

Abstract
AIM
To ascertain whether cholestasis affects the expression of two CYP3A isoforms (CYP3A1 and CYP3A2) and of pregnane X receptor (PXR) and constitutive androstane receptor (CAR).
METHODS
Cholestasis was induced by bile duct ligation in 16 male Wistar rats; whereas 8 sham-operated rats were used as controls. Severity of cholestasis was assessed on histological examination of liver sections, and serum concentrations of albumin, AST, ALT, GGT, ALPK and bilirubin. Gene and protein expressions of PXR, CAR, CYP3A1 and CYP3A2 were assessed by means of qRT-PCR and Western blot, respectively. Alterations in CYP3A activity were measured by calculating the kinetic parameters of 4-OH and 1’-OH-midazolam hydroxylation, marker reactions for CYP3A enzymes.
RESULTS
The mRNA and protein expression of CYP3A1 increased significantly in mild cholestasis (P < 0.01). At variance, mRNA and protein expression of CYP3A2 didn’t change in mild cholestasis, whereas the expression and activity of both CYP3A1 and CYP3A2 decreased dramatically when cholestasis became severe. Consistently with these observations, the nuclear expression of both PXR and CAR, which was measured because they both translocate into the cell nucleus after their activation, virtually disappeared in the late stage of cholestatic injury, after an initial increase. These results indicate that early- and late-stage cholestasis affects CYP3A-mediated drug metabolism differently, probably as consequence of the different activation of PXR and CAR.
CONCLUSION
Early- and late-stage cholestasis affects CYP3A-mediated drug metabolism differently. PXR and CAR might be targeted therapeutically to promote CYP3A-mediated liver detoxification.
Keywords: Cholestasis, CYP3A, Drug metabolism, Pregnane X receptor, Constitutive androstane receptor
Core tip: We demonstrated that early- and late-stage cholestasis affects CYP3A-mediated metabolism differently, probably as consequence of the different activation of pregnane X receptor (PXR) and constitutive androstane receptor (CAR). As a consequence, cholestatic patients may have an altered drug metabolism: in the early stage due to the induction of CYP3A enzymes; and in the late stage due to the high deposition of fibrotic liver and consequent hepatocyte loss. Secondly, since PXR activation is known to induce alternative hepatic export routes and detoxification enzymes, the induction of these cellular pathways with PXR and/or CAR agonists could be exploited as a therapeutic strategy for the management of cholestatic diseases.
INTRODUCTION
Cholestasis can be defined as a bile flow impairment such that insufficient amounts of bile reach the duodenum[1]. Cholestatic liver diseases comprise a heterogeneous group of disorders resulting from bile duct injury caused by genetic defects, mechanical obstruction, toxins, or immune system dysregulation, and giving rise to bile and liver tissue damage. Primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are the two main classes of cholestatic disease, and chronic cholestasis and liver inflammation are their main pathophysiological components[2]. Common clinical manifestations and pathogenic features of cholestatic disease include the responses of cholangiocytes and hepatocytes to injury. In an early stage, the accumulation of potentially cytotoxic bile acids (BAs) in the liver can lead to hepatocellular damage; this is followed by inflammation, which causes hepatic cell death, biliary fibrosis and liver cirrhosis[3]. To escape the deleterious effects of high concentrations of BAs, the liver triggers an adaptive response to cholestasis, activating a complex network of nuclear receptors (NRs) that tightly regulate the BA transporters to maintain proper BA homeostasis. The BAs act as agonists/activators of the NRs, which are ligand-activated transcription factors regulating a broad array of key hepatic processes, such as hepatobiliary excretory function, hepatic glucose and lipid metabolism, inflammation, regeneration, fibrosis, and tumorigenesis[4]. In addition to BAs, there are other biliary constituents that can activate the NRs, such as bilirubin. The NRs include the constitutive androstane receptor (CAR), and the pregnane X receptor (PXR), which control the expression of CYP3A drug-metabolizing enzymes. PXR and CAR act as xenobiotic sensors, as one of their main functions is to regulate the expression of enzymes and transporters involved in xenobiotic elimination. These NRs are also involved in controlling hepatic processes closely related to the progression of cholestatic diseases, such as BA homeostasis, lipid metabolism, fibrosis and inflammation[5]. Changes in CAR and PXR expression have already been identified in the course of cholestatic liver disease, but different effects have been documented depending on the etiology of the cholestasis. In particular, a pronounced increase in PXR and CAR expression was documented in patients with obstructive cholestasis[5], as opposed to a moderate reduction of their expression in primary biliary cholangitis[6]. In an animal model of cholestasis, it was demonstrated that PXR has a protective effect, reducing inflammation and fibrosis[7].
There is general consensus that liver disease impairs various pathways of hepatic drug metabolism. Both in vitro and in vivo studies have shown a decrease in CYP3A activity in cholestatic liver disease, but a clear demonstration of the mechanism(s) responsible for it is still lacking[8-13]. To our knowledge, no studies published to date simultaneously analyzed CYP3A enzyme expression and activity and the activation of NRs responsible for their transcriptional control in different stages of cholestatic disease. CYP3A1 and CYP3A2 are regarded as the metabolically most important CYP3A isoforms in male rats. The former is the isoform mainly susceptible to induction, while the latter is the isoform expressed at the highest constitutive level[14,15]. CYP3A enzymes are the most abundant CYPs in human beings, and the most important enzymes in terms of drug metabolism, because they have a key role in the first-pass and systemic metabolism of many drugs[16,17].
The aim of the present study was to analyze the gene and protein expression, and the enzymatic activity of CYP3A1 and CYP3A2, as well as the nuclear expression of CAR and PXR. For this purpose, we used a validated animal model of cholestasis based on the bile duct ligation technique in animals rigorously stratified by degree of liver injury.
MATERIALS AND METHODS
Reagents
Midazolam, 4-hydroxymidazolam (4OH-MDZ), nicotinamide adenine dinucleotide 2′-phosphate (NADPH), dimethylsulfoxide (DMSO), 40% acrylamide solution, sodium dodecyl sulfate (SDS), and Tween 20 were purchased from Sigma-Aldrich Italy (Milan, Italy). 1’-hydroxymidazolam (1’OH-MDZ) was purchased from SPIBio Bertin Pharma (Montigny le Bretonneux, France). Ultrapure water was obtained with Pure-Lab Option Q apparatus (Elga Lab Water, High Wycombe, United Kingdom). Sucrose, Tris and MgCl2 were purchased from Applichem (Chicago, IL, United States). HPLC-grade methanol was purchased from Scharlau (Barcelona, Spain). Rabbit polyclonal anti-PXR, rabbit polyclonal anti-CAR, mouse anti-CYP3A1, rabbit anti-CYP3A2 and HRP-conjugated anti-mouse IgG antibodies were obtained from Abcam (Cambridge, United Kingdom). HRP-conjugated anti-rabbit IgG were obtained from Millipore (Billerica, MA, United States), and HRP-conjugated anti-goat IgG from Jackson Immuno Research (West Grove, PA, United States). Rabbit polyclonal anti-calnexin, rabbit polyclonal anti-GAPDH and goat polyclonal anti-acetyl histone H3 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, United States).
Animal care and use statement
The procedures involving the animals were managed in accordance with national and international laws and policies (Directive 2010/63/EU on the protection of animals used for scientific purposes). The study design was approved by the Ethics Committee of University of Padova, and by the Italian Ministry for the care and use of laboratory animals (Prot. no. 24, 2015). Rats were kept in standard conditions (24 °C, 12 h/12 h day/night cycle), and they have ad libitum access to food and water. Protocols involving animals were designed to minimize their pain, and each treatment was performed under isoflurane anesthesia.
Animal treatments
Cholestasis was induced in 16 male Wistar Kyoto rats (Charles River, Boston, MA, United States) by bile duct ligation and resection, as previously described[18,19]. Eight sham-operated rats were used as control animals. Eight bile-duct-ligated (BDL) animals were sacrificed two weeks after surgery to obtain a model of mild cholestasis, and the other 8 four weeks after surgery to obtain a model of severe liver injury. Blood was collected at the time of their sacrifice by cardiac puncture to perform biochemical liver function tests: alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALKP), γ-glutamyl transferase (GGT), serum albumin, total and conjugated bilirubin. After sacrificing the animals, their livers were promptly removed and weighed, and a piece was excised, fixed in 4% formalin and embedded in paraffin for histological examination. Bile acid concentration was measured on liver tissue omogenates by means of the Bile Acid Assay kit obtained from Sigma-Aldrich Italy (Milan, Italy), following manufacturer’s instructions. The remaining liver tissue was washed in a buffer containing 50 mmol/L Tris, 150 mmol/L KCl, 2 mmol/L EDTA, (pH 7.4), frozen and stored at -80 °C.
Histological examination
Two 4-micron sections were cut from the paraffin-embedded liver tissue, and stained with hematoxylin-eosin and fuchsin-picric acid (van Gieson staining), as previously described[20,21]. Images were obtained with a Leica SCN400 slide scanner. The Ishak system was adopted to quantify the extent of liver damage, scoring livers from 0 to 6 by severity of fibrosis[22].
Determination of mRNA levels by qRT-PCR
Total RNA was extracted from liver tissue after homogenization and purified with the SV Total RNA Isolation System (Promega Corporation, Madison, WI, United States). Liver tissue was homogenized in lysis buffer and mRNA was purified by means of silica-gel-based columns, according to the manufacturer’s instructions. A DNAse treatment was used during RNA extraction to prevent genomic DNA contamination. Purified RNA was eluted in a final volume of 100 μL RNAse-free water. Aliquots were stored at -80 °C until use. qRT-PCR was carried out with the commercial One Step SYBR PrimeScript RT-PCR Kit (Takara, Mountain View, CA, United States). The reaction was carried on in a volume of 10 μL as recommended by the manufacturer, using the PCR primers listed in Table 1. The qRT-PCR thermal program was run as follows: 15 min at 50 °C and 2 min at 95 °C for the reverse transcription, 40 cycles of 15 s at 95 °C and 60 s at 60 °C for the PCR reaction, and then 15 s at 95 °C, 15 s at 55 °C and 15 s at 95 °C for the dissociation curve. During the exponential phase, the fluorescence signal threshold was calculated, and the cycle threshold (Ct) was ascertained. The Ct values were used to calculate the relative mRNA expression, according to the mathematical quantification model proposed by Pfaffl[23]. All samples were tested in triplicate and β-actin was used as the housekeeping gene.
Table 1.
Primer sequences used in the qRT-PCR analysis and NCBI reference sequences
| Gene | Forward primer (5’-3’) | Reverse primer (5’-3’) | RefSeq |
| CYP3A1 | CCATCACGGACACAGAAATG | CTTTCCCCATAATCCCCACT | NM013105 |
| CYP3A2 | AGTGGGGATTATGGGGAAAG | CAATGATGGGGGAACATCTCC | NM153312 |
| PXR | CAAATCTGCCGTGTATGTGG | GTTTCATGGCCCTTCTGAAA | NM052980 |
| CAR | GAGGAAAGACATGATACTGTCAG | GTCTGGATGAGCTCTTTCTGCT | NM022941 |
| β-actin | GCCACCAGTTCGCCATGGA | TTCTGACCCATACCCACCAT | NM031144 |
Preparation of hepatic nuclear fraction
Nuclear and cytoplasmic fractions were obtained according to the method described by Dimauro et al[24] with minor modifications. Briefly, 150 mg of liver tissue was allowed to thaw in STM buffer containing 250 mmol/L sucrose, 50 mmol/L Tris-HCl pH 7.4, 5 mmol/L MgCl2 and Complete Protease Inhibitor Cocktail (Roche, Milan, Italy). The tissue was then homogenized for 1 min at 1000 rpm with an IKA T25 Ultra-Turrax disperser. The homogenate was maintained in ice for 30 min, vortexed and centrifuged at 800 g for 15 min. The pellet was resuspended in STM buffer, vortexed and then centrifuged at 500 g for 15 min. The supernatant was discarded and the pellet was washed in STM buffer, vortexed and centrifuged at 1000 g for 15 min. The resulting pellet, containing the nuclei, was resuspended in NET buffer, containing 20 mmol/L HEPES, 1.5 mmol/L MgCl2, 0.5 mol/L NaCl, 0.2 mmol/L EDTA, 20% glycerol, 1% Triton-X-100 and Complete Protease Inhibitor Cocktail. It was vortexed for 15 seconds and maintained in ice for 30 min. Then, it was sonicated twice with a Soniprep 150 MSE (London, United Kingdom) and centrifuged at 9000 g for 30 min at 4 °C. The resulting supernatant was the “nuclear fraction”.
Preparation of hepatic microsomal fraction
Microsomal fractions were obtained as reported previously by Floreani et al[21]. The pellet containing the microsomal fraction was resuspended in 250 mmol/L sucrose, aliquoted and stored at -8 °C. The protein content of the nuclear and microsomal fractions was measured with a commercially available kit (Thermo Fisher BCA Protein Assay kit, Rockford, IL, United States) using bovine serum albumin for the calibration curve.
Western blot analyses
Western blot analyses to ascertain the protein expression of CYPs and the nuclear protein expression of PXR and CAR were performed using 30 µg per lane of nuclear or microsomal fractions, as previously described[20]. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed on 8% polyacrylamide gels in reducing-denaturing conditions, and proteins were transferred to a 0.45 µm nitrocellulose membrane (BioRad, Hercules, CA, United States). Anti-PXR, anti-CAR, anti-CYP3A1 and anti-CYP3A2 primary antibodies were used to detect PXR, CAR, CYP3A1 and CYP3A2 proteins in the hepatic fractions, as previously described by De Martin et al[20]. The signal intensity of the immunoreactive bands was analyzed with the Quantity One software (Bio-Rad Laboratories, Hercules, CA, United States), and normalized to that of the acetyl-histone H3 or the calnexin bands, for the nuclear or microsomal fractions, respectively.
Measurements of 4- and 1’-midazolam hydroxylation
To analyze the formation of 4OH-MDZ and 1’OH-MDZ, microsomal preparations were incubated with increasing concentrations of midazolam (between 0.5 pmol/L and 20 pmol/L, n = 9) in a total volume of 200 µL, as previously described[20]. Ten µg of microsomal proteins obtained from sham and rats with mild cholestasis were used, and 80 µg obtained from rats with severe cholestasis. The reaction was run for 5 or 10 min at 37 °C in 0.1 mol/L KH2PO4, adding 0.5 mmol/L of NADPH, and then the reaction was stopped by adding 100 µL of cold methanol. The samples were centrifuged at 20000 g for 10 min and the supernatants were analyzed by HPLC using a Shimadzu system equipped with a UV detector. The two metabolites were separated chromatographically by means of a Kinetex EVO C18 column (5 µm, 150 mm × 4.6 mm, Phenomenex, Castel Maggiore, Italy). The mobile phase was water and acetonitrile (67:33). Isocratic elution was conducted with a constant flow of 1 mL/min for 15 min. The UV detector was set up at 220 nm, eluting 4OH-MDZ after 4.7 min, and 1'OH-MDZ after 5.2 min. The 4OH-MDZ and 1'OH-MDZ were quantified with standard calibration curves obtained with authentic 4OH-MDZ and 1'OH-MDZ at concentrations ranging from 1 to 50 nmol/mL (n = 8), processed in exactly the same way as the samples obtained from the kinetic experiments. The calibration curves were linear in this concentration range (r2 = 0.99), the lowest value in the range representing the quantification limit of the assay. Both inter- and intra-assay CVs for the 4OH-MDZ and 1'OH-MDZ determinations (n = 5) were lower than 5% at 1 nmol/mL, and lower than 3% at 50 nmol/mL. MDZ 4- and 1’-hydroxylase activity was expressed as pmol of 4OH-MDZ and 1'OH-MDZ produced per mg of protein per min.
Kinetic and statistical analyses
The F-test was used to judge the best-fitting kinetic model for 4- and 1’-hydroxylation of midazolam (hyperbolic Michaelis-Menten model or Michaelis-Menten model with substrate inhibition). Kinetic parameters were estimated by non-linear regression analysis of untransformed initial velocity data (GraphPad Prism software) using the appropriate equation. The following kinetic parameters were calculated: Vmax (maximum velocity of the reaction), and Km (substrate concentration yielding 50% of Vmax).
Statistical analyses were performed with the GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, United States). Unless specified otherwise, the data are presented as mean values ± SD. The experimental results were compared by one-way analysis of variance (ANOVA) or Student’s t-test, as appropriate. In the case of statistically significant differences (α = 0.05), the analysis of variance was followed by the Newman-Keuls post-hoc test. A P value < 0.05 was considered statistically significant.
RESULTS
Biochemical findings and histology of rat livers
The animal model of cholestasis was obtained by ligating the common bile duct[25]. The presence of cholestasis was confirmed after sacrificing the animal on the basis of plasma biochemical tests and histological examination. The latter revealed a normal liver architecture in the sham-operated animals (Figure 1A). The livers from rats sacrificed after 2 wk showed mild cholestasis (Figure 1B, Ishak score 1-3), and fibrous septa. Rats sacrificed after 4 wk showed severe cholestasis with bridging fibrosis surrounding cirrhotic nodules, and the original liver structure completely destroyed (Figure 1C, Ishak score 4-6). The degree of liver dysfunction was also assessed on the basis of serum biochemistry (albumin, AST, ALT, total and conjugated bilirubin, alkaline phosphatase and γGT), as shown in Table 2. The rats with mild cholestasis had the same serum albumin levels as the healthy control animals, whereas the levels were significantly lower in the rats with severe cholestasis (P < 0.05 vs control and mildly cholestatic rats). All other serum markers of liver injury were significantly increased in rats with severe cholestasis by comparison with either the controls or the animals with mild cholestasis. Liver bile acid concentration increased as liver function worsens (252 ± 32 nmol/g in sham-operated vs 404 ± 51 nmol/g in mildly cholestatic vs 515 ± 62 nmol/g liver tissue in severely cholestatic rats).
Figure 1.
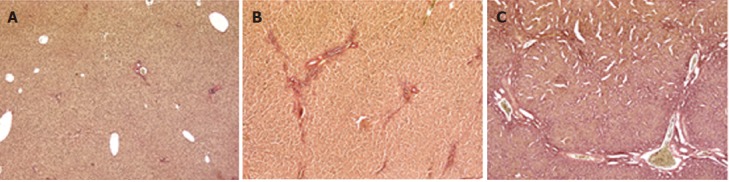
Histological analyses of rat livers. Representative photomicrographs of liver sections taken from sham-operated (A), mildly cholestatic (B) and severely cholestatic rats (C), stained with van Gieson to detect liver fibrosis (magnification × 5).
Table 2.
Biochemical liver function parameters and Ishak scores
| Sham-operated rats | Mildly cholestatic rats | Severely cholestatic rats | |
| Albumin (g/L) | 36.1 ± 3.3 | 35.8 ± 3.8 | 31.2 ± 3.7ad |
| ALT (U/L) | 43.7 ± 8.1 | 44.2 ± 8.8 | 115.3 ± 32.4cf |
| AST (U/L) | 92.2 ± 41.6 | 103.5 ± 33.0 | 402.7 ± 136.6cf |
| γGT (U/L) | 0.1 ± 0.6 | 0.5 ± 0.5 | 52.4 ± 34.8be |
| ALKP (U/L) | 133.9 ± 47.1 | 150.8 ± 38.0 | 320.1 ± 107.2cf |
| Conjugated bilirubin (mmol/L) | 0.8 ± 0.3 | 0.7 ± 0.3 | 133.1 ± 56.9cf |
| Total bilirubin (mmol/L) | 1.1 ± 0.6 | 1.0 ± 0.3 | 151.2 ± 64.1cf |
| Ishak score | 0 | 1-3 (1) | 4-6 (5) |
The results are given as mean ± SD obtained from 8 rats of each group. Ishak scores are given as range (median).
P < 0.05;
P < 0.01;
P < 0.001 vs sham-operated rats;
P < 0.05;
P < 0.01;
P < 0.001 vs mildly cholestatic rats.
mRNA and protein expression of CYP3A1 and CYP3A2
The mRNA expressions of CYP3A1 and CYP3A2 are shown in Figure 2A and B. We found CYP3A1 gene expression significantly increased in mild cholestasis (P < 0.01 vs controls), while CYP3A2 gene expression tended to increase with respect to controls, but not to any significant degree. In contrast, there was a significant drop in the gene expression of both CYP3A1 (P < 0.05 vs controls, and P < 0.001 vs rats with mild cholestasis) and CYP3A2 (P < 0.05 vs controls, and P < 0.01 vs rats with mild cholestasis) when cholestasis became severe.
Figure 2.
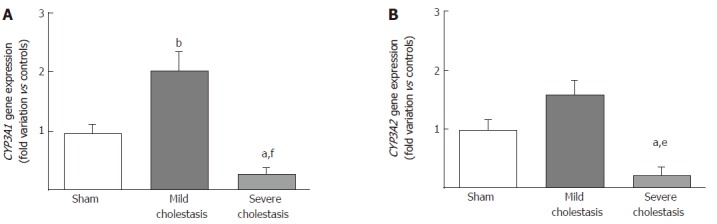
mRNA expression of CYP3A isoforms in rat livers. Gene expression of CYP3A1 (A) and CYP3A2 (B). Results are mean ± SEM, obtained from 8 rats of each group. aP < 0.05, bP < 0.01 vs sham-operated rats; eP < 0.01 and fP < 0.001 vs rats with mild cholestasis.
Figure 3 shows the protein expression of the two CYP3A isoforms (assessed on liver microsomal fractions). Like the mRNA, CYP3A1 protein expression increased significantly in mild cholestasis (P < 0.01 vs controls), then decreased significantly in severe cholestasis (P < 0.05 vs controls, and P < 0.001 vs rats with mild cholestasis). CYP3A2 protein expression only decreased when cholestasis became severe without first showing any significant increase in mild cholestasis (P < 0.05 vs controls, and P < 0.01 vs rats with mild cholestasis).
Figure 3.
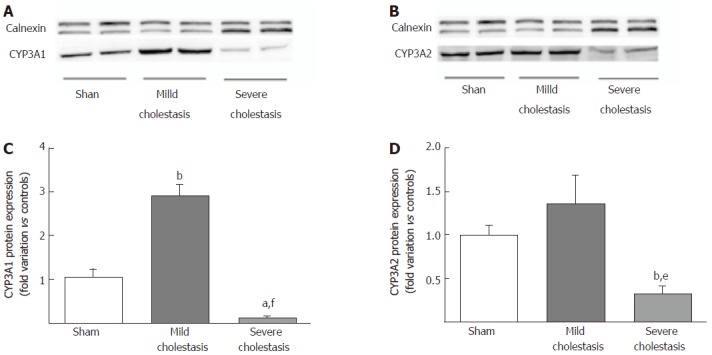
Microsomal protein expression of CYP3A isoforms in rat livers. A representative Western blot experiment showing CYP3A1 (A) and CYP3A2 (B) protein expression in microsomal fractions obtained from sham-operated and cholestatic rats. Densitometric analysis of CYP3A1 (C) and CYP3A2 (D) protein bands. Results are mean ± SEM obtained from 8 rats of each group. Each experiment was conducted in triplicate. aP < 0.05, bP < 0.01 vs sham rats; eP < 0.01, fP < 0.001 vs mildly cholestatic rats.
Kinetic analysis of 4- and 1’-hydroxylation activities
Midazolam hydroxylation in positions 1' and 4 was used as a marker reaction to assess CYP3A1 and CYP3A2 enzyme activity in the rat liver microsomal fractions[26,27]. As shown in Figure 4, both reactions followed the classical Michaelis-Menten kinetic model. Table 3 shows the kinetic parameters of the midazolam hydroxylation reactions in the three groups of rats. For both reactions, there was a slight, statistically insignificant increase in Vmax in the rats with mild cholestasis, while the Vmax of both reactions decreased in the animals with severe cholestatic injury (P < 0.001 vs controls and rats with mild cholestasis). In both hydroxylation reactions, there were no significant inter-group differences in the Km values.
Figure 4.
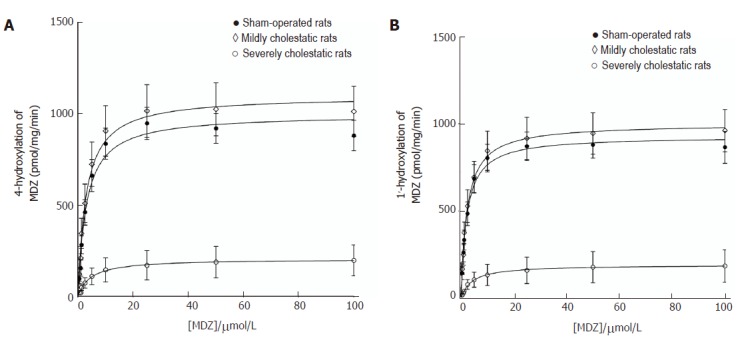
Enzymatic activity of CYP3A in microsomes obtained from sham-operated, mildly and severely cholestatic rats. Kinetics of 4- and 1’- hydroxylation activities of liver microsomes obtained from sham-operated, mildly cholestatic and severely cholestatic rats. Results are means ± SEM of data obtained from 8 rats per group. For each rat, enzymatic activity was tested in duplicate.
Table 3.
Kinetic parameters for 4- and 1’-hydroxylation activities of microsomal preparations obtained from sham-operated and cholestatic rats
|
4-OH MDZ |
1’-OH MDZ |
|||
| Vmax (pmol/mg/min) | Km (μmol/L) | Vmax (pmol/mg/min) | Km (μmol/L) | |
| Sham-operated rats | 1009.9 ± 269.4 | 3.4 ± 1.1 | 946.7 ± 292.2 | 2.5 ± 0.9 |
| Mildly cholestatic rats | 1116.0 ± 333.7 | 3.5 ± 1.4 | 1017.0 ± 317.4 | 2.8 ± 1.6 |
| Severely cholestatic rats | 218.4 ± 88.1cf | 6.1 ± 4.7 | 200.1 ± 68.7cf | 4.3 ± 2.6 |
The results are reported as mean ± SD of 8 rats per group.
P < 0.001 vs sham-operated rats;
P < 0.001 vs mildly cholestatic rats.
Expression of nuclear receptors PXR and CAR
To see whether cholestasis influences the expression of CYP3A enzymes by modifying the nuclear levels of the two NRs mainly responsible for regulating their transcription, we measured PXR and CAR mRNA and protein nuclear expression (because they both translocate into the cell nucleus after their activation). As shown in Figures 5 and 6, a significant increase in PXR mRNA and protein expression was observed in the rats with mild cholestasis (P < 0.001), whereas a significant reduction was evident in those with severe cholestasis (P < 0.05 and P < 0.001 vs controls and rats with mild cholestasis, respectively). The mRNA expression of CAR rose in mild cholestasis too (P < 0.05), and so did its nuclear protein expression, and they both dropped significantly in severe cholestasis (P < 0.05 vs mild cholestasis).
Figure 5.
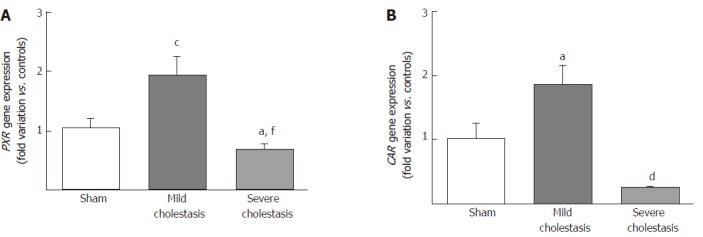
mRNA expression of nuclear receptors in rat livers. Pregnane x receptor (A) and constitutive androstane receptor (B) gene expression in sham-operated and cholestatic rats reported as fold variations compared with sham rats. Results are mean ± SEM obtained from 8 rats in each group. aP < 0.05, cP < 0.001 vs sham rats; dP < 0.05, fP < 0.01 vs mildly cholestatic rats.
Figure 6.
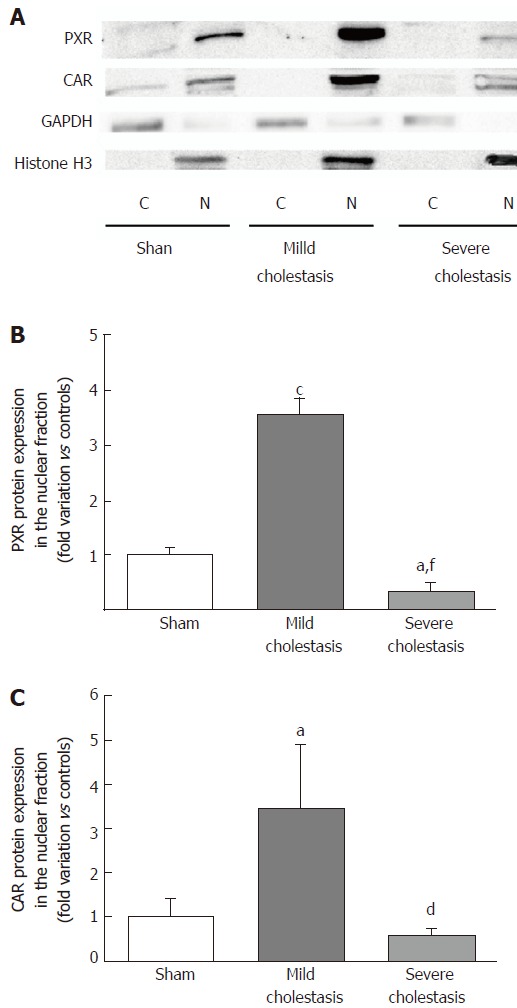
Nuclear protein expression of nuclear receptors in rat livers. Representative Western blot membrane showing pregnane x receptor and constitutive androstane receptor protein expression in the cytoplasmic and nuclear fractions of sham and cholestatic rats (A). GAPDH and Histone H3 were used to assess the purity of the cytoplasmic and nuclear fractions, respectively. Densitometric analysis of proteins in the nuclear fraction representing the PXR (B) and CAR (C) nuclear expression. Results are expressed as mean ± SEM obtained from 8 rats in each group. aP < 0.05, cP < 0.001 vs sham rats; dP < 0.05, fP < 0.001 vs mildly cholestatic rats.
DISCUSSION
In this study we investigated whether cholestasis affects the gene and protein expression, and the enzymatic activity of CYP3A1 and CYP3A2 enzymes, as well as the activation of CAR and PXR, the nuclear receptors controlling their transcription. For this purpose, we used a validated animal model of cholestasis induced by bile duct ligation, and rigorously stratified the animals on the basis of the severity of their liver injury. We found that mRNA and protein expression of CYP3A1 increased significantly in mild cholestasis (P < 0.01), whereas the expression and activity of both CYP3A1 and CYP3A2 decreased dramatically when cholestasis became severe. Alterations of the Vmax of 4-OH and 1’-OH-midazolam hydroxylation (marker reactions of CYP3A activity) were also identified. Consistently with these findings, the nuclear expression of both PXR and CAR rose initially, then virtually disappeared in the late stage of cholestatic injury.
Liver disease is known to impair various pathways of hepatic drug metabolism. Animal and clinical in vivo studies have shown that drug metabolism due to the 3A subfamily of CYP enzymes is significantly altered in severe liver disease[28,29]. In vitro studies have shown a decrease in CYP3A activity in cholestatic liver disease[30], though the mechanism(s) behind it have not been clarified.
Various studies, as reviewed in Chen et al[31] for instance, focused on the NRs controlling the expression of drug-metabolizing enzymes, analyzing their regulation of gene transcription. Since these events are crucial in the detoxification and elimination of the potentially toxic biliary constituents accumulating in cholestasis, NRs represent attractive targets of pharmacotherapy for cholestatic disorders. CAR and PXR control the expression of CYP3A, and are known to facilitate adaptation to the higher intracellular bile acid concentrations of cholestasis by upregulating alternative hepatic export routes (MRP3 and MRP4), and inducing detoxification enzymes (SULTs, UGTs and CYPs)[5]. In the present study, we examined how different degrees of cholestasis influenced the expression of CYP3A1 and CYP3A2, the main CYP3A isoforms involved in drug metabolism in the rat liver. We demonstrated that cholestasis-induced changes in their expression levels correlate with the observed changes in nuclear PXR and CAR expression, which are probably due in turn to the fact that they are activated by compounds, such as BAs, which increase in the cholestatic liver[5,7,32]. Our finding that only the mRNA and protein levels of CYP3A1, but not CYP3A2, increased significantly in mild cholestasis is consistent with the significant increase in nuclear PXR and CAR expression, and probably due to CYP3A1 being much more susceptible to induction[15]. The significant increase in CYP3A1 mRNA and protein expressions in the early stages of cholestasis prompts a slight, but statistically insignificant increase in the CYP3A-mediated metabolism of midazolam, probably because the constitutive expression of CYP3A1 in the rat liver is much lower than that of CYP3A2[20], and its protein expression is only doubled in rats with mild cholestasis. It is worth noting that a protective role of PXR had already been identified in an animal model of cholestasis, in which this NR was able to modulate inflammation and fibrosis[7]. Indeed, hepatic damage from bile acid accumulation was increased in PXR-/- mice, and, on the basis of gene expression analyses, it has been suggested that, in response to cholestasis, PXR repressed and induced the expression of the hepatic membrane transporters OATP-C and OATP2 (OATP1B1), respectively[33]. Accordingly, Xie et al[34], demonstrated that the PXR agonist PCN could not reduce lithocholic acid (LCA)-induced toxicity in PXR-/- mice. Indeed, Saini and collaborators[35] have demonstrated that CAR activation is both necessary and sufficient to confer resistance to the hepatotoxicity of LCA. Our findings support these observations, suggesting that this protective role of PXR is lost in the late stages of cholestasis due to a downregulation of its expression. This would also be the cause behind the associated decline in drug-metabolizing activity. The changes in CYP3A enzymes can be explained by bearing in mind that BAs start to accumulate in the liver right from the beginning of cholestatic disease, activating the PXR and CAR receptors. This prompts an increase in the expression of transporters and enzymes, such as CYP3A1, to promote the elimination of endogenous and exogenous substances. This compensatory mechanism is lost when liver function deteriorates because the associated reduction in NRs gives rise to a dramatic decrease in both CYP3A isoforms, with a consequent further accumulation of hepatic BAs. There is a well-known cross-talk operating between these NRs[36,37], but the molecular mechanisms behind it remain to be clarified. It has recently been observed that some NRs share the same response elements in transactivation of their target genes. This is the mechanistic molecular base for the so called “cross-talk of NRs”[38]. We know that PXR cross-talks with other NRs (in particular, CAR and FXR) to regulate intermediate metabolism through the trans-activation and trans-repression of genes controlling cholesterol, bile acid, bilirubin, glucose and lipid homeostasis[38]. FXR, CAR and PXR share many regulatory effects and their functionality in the context of regulation of liver detoxification and bile acid metabolism is largely overlapping. Studies performed in animal models of cholestasis have already shown that FXR and PXR agonists show overlapping activities[39]. On the other hand, it has been shown that FXR functions as a CAR antagonist in regulating the expression of the basolateral transporter MRP4[40]. It has also been suggested that PXR may play a role in the regulation of CYP7A1, the rate-limiting enzyme of cholesterol catabolism and bile acid formation, which is transcriptionally regulated by FXR[41].
On the basis of the results obtained in this study, we can hypothesize that PXR and CAR work synergistically to maintain CYP3A induction in the early stages of cholestasis, and their detoxification function fails when the liver dysfunction becomes severe. This finding can have two clinical consequences. For start, cholestatic patients may have an altered drug metabolism: in the early stage due to the induction of CYP3A enzymes; and in the late stage due to the high deposition of fibrotic liver and consequent hepatocyte loss. This effect could be particularly relevant in humans because a single isoform, i.e., CYP3A4, is responsible for metabolizing more than 50% of the drugs used in medical practice[42]. Secondly, since PXR activation is known to induce alternative hepatic export routes (MRP3 and MRP4) and detoxification enzymes (SULTs, UGTs and CYPs), the induction of these cellular pathways with PXR and/or CAR agonists could be exploited as a therapeutic strategy for the management of cholestatic diseases.
In conclusion, our findings clearly demonstrate that early- and late-stage cholestasis affects CYP3A-mediated drug metabolism differently. Since changes in the BA detoxification routes play a pivotal part in cholestatic liver injury, the regulation of PXR and CAR could be targeted therapeutically with the aim of ameliorating cholestatic liver injury by regulating CYP3A-mediated metabolism of BA.
ARTICLE HIGHLIGHTS
Research background
Primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are the two main classes of cholestatic disease, and chronic cholestasis and liver inflammation are their main pathophysiological components[2]. To escape the deleterious effects of high concentrations of bile acids (BAs), a peculiar feature of cholestatic disease, the liver triggers an adaptive response to cholestasis, activating a complex network of nuclear receptors (NRs) that tightly regulate the BA transporters to maintain proper BA homeostasis. Among liver NRs, both the constitutive androstane receptor (CAR) and the pregnane X receptor (PXR), which control the expression of CYP3A drug-metabolizing enzymes, play a major role in these adaptive responses. PXR and CAR act as xenobiotic sensors, as one of their main functions is to regulate the expression of enzymes and transporters involved in xenobiotic elimination. These NRs are also involved in controlling hepatic processes closely related to the progression of cholestatic diseases, such as BA homeostasis, lipid metabolism, fibrosis and inflammation[5]. Changes in CAR and PXR, and CYP3A expression have already been identified in the course of cholestatic liver disease, but different effects have been documented depending on the etiology and severity of the cholestasis. The aim of this study was to analyze expression and enzymatic activity of CYP3A1 and CYP3A2, as well as the nuclear expression of CAR and PXR in a validated animal model of cholestasis rigorously stratified on the basis of the degree of liver dysfunction.
Research motivation
Both in vitro and in vivo studies have shown a decrease in CYP3A activity in cholestatic liver disease, but a clear demonstration of the mechanism(s) responsible for it is still lacking. To our knowledge, no studies published to date simultaneously analyzed CYP3A enzyme expression and activity and the activation of NRs responsible for their transcriptional control in different stages of cholestatic disease. CYP3A enzymes are the most abundant CYPs in human beings, and the most important enzymes in terms of drug metabolism, because they have a key role in the first-pass and systemic metabolism of many drugs. On the basis of these considerations, the aim of this study was to analyze expression and enzymatic activity of CYP3A1 and CYP3A2, as well as the nuclear expression of CAR and PXR. For this purpose, we used a validated animal model of cholestasis based on the bile duct ligation technique in animals rigorously stratified by degree of liver injury.
Research objectives
In this study we investigated whether cholestasis affects the gene and protein expression, and the enzymatic activity of CYP3A1 and CYP3A2 enzymes, as well as the activation of CAR and PXR, the nuclear receptors controlling their transcription. Our results let us hypothesize that PXR and CAR work synergistically to maintain CYP3A induction in the early stages of cholestasis, and their detoxification function fails when the liver dysfunction becomes severe.
Research methods
The procedures involving the animals were managed in accordance with national and international laws and policies (Directive 2010/63/EU on the protection of animals used for scientific purposes). The study design was approved by the Ethics Committee of University of Padova, and by the Italian Ministry for the care and use of laboratory animals (Prot. no. 24, 2015). Gene and protein expressions of PXR, CAR, CYP3A1 and CYP3A2 were assessed by means of qRT-PCR and Western blot, respectively. Alterations in CYP3A activity were measured by calculating the kinetic parameters of marker reactions for CYP3A enzymes. Statistical analyses were performed with the GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, United States). The experimental results were compared by one-way analysis of variance or Student’s t-test, as appropriate. In the case of statistically significant differences (α = 0.05), the analysis of variance was followed by the Newman-Keuls post-hoc test. A P value < 0.05 was considered statistically significant.
Research results
On the basis of the results obtained in this study, we could hypothesize that PXR and CAR work synergistically to maintain CYP3A induction in the early stages of cholestasis, and their detoxification function fails when the liver dysfunction becomes severe. The mechanism by which cholestasis affects “cross-talk” between PXR, CAR and other NRs in the liver remains to be described in detail.
Research conclusions
The findings of this study can have two clinical consequences. For start, cholestatic patients may have an altered drug metabolism: in the early stage due to the induction of CYP3A enzymes; and in the late stage due to the high deposition of fibrotic liver and consequent hepatocyte loss. This effect could be particularly relevant in humans because a single isoform, i.e., CYP3A4, is responsible for metabolizing more than 50% of the drugs used in medical practice. Secondly, since PXR activation is known to induce alternative hepatic export routes (MRP3 and MRP4) and detoxification enzymes (SULTs, UGTs and CYPs), the induction of these cellular pathways with PXR and/or CAR agonists could be exploited as a therapeutic strategy for the management of cholestatic diseases.
Research perspectives
On the basis of the results obtained in this study, we hypothesized that cholestatic patients may have an altered drug metabolism and suggested the induction of liver detoxification by means of PXR and/or CAR agonists as a therapeutic strategy for the management of cholestatic diseases. To test the first hypothesis, clinical studies analyzing the pharmacokinetic parameters of cholestatic patients stratified by degree of liver injury can be performed after the administration of selected drugs. Preclinical studies analyzing the effects of the administration of PXR/CAR agonists on the cholestatic liver are currently in progress in our laboratory.
ACKNOWLEDGEMENTS
We thank Professor Pietro Palatini for kindly revising this manuscript, Mr. Mauro Berto for his skillful technical assistance, and Professor Monica Montopoli for her assistance in BA measurement.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): D
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Institutional Review board of the “Organismo Preposto al Benessere Animale (OPBA)” of the University of Padova.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Ethics Committee of University of Padova, and by the Italian Ministry for the care and use of laboratory animals [Prot. no. 24, 2015].
Conflict-of-interest statement: The authors have no conflict of interest to report.
Data sharing statement: Statistical analyses and dataset available from the corresponding author at sara.demartin@unipd.it.
Peer-review started: August 7, 2017
First decision: August 30, 2017
Article in press: October 17, 2017
P- Reviewer: Chen CJ, Hori T, Paumgartner G S- Editor: Gong ZM L- Editor: A E- Editor: Ma YJ
Contributor Information
Daniela Gabbia, Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova 35131, Italy.
Arianna Dalla Pozza, Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova 35131, Italy.
Laura Albertoni, Department of Medicine, General Pathology and Cytopathology Unit, University of Padova, Padova 35131, Italy.
Roberta Lazzari, Department of Cardiac, Thoracic, and Vascular Sciences, Hygiene and Public Health Unit, University of Padova, Padova 35131, Italy.
Giorgia Zigiotto, Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova 35131, Italy.
Maria Carrara, Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova 35131, Italy.
Vincenzo Baldo, Department of Cardiac, Thoracic, and Vascular Sciences, Hygiene and Public Health Unit, University of Padova, Padova 35131, Italy.
Tatjana Baldovin, Department of Cardiac, Thoracic, and Vascular Sciences, Hygiene and Public Health Unit, University of Padova, Padova 35131, Italy.
Annarosa Floreani, Department of Surgery, Oncology and Gastroenterology, University of Padova, Padova 35131, Italy.
Sara De Martin, Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova 35131, Italy. sara.demartin@unipd.it.
References
- 1.Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339:1217–1227. doi: 10.1056/NEJM199810223391707. [DOI] [PubMed] [Google Scholar]
- 2.Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology. 2010;139:1481–1496. doi: 10.1053/j.gastro.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin Res Hepatol Gastroenterol. 2012;36 Suppl 1:S3–S12. doi: 10.1016/S2210-7401(12)70015-3. [DOI] [PubMed] [Google Scholar]
- 4.Trauner M, Halilbasic E. Nuclear receptors as new perspective for the management of liver diseases. Gastroenterology. 2011;140:1120–1125.e1-12. doi: 10.1053/j.gastro.2011.02.044. [DOI] [PubMed] [Google Scholar]
- 5.Halilbasic E, Baghdasaryan A, Trauner M. Nuclear receptors as drug targets in cholestatic liver diseases. Clin Liver Dis. 2013;17:161–189. doi: 10.1016/j.cld.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zollner G, Wagner M, Fickert P, Silbert D, Gumhold J, Zatloukal K, Denk H, Trauner M. Expression of bile acid synthesis and detoxification enzymes and the alternative bile acid efflux pump MRP4 in patients with primary biliary cirrhosis. Liver Int. 2007;27:920–929. doi: 10.1111/j.1478-3231.2007.01506.x. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Sanchez E, Firrincieli D, Housset C, Chignard N. Nuclear Receptors in Acute and Chronic Cholestasis. Dig Dis. 2015;33:357–366. doi: 10.1159/000371688. [DOI] [PubMed] [Google Scholar]
- 8.Hrycay E, Forrest D, Liu L, Wang R, Tai J, Deo A, Ling V, Bandiera S. Hepatic bile acid metabolism and expression of cytochrome P450 and related enzymes are altered in Bsep (-/-) mice. Mol Cell Biochem. 2014;389:119–132. doi: 10.1007/s11010-013-1933-y. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Murray M, Liddle C, Jiang XM, Farrell GC. Downregulation of male-specific cytochrome P450s 2C11 and 3A2 in bile duct-ligated male rats: importance to reduced hepatic content of cytochrome P450 in cholestasis. Hepatology. 1995;22:580–587. [PubMed] [Google Scholar]
- 10.Chen J, Farrell GC. Bile acids produce a generalized reduction of the catalytic activity of cytochromes P450 and other hepatic microsomal enzymes in vitro: relevance to drug metabolism in experimental cholestasis. J Gastroenterol Hepatol. 1996;11:870–877. doi: 10.1111/j.1440-1746.1996.tb00095.x. [DOI] [PubMed] [Google Scholar]
- 11.Bastien MC, Leblond F, Pichette V, Villeneuve JP. Differential alteration of cytochrome P450 isoenzymes in two experimental models of cirrhosis. Can J Physiol Pharmacol. 2000;78:912–919. [PubMed] [Google Scholar]
- 12.Agüero RM, Favre C, Rodriguez-Garay EA. Inhibitory effect of short-term bile duct ligation on hepatic cytochrome P450 of bile acid-depleted rats. Pathobiology. 2001;69:30–35. doi: 10.1159/000048755. [DOI] [PubMed] [Google Scholar]
- 13.Fukushima S, Okuno H, Shibatani N, Nakahashi Y, Seki T, Okazaki K. Effect of biliary obstruction and internal biliary drainage on hepatic cytochrome P450 isozymes in rats. World J Gastroenterol. 2008;14:2556–2560. doi: 10.3748/wjg.14.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosal A, Satoh H, Thomas PE, Bush E, Moore D. Inhibition and kinetics of cytochrome P4503A activity in microsomes from rat, human, and cdna-expressed human cytochrome P450. Drug Metab Dispos. 1996;24:940–947. [PubMed] [Google Scholar]
- 15.Jan YH, Mishin V, Busch CM, Thomas PE. Generation of specific antibodies and their use to characterize sex differences in four rat P450 3A enzymes following vehicle and pregnenolone 16alpha-carbonitrile treatment. Arch Biochem Biophys. 2006;446:101–110. doi: 10.1016/j.abb.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Palatini P, De Martin S, Pegoraro P, Orlando R. Enzyme inhibition and induction in liver disease. Curr Clin Pharmacol. 2008;3:56–69. doi: 10.2174/157488408783329896. [DOI] [PubMed] [Google Scholar]
- 17.Palatini P, De Martin S. Pharmacokinetic drug interactions in liver disease: An update. World J Gastroenterol. 2016;22:1260–1278. doi: 10.3748/wjg.v22.i3.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarcin O, Basaranoglu M, Tahan V, Tahan G, Sücüllü I, Yilmaz N, Sood G, Snyder N, Hilman G, Celikel C, et al. Time course of collagen peak in bile duct-ligated rats. BMC Gastroenterol. 2011;11:45. doi: 10.1186/1471-230X-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verbeke L, Farre R, Trebicka J, Komuta M, Roskams T, Klein S, Elst IV, Windmolders P, Vanuytsel T, Nevens F, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59:2286–2298. doi: 10.1002/hep.26939. [DOI] [PubMed] [Google Scholar]
- 20.De Martin S, Gabbia D, Albertin G, Sfriso MM, Mescoli C, Albertoni L, Paliuri G, Bova S, Palatini P. Differential effect of liver cirrhosis on the pregnane X receptor-mediated induction of CYP3A1 and 3A2 in the rat. Drug Metab Dispos. 2014;42:1617–1626. doi: 10.1124/dmd.114.058511. [DOI] [PubMed] [Google Scholar]
- 21.Floreani M, De Martin S, Gabbia D, Barbierato M, Nassi A, Mescoli C, Orlando R, Bova S, Angeli P, Gola E, et al. Severe liver cirrhosis markedly reduces AhR-mediated induction of cytochrome P450 in rats by decreasing the transcription of target genes. PLoS One. 2013;8:e61983. doi: 10.1371/journal.pone.0061983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, Denk H, Desmet V, Korb G, MacSween RN. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22:696–699. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- 23.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dimauro I, Pearson T, Caporossi D, Jackson MJ. A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes. 2012;5:513. doi: 10.1186/1756-0500-5-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tag CG, Sauer-Lehnen S, Weiskirchen S, Borkham-Kamphorst E, Tolba RH, Tacke F, Weiskirchen R. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp. 2015:96. doi: 10.3791/52438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi K, Urashima K, Shimada N, Chiba K. Substrate specificity for rat cytochrome P450 (CYP) isoforms: screening with cDNA-expressed systems of the rat. Biochem Pharmacol. 2002;63:889–896. doi: 10.1016/s0006-2952(01)00843-7. [DOI] [PubMed] [Google Scholar]
- 27.Liu YT, Hao HP, Liu CX, Wang GJ, Xie HG. Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab Rev. 2007;39:699–721. doi: 10.1080/03602530701690374. [DOI] [PubMed] [Google Scholar]
- 28.Farrell GC, Zaluzny L. Microsomal protein synthesis and induction of cytochrome P-450 in cirrhotic rat liver. Aust J Exp Biol Med Sci. 1984;62(Pt 3):291–301. doi: 10.1038/icb.1984.29. [DOI] [PubMed] [Google Scholar]
- 29.Ene MD, Roberts CJ. Pharmacokinetics of nifedipine after oral administration in chronic liver disease. J Clin Pharmacol. 1987;27:1001–1004. doi: 10.1002/j.1552-4604.1987.tb05604.x. [DOI] [PubMed] [Google Scholar]
- 30.George J, Liddle C, Murray M, Byth K, Farrell GC. Pre-translational regulation of cytochrome P450 genes is responsible for disease-specific changes of individual P450 enzymes among patients with cirrhosis. Biochem Pharmacol. 1995;49:873–881. doi: 10.1016/0006-2952(94)00515-n. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, Tang Y, Guo C, Wang J, Boral D, Nie D. Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem Pharmacol. 2012;83:1112–1126. doi: 10.1016/j.bcp.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudraiah S, Zhang X, Wang L. Nuclear Receptors as Therapeutic Targets in Liver Disease: Are We There Yet? Annu Rev Pharmacol Toxicol. 2016;56:605–626. doi: 10.1146/annurev-pharmtox-010715-103209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stedman CA, Liddle C, Coulter SA, Sonoda J, Alvarez JG, Moore DD, Evans RM, Downes M. Nuclear receptors constitutive androstane receptor and pregnane X receptor ameliorate cholestatic liver injury. Proc Natl Acad Sci USA. 2005;102:2063–2068. doi: 10.1073/pnas.0409794102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci USA. 2001;98:3375–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saini SP, Sonoda J, Xu L, Toma D, Uppal H, Mu Y, Ren S, Moore DD, Evans RM, Xie W. A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol Pharmacol. 2004;65:292–300. doi: 10.1124/mol.65.2.292. [DOI] [PubMed] [Google Scholar]
- 36.Staudinger JL, Woody S, Sun M, Cui W. Nuclear-receptor-mediated regulation of drug- and bile-acid-transporter proteins in gut and liver. Drug Metab Rev. 2013;45:48–59. doi: 10.3109/03602532.2012.748793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oladimeji P, Cui H, Zhang C, Chen T. Regulation of PXR and CAR by protein-protein interaction and signaling crosstalk. Expert Opin Drug Metab Toxicol. 2016;12:997–1010. doi: 10.1080/17425255.2016.1201069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pavek P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front Pharmacol. 2016;7:456. doi: 10.3389/fphar.2016.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jonker JW, Liddle C, Downes M. FXR and PXR: potential therapeutic targets in cholestasis. J Steroid Biochem Mol Biol. 2012;130:147–158. doi: 10.1016/j.jsbmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Renga B, Migliorati M, Mencarelli A, Cipriani S, D’Amore C, Distrutti E, Fiorucci S. Farnesoid X receptor suppresses constitutive androstane receptor activity at the multidrug resistance protein-4 promoter. Biochim Biophys Acta. 2011;1809:157–165. doi: 10.1016/j.bbagrm.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 41.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352:2211–2221. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]