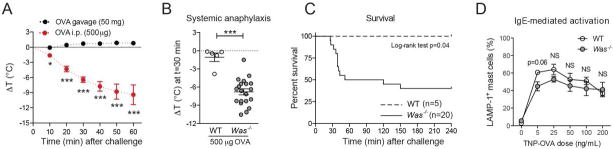
Systemic anaphylaxis in Was−/− mice. (A) Core temperature of Was−/− mice after oral or systemic challenge. (B) Anaphylaxis in Was−/− mice after systemic challenge. (C) Kaplan-Meier survival curves. (D), IgE-mediated mast cell degranulation in WT (open circles) and Was−/− MCs (gray circles). Error bars depict SEM. *p<0.05; **p<0.01; ***p<0.001; NS: not significant; t-test or Mann-Whitney U test.
Spontaneous oral sensitization occurs independently of FcεRI-mediated IgE signals
Recent studies implicated a role for IgE/FcεRI-mediated signals in the expansion of mucosal mast cells and the pathogenesis of food allergy [28, 38]. To address whether IgE/FcεRI signals contribute to spontaneous oral sensitization and the development of IgE-mediated food allergy in Was−/− mice, we generated FcεRI-deficient Was−/−Fcer1a−/− mice. Successful ablation of FcεRI was confirmed by the absence of surface-bound IgE on basophils (Fig. 5A and S5). Was−/−Fcer1a−/− mice presented with a non-significant reduction in basophil numbers but had no other apparent phenotype (Fig. 5B). Serum levels of total IgE and IgG1 with reactivity to major chow antigens (soy and wheat) were similar in Was−/−Fcer1a−/− and Was−/−Fcer1a+/− mice (Fig. S5). Adjuvant free OVA gavages induced similar levels of OVA-specific IgE and IgG1 in Was−/−Fcer1a−/− and Was−/−Fcer1a+/− littermates (Fig. 5C and 5D). Serum MCPT1 in Was−/−Fcer1a−/− mice was elevated to a similar degree as FcεRI-expressing Was−/− mice indicating that IgE/FcεRI-mediated signals are dispensable for mucosal mast cell activation in the spontaneous Was−/− model (Fig. 5E). When comparing serum MCPT1 in the OVA/alum model in WT and Fcer1a−/− mice significantly lower titers were found in Fcer1a-deficient animals (Fig. 5E) as also recently described by others [38].
Figure 5.
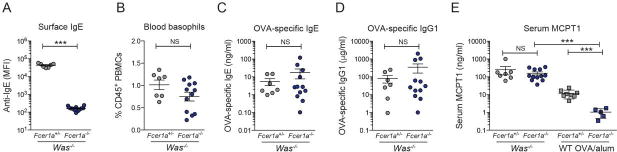
Adjuvant-free oral sensitization in FcεRI-deficient animals. (A) Absence of surface on Was−/−Fcer1a−/− basophils. (B) Basophil numbers in Was−/−Fcer1a+/− (gray) and Was−/−Fcer1a−/− (black). (C) OVA-specific IgE and (D) IgG1 levels. (E) Serum MCPT1 in Was−/−Fcer1a+/− and Was−/−Fcer1a−/− littermates compared to OVA/alum-sensitized Fcer1a+/−or WT. Error depict as SEM. ***p<0.001; NS: not significant as determined by t-test.
Early studies in Fcer1a−/− animals demonstrated that anaphylactic responses can occur in the absence of FcεRI [25], which is mediated primarily through activation of basophils, macrophages, and neutrophils via IgG/antigen immune complexes and crosslinking of FcγRs [39]. Since OVA-specific IgG1 responses are induced in Was−/−Fcer1a+/− mice (Fig. S5), we investigated IgG-mediated anaphylaxis. Systemic OVA challenge of Was−/−Fcer1a−/− resulted in anaphylactic shock with similar survival curves as seen in Was−/−Fcer1a+/− littermates (Fig. S5) indicating that IgG-mediated cell activation can compensate for the absence of IgE signals on MC and basophils in Was−/−Fcer1a−/− mice.
Discussion
Experimental murine models provide important tools to study the pathology of allergic reactions and to develop new therapeutic intervention strategies for food allergy; a condition that is currently only managed by allergen avoidance and emergency management with epinephrine injections. In the present work, we characterized WASP deficiency as an experimental model of spontaneous IgE-mediated small intestinal food allergy. WASP deficiency in mice of either the Th2-prone BALB/c or the Th2-resistent C57BL/6 background exhibits spontaneous sensitization to orally administered antigens in the absence of systemic or intestinal adjuvants. These mice provide a valuable addition to the experimental strategies of allergy research for understanding naturally occurring disease. WASP-deficient strains will enable studies that seek to define intervention strategies for food allergy in the absence of inflammatory adjuvants, which are confounding factors of isomorphic murine models [21].
When comparing food allergy in pediatric WAS patients to patients with common food allergies in the general pediatric population, we found that sera from patients with either condition showed similarly increased serum IgE. Analysis of the IgE and IgG4 reactivity patterns showed highly comparable specificity profiles for food allergens. Both food allergic patient groups were polysensitized and IgE reactivity was frequently directed against common allergy-inducing foods [2, 11, 40]. These observations indicate that the antigenicity of food is similar in WAS and food allergic patients. Comparable to WAS patients, Was−/− mice mount strong IgE responses against soy and wheat [23]. Polysensitization is a hallmark of human food allergy that is not found in the adjuvant-based murine models that rely on mono-sensitization to a single model antigen. Food allergy in the spontaneous Was−/− model, however, involves IgE reactivity to multiple dietary antigens. Therefore, WASP-deficient mice can be utilized as a model to define mechanisms underlying polysensitization.
Was−/− mice also provide a unique opportunity to study de novo intestinal sensitization in a pre-sensitized host. Adjuvant-based experimental models are not designed to address this question. We observed that the fraction of OVA-specific IgE was as high as 80% of total IgE in food allergic mice following systemic sensitization with OVA/alum. Most food allergic patients do not present with high serum levels of antigen specific IgE. Spontaneous oral sensitization in Was−/− mice only induced OVA-specific IgE titers that constituted on average <10%, and in some mice even <1%, of total IgE. In vitro mast cell degranulation assays and in vivo antigen challenge showed, however, that minor fractions of antigen-specific IgE are sufficient to mediate adequate mast cell activation. The discrepancy between low antigen-specific serum IgE and severe anaphylactic responses is well appreciated in food allergic patients [11, 41]. These observations further demonstrate that spontaneous sensitization occurring in food allergic patients is similar in the Was−/− mouse model. In addition, results from our ImmunoCAP studies revealed that serum obtained from 4 out of 5 WAS patients showed IgE reactivity against at least one aeroallergen (data not shown). Although we have not yet studied sensitization against non-food allergens in Was−/− mice, it is conceivable that the animals will also have utility for studies that pertain to sensitization and allergic responses against aeroallergens.
WASP is expressed in all hematopoietic lineages and is functionally linked to a large variety of immune effector functions [42]. In our previous study, we have demonstrated that loss of WASP from FOXP3+ Tregs alone is sufficient for spontaneous Th2-mediated intestinal inflammation to occur [23]. However, in Was−/− mice and Wiskott-Aldrich syndrome patients, WASP is lacking from every immune compartment that is involved in the allergic response. In the complete absence of WASP from leukocytes, we nevertheless found robust type I hypersensitivity responses in Was−/− animals on the BALB/c background, which developed allergic diarrhea following allergen ingestion in comparable frequency to WT mice that were subjected to a traditional OVA/Alum experimental model. Although it is possible – and even likely – that WASP-deficiency in immune cells affects the outcome of food allergic responses in vivo, our results demonstrate that such effects do not preclude the study of type I hypersensitivity in this model. The absence of anaphylaxis after oral challenge is not surprising as it is known to be particularly difficult to model in mice and also does not occur following OVA/Alum sensitization and challenge of WT mice [21, 43, 44].
IgE-independent anaphylaxis has recently been recognized as a regulator of the severity of anaphylactic reactions in humans [45]. Interestingly, spontaneous food allergy and food anaphylaxis also develops in Was−/−Fcer1a−/− mice demonstrating that IgE/FcεRI-inflammatory signals are not required for food sensitization. It is important to note that IgE/FcεRI-independency in this model does not mean that sensitization occurred without IgE-mediated cell activation. In fact, MCPT1, a classical readout of IgE-mediated anaphylaxis [39], was surprisingly high in FcεRI-deficient animals without additional antigen challenge. This observation indicates that food-antigen specific IgE result in FcεRI-independent activation of mast cells. One possibility is that IgE activates CD16 in Fcer1a−/− mice as previously described [25]. Alternatively, but not mutually exclusively, the lack of survival signals via IgE/FcεRI might result in an unstable mast cell phenotype and more antigen- and immunoglobulin-independent MCPT1 release [8, 46]. A contribution of food-specific IgG signals as triggers of spontaneous sensitization is equally possible. In conclusion, studies on spontaneous food allergy in Was−/−Fcer1a−/− mice have the potential to provide a better understanding of IgE/FcεRI-independent mechanisms of sensitization and anaphylaxis. However, to address the question of whether spontaneous sensitization can occur in a strictly IgE-independent fashion, it will be necessary to generate IgE-deficient Was−/− mice.
In summary, we have identified Was−/− mice as an experimental tool to model spontaneous food allergy. Sensitization to food antigens occurred in Was−/− mice as well as WAS patients and is directed against allergens relevant to common food allergy in humans. In addition, polysensitization was observed in both Was−/− mice and patients with food allergy. Combined, the pathology of food allergy in Was−/− mice supports their utility as a model of allergic sensitization in humans. Studies of food allergy in WASP-deficient mice will likely reveal new insights in to the etiology of human food allergy and inspire new therapeutic approaches that interfere with spontaneous onset of this detrimental condition.
Supplementary Material
Acknowledgments
This work was supported by grant DK034854 (to SBS) from the National Institutes of Health. EF is supported by a Bridge Grant from the Research Council of Boston Children’s Hospital and a FARE New Investigator Award. This work was further supported by a senior research grant of the Crohn’s and Colitis Foundation (to EF), unrestricted gifts from the Mead Johnson Nutrition Company (to WSL and EF), and by the Harvard Digestive Diseases Center Grant P30DK034854 (Cores B and C). We thank Drs. Hans Oettgen and Oliver Burton for their suggestions and helpful advice. We also thank the Fiebiger and the Snapper laboratories for discussions and technical assistance.
Abbreviations
- BM
bone marrow derived
- BSA
bovine serum albumin
- FcεRI
high-affinity IgE Fc receptor
- Ig
immunoglobulin
- MC
mast cells
- OVA
ovalbumin
- Treg
regulatory T cell
- WAS
Wiskott-Aldrich syndrome
- WASP
Wiskott-Aldrich syndrome protein
Footnotes
PROF. ERIKA JENSEN-JAROLIM (Orcid ID : 0000-0003-4019-5765)
DR. EDDA FIEBIGER (Orcid ID : 0000-0003-3385-0100)
Authors’ contributions
WSL, JAG, KM, BFS, SBS, and EF designed experiments, performed research, and analyzed data. WSL, KM, EHHMR, EJJ, SN, and SBS were involved in patient recruitment and analysis of human data. WSL, JAG, SBS, and EF wrote the first draft of manuscript. All co-authors contributed to writing the final manuscript and approved its last version.
Conflicts of interest
The authors declare that they have no conflict of interest.
References
- 1.Kattan JD, Sicherer SH. Optimizing the diagnosis of food allergy. Immunol Allergy Clin North Am. 2015;35(1):61–76. doi: 10.1016/j.iac.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sicherer SH, Sampson HA. Food allergy: Epidemiology, pathogenesis, diagnosis, and treatment. J Allergy Clin Immunol. 2014;133(2):291–307. doi: 10.1016/j.jaci.2013.11.020. quiz 308. [DOI] [PubMed] [Google Scholar]
- 3.Berin MC. Pathogenesis of IgE-mediated food allergy. Clin Exp Allergy. 2015;45(10):1483–96. doi: 10.1111/cea.12598. [DOI] [PubMed] [Google Scholar]
- 4.Berin MC, Sampson HA. Mucosal immunology of food allergy. Curr Biol. 2013;23(9):R389–400. doi: 10.1016/j.cub.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston LK, Chien KB, Bryce PJ. The immunology of food allergy. J Immunol. 2014;192(6):2529–34. doi: 10.4049/jimmunol.1303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longo G, et al. IgE-mediated food allergy in children. Lancet. 2013;382(9905):1656–64. doi: 10.1016/S0140-6736(13)60309-8. [DOI] [PubMed] [Google Scholar]
- 7.Boden SR, Burks A Wesley. Anaphylaxis: a history with emphasis on food allergy. Immunol Rev. 2011;242(1):247–57. doi: 10.1111/j.1600-065X.2011.01028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18(5):693–704. doi: 10.1038/nm.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henson M, Burks AW. The future of food allergy therapeutics. Semin Immunopathol. 2012;34(5):703–14. doi: 10.1007/s00281-012-0319-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer RN, et al. The future of biologics: applications for food allergy. J Allergy Clin Immunol. 2015;135(2):312–23. doi: 10.1016/j.jaci.2014.12.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampson HA, et al. Food allergy: a practice parameter update-2014. J Allergy Clin Immunol. 2014;134(5):1016–25 e43. doi: 10.1016/j.jaci.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 12.Berin MC, Shreffler WG. Mechanisms Underlying Induction of Tolerance to Foods. Immunol Allergy Clin North Am. 2016;36(1):87–102. doi: 10.1016/j.iac.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Bryce PJ. Balancing Tolerance or Allergy to Food Proteins. Trends Immunol. 2016 doi: 10.1016/j.it.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noval Rivas M, Chatila TA. Regulatory T cells in allergic diseases. J Allergy Clin Immunol. 2016;138(3):639–52. doi: 10.1016/j.jaci.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8(3):205–17. doi: 10.1038/nri2273. [DOI] [PubMed] [Google Scholar]
- 16.Oettgen HC, Burton OT. IgE and Mast Cells: The Endogenous Adjuvant. Adv Immunol. 2015;127:203–56. doi: 10.1016/bs.ai.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Benede S, et al. The rise of food allergy: Environmental factors and emerging treatments. EBioMedicine. 2016;7:27–34. doi: 10.1016/j.ebiom.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pali-Scholl I, Jensen-Jarolim E. Anti-acid medication as a risk factor for food allergy. Allergy. 2011;66(4):469–77. doi: 10.1111/j.1398-9995.2010.02511.x. [DOI] [PubMed] [Google Scholar]
- 19.Plunkett CH, Nagler CR. The Influence of the Microbiome on Allergic Sensitization to Food. J Immunol. 2017;198(2):581–589. doi: 10.4049/jimmunol.1601266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wesemann DR, Nagler CR. The Microbiome, Timing, and Barrier Function in the Context of Allergic Disease. Immunity. 2016;44(4):728–38. doi: 10.1016/j.immuni.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oyoshi MK, et al. Food allergy: Insights into etiology, prevention, and treatment provided by murine models. J Allergy Clin Immunol. 2014;133(2):309–17. doi: 10.1016/j.jaci.2013.12.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berin MC, Mayer L. Immunophysiology of experimental food allergy. Mucosal Immunol. 2009;2(1):24–32. doi: 10.1038/mi.2008.72. [DOI] [PubMed] [Google Scholar]
- 23.Lexmond WS, et al. FOXP3+ Tregs require WASP to restrain Th2-mediated food allergy. J Clin Invest. 2016;126(10):4030–4044. doi: 10.1172/JCI85129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lexmond WS, et al. Accuracy of digital mRNA profiling of oesophageal biopsies as a novel diagnostic approach to eosinophilic oesophagitis. Clin Exp Allergy. 2015;45(8):1317–27. doi: 10.1111/cea.12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dombrowicz D, et al. Absence of Fc epsilonRI alpha chain results in upregulation of Fc gammaRIII-dependent mast cell degranulation and anaphylaxis. Evidence of competition between Fc epsilonRI and Fc gammaRIII for limiting amounts of FcR beta and gamma chains. J Clin Invest. 1997;99(5):915–25. doi: 10.1172/JCI119256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Platzer B, et al. Dendritic cell-bound IgE functions to restrain allergic inflammation at mucosal sites. Mucosal Immunol. 2015;8(3):516–32. doi: 10.1038/mi.2014.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandt EB, et al. Mast cells are required for experimental oral allergen-induced diarrhea. J Clin Invest. 2003;112(11):1666–77. doi: 10.1172/JCI19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burton OT, et al. Immunoglobulin E signal inhibition during allergen ingestion leads to reversal of established food allergy and induction of regulatory T cells. Immunity. 2014;41(1):141–51. doi: 10.1016/j.immuni.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jong AB, Dikkeschei LD, Brand PL. Sensitization patterns to food and inhalant allergens in childhood: a comparison of non-sensitized, monosensitized, and polysensitized children. Pediatr Allergy Immunol. 2011;22(2):166–71. doi: 10.1111/j.1399-3038.2010.00993.x. [DOI] [PubMed] [Google Scholar]
- 30.Liu AH, et al. National prevalence and risk factors for food allergy and relationship to asthma: results from the National Health and Nutrition Examination Survey 2005–2006. J Allergy Clin Immunol. 2010;126(4):798–806 e13. doi: 10.1016/j.jaci.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aalberse RC, Platts-Mills TA, Rispens T. The Developmental History of IgE and IgG4 Antibodies in Relation to Atopy, Eosinophilic Esophagitis, and the Modified TH2 Response. Curr Allergy Asthma Rep. 2016;16(6):45. doi: 10.1007/s11882-016-0621-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burton OT, et al. Oral immunotherapy induces IgG antibodies that act through FcgammaRIIb to suppress IgE-mediated hypersensitivity. J Allergy Clin Immunol. 2014 doi: 10.1016/j.jaci.2014.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sicherer SH, et al. The natural history of egg allergy in an observational cohort. J Allergy Clin Immunol. 2014;133(2):492–9. doi: 10.1016/j.jaci.2013.12.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kubota T, et al. Different stabilities of the structurally related receptors for IgE and IgG on the cell surface are determined by length of the stalk region in their alpha-chains. J Immunol. 2006;176(11):7008–14. doi: 10.4049/jimmunol.176.11.7008. [DOI] [PubMed] [Google Scholar]
- 35.Spergel JM, et al. Food Allergy in Infants With Atopic Dermatitis: Limitations of Food-Specific IgE Measurements. Pediatrics. 2015;136(6):e1530–8. doi: 10.1542/peds.2015-1444. [DOI] [PubMed] [Google Scholar]
- 36.Lemon-Mule H, et al. Pathophysiology of food-induced anaphylaxis. Curr Allergy Asthma Rep. 2008;8(3):201–8. doi: 10.1007/s11882-008-0034-6. [DOI] [PubMed] [Google Scholar]
- 37.Pivniouk VI, et al. Impaired signaling via the high-affinity IgE receptor in Wiskott-Aldrich syndrome protein-deficient mast cells. Int Immunol. 2003;15(12):1431–40. doi: 10.1093/intimm/dxg148. [DOI] [PubMed] [Google Scholar]
- 38.Chen CY, et al. Induction of Interleukin-9-Producing Mucosal Mast Cells Promotes Susceptibility to IgE-Mediated Experimental Food Allergy. Immunity. 2015;43(4):788–802. doi: 10.1016/j.immuni.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finkelman FD. Anaphylaxis: lessons from mouse models. J Allergy Clin Immunol. 2007;120(3):506–15. doi: 10.1016/j.jaci.2007.07.033. quiz 516–7. [DOI] [PubMed] [Google Scholar]
- 40.Burks AW, et al. ICON: food allergy. J Allergy Clin Immunol. 2012;129(4):906–20. doi: 10.1016/j.jaci.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Steckelbroeck S, Ballmer-Weber BK, Vieths S. Potential, pitfalls, and prospects of food allergy diagnostics with recombinant allergens or synthetic sequential epitopes. J Allergy Clin Immunol. 2008;121(6):1323–30. doi: 10.1016/j.jaci.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat Rev Immunol. 2010;10(3):182–92. doi: 10.1038/nri2724. [DOI] [PubMed] [Google Scholar]
- 43.Bryce PJ, et al. Humanized mouse model of mast cell-mediated passive cutaneous anaphylaxis and passive systemic anaphylaxis. J Allergy Clin Immunol. 2016;138(3):769–79. doi: 10.1016/j.jaci.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burton OT, et al. A humanized mouse model of anaphylactic peanut allergy. J Allergy Clin Immunol. 2016 doi: 10.1016/j.jaci.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finkelman FD, Khodoun MV, Strait R. Human IgE-independent systemic anaphylaxis. J Allergy Clin Immunol. 2016;137(6):1674–80. doi: 10.1016/j.jaci.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kashiwakura J, Otani IM, Kawakami T. Monomeric IgE and mast cell development, survival and function. Adv Exp Med Biol. 2011;716:29–46. doi: 10.1007/978-1-4419-9533-9_3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.