

Abstract
AIM
To investigate the diversity of bacterial lactase genes in the intestinal contents of mice with antibiotics-induced diarrhea.
METHODS
Following 2 d of adaptive feeding, 12 specific pathogen-free Kunming mice were randomly divided into the control group and model group. The mouse model of antibiotics-induced diarrhea was established by gastric perfusion with mixed antibiotics (23.33 mL·kg-1·d-1) composed of gentamicin sulfate and cephradine capsules administered for 5 days, and the control group was treated with an equal amount of sterile water. Contents of the jejunum and ileum were then collected and metagenomic DNA was extracted, after which analysis of bacterial lactase genes using operational taxonomic units (OTUs) was carried out after amplification and sequencing.
RESULTS
OTUs were 871 and 963 in the model group and control group, respectively, and 690 of these were identical. There were significant differences in Chao1 and ACE indices between the two groups (P < 0.05). Principal component analysis, principal coordination analysis and nonmetric multidimensional scaling analyses showed that OTUs distribution in the control group was relatively intensive, and differences among individuals were small, while in the model group, they were widely dispersed and more diversified. Bacterial lactase genes from the intestinal contents of the control group were related to Proteobacteria, Actinobacteria, Firmicutes and unclassified bacteria. Of these, Proteobacteria was the most abundant phylum. In contrast, the bacterial population was less diverse and abundant in the model group, as the abundance of Bradyrhizobium sp. BTAi1, Agrobacterium sp. H13-3, Acidovorax sp. KKS102, Azoarcus sp. KH32C and Aeromonas caviae was lower than that in the control group. In addition, of the known species, the control group and model group had their own unique genera, respectively.
CONCLUSION
Antibiotics reduce the diversity of bacterial lactase genes in the intestinal contents, decrease the abundance of lactase gene, change the lactase gene strains, and transform their structures.
Keywords: Antibiotics-induced diarrhea, Lactase genes, Gene diversity, Intestinal bacteria, High-throughput sequencing
Core tip: The mechanism of antibiotics-induced diarrhea has been studied in a wide range of diverse microbes, but less on functional enzymes. The current study aimed to determine the mechanism of lactase activity from genetic diversity and provide a basis for antibiotics-induced diarrhea. Alpha/Beta diversity analysis showed that there were significant differences between the control mice and model mice in types of lactase genes expressed and their activities. Following the antibiotics-induced diarrhea symptoms, the intestinal lactase genes changed, the number of strains was reduced and the abundance decreased, indicating changes in community structure and decreased diversity of lactase genes.
INTRODUCTION
Diarrhea is a common complication of antibiotic therapy and any antibiotic may disrupt the intestinal microbiota leading to diarrhea[1]. Antibiotics are usually used to treat diarrhea in children. Most studies on the impact of antibiotics on diarrhea focus on the occurrence of antibiotics-associated diarrhea (AAD)[2]. AAD can be induced by almost all antibacterial agents following the administration of mixed or individual antibiotics for several days. To date, the mechanism of AAD is unknown. However, AAD can cause severe side effects, as it is associated with damage due to intestinal micro-organisms, and disorders in intestinal function and flora[3].
Previous studies have shown that drug- or antibiotics-induced diarrhea is associated with intestinal lactase dysfunction, due to loss of its activity[4]. Antibiotics can destroy or inhibit intestinal lactase activity, leading to diarrhea. Data show that 62.5% of infantile diarrhea is related to intolerance of lactose activity[5]. Similar observations were also found in other drug-induced diarrhea, and treatment with lactase supplements is a good option for most types of diarrhea due to the importance of lactase activity for the control of intestinal function[6].
Lactase, an enzyme also known as β-D-galactosidase (EC3.2.1.23), catalyzes lactose hydrolysis to glucose or galactose[7]. Clinically, various symptoms have been associated with lactose intolerance, such as abdominal pain, abdominal distension, and diarrhea[6,8]. Individuals may be lactose intolerant to varying degrees, depending on the severity of these symptoms. Lactase is mainly produced by bacteria living in the intestinal tract of animals, for example Lactobacillus sp., Bifidobacterium sp., Bacillus sp., Escherichia coli, Streptococcus thermophilus, and Enterobacter aerogenes[9,10].
Lactase activity is tightly regulated by its expression and environment, which can alter and modify the gene expression of lactase isoforms. Various isoforms of the lactase gene have been identified and reported to be widely expressed in the intestinal tract, with diverse enzyme activities. In addition, the expression, protein modification and isoforms can change in different microenvironments[11]. Studies have shown that AAD is not only associated with dysbacteriosis, but also damage to intestinal lactase activity, leading to diarrhea[4].
In the present study, we found that the activity of lactase in intestinal contents was significantly reduced in mice with antibiotics-induced diarrhea. In order to provide a basis for the mechanism of antibiotics-induced diarrhea, we investigated whether the change in lactase activity was caused by altered gene expression. Therefore, we compared the diversity of bacterial lactase genes expressed in control mice and in model mice with antibiotics-induced diarrhea.
MATERIALS AND METHODS
Materials
Animals: Twelve mature specific pathogen-free Kunming mice (six males and six females) weighing 18-22 g were purchased from Hunan Slaccas Jingda Laboratory Animal Co., Ltd (Hunan, China) with license number SCXK (Xiang) 2013-0004.
Reagents: Chemicals were purchased from Yichang Renfu Pharmaceutical Co., Ltd. and Suzhou Zhonghua Pharmaceutical Industry Co., Ltd., including gentamicin sulfate for injection and cephradine capsules. Solutions including protease K, lysozyme, Tris saturated phenol-chloroform-isoamyl alcohol (25:24:1), TE buffer and acetone were purchased from Beijing Ding-Guo Biotechnology Co., Ltd. Other solutions, such as 0.1 mol/L phosphate buffer solution (PBS), hexadecyl trimethyl ammonium bromide (CTAB)/NaCl, 10% sodium dodecyl sulfate (SDS), 5 mol/L NaCl, chloroform-isoamyl alcohol (24:1), 3 mol/L sodium acetate and 70% ethanol, were prepared in the laboratory.
Methods
The mice were randomly allocated to the control and model groups, with six mice in each group. The mice in the model group were administered 0.35 mL antibiotic mixture at a concentration of 62.5 g/L[12], twice a day for 5 d. The mice in the control group were treated in the same way, but received distilled water. The animals were maintained under controlled conditions (23-25 °C, humidity 50%-70%). Animal surgery followed international regulations and standards.
Collection of intestinal contents
Following the development of antibiotics-induced diarrhea symptoms, the mice were sacrificed using cervical vertebra dislocation, and their intestinal contents from the jejunum and ileum were collected and immediately frozen until analysis[12].
Metagenome extraction
According to a previous report[13], 2.0 g of intestinal contents was collected in a sterile environment, placed in a 50 mL germ-free centrifuge tube and homogenized in 30 mL of 0.1 mol/L PBS, followed by centrifugation at 200 × g for 2 min. After washing twice with PBS, the supernatant was transferred into fresh germ-free tubes and centrifuged for 8 min at 10000 × g. The sediment was collected, washed once with PBS, twice with acetone, and three times with PBS, then resuspended in 4 mL TE buffer. After sample pretreatment, 500 μL of bacterial suspension was added with 5 μL proteinase K, 20 μL lysozyme and 45 μL TE buffer, and homogenized in 1.5 mL sterile Eppendorf tubes. Samples were incubated at 37 °C for 30 min and 30 μL 10% SDS was added and mixed well, followed by incubation at 37 °C for 40 min, with vortexing once every 10 min. The mixture was vortexed at 65 °C for 10 min after adding 80 μL of CTAB/NaCl and 100 μL of 5 mol/L NaCl. An equal volume of Tris saturated phenol-chloroform-isoamyl alcohol (25:24:1) was then added to the sample, mixed well and centrifuged at 10000 × g for 3 min. The supernatant was transferred to new sterile tubes, mixed with an equal volume of chloroform-isoamyl alcohol (24:1), and centrifuged at 10000 × g for 3 min. The supernatant was transferred into new sterile tubes and mixed with an equal volume of chloroform-isoamyl alcohol (24:1). The supernatant was transferred into fresh sterile tubes after centrifugation at 10000 × g for 3 min, a double volume of absolute ethyl alcohol and 1/10 volume of 3 mol/L sodium acetate were added, and precipitated at -20 °C for approximately 12 h. Samples were centrifuged at 10000 × g for 3 min. The sediment was collected and washed with 70% ethanol, dried and then dissolved in 50 μL TE buffer for DNA extraction.
PCR amplification and sequencing
To amplify the DNA, universal primers for bacterial lactase genes were designed and purchased from Shanghai Personal Biotechnology Co., Ltd. The sequence for the upstream primer was: 5′-TRRGC AACGAATACGGSTG-3′, and the downstream primer was: 5′-ACCATGAARTTSGTGGTSARCGG-3′. The PCR amplification system (25 μL) contained 11.25 μL sterilized ultrapure water, 0.25 μL Q5 high-fidelity DNA polymerase, 5 μL 5 × reaction buffer, 5 μL 5 × high GC buffer, 0.5 μL dNTP (10 mmol/L), 1 μL template DNA, 1 μL upstream primer (10 μmol/L) and 1 μL downstream primer (10 μmol/L). PCR conditions were as follows: 98 °C for 30 s, followed by 32 cycles at 98 °C for 15 s, 46 °C for 30 s (annealing) and 72 °C for 30 s (extension), and then 72 °C for 5 min[14]. The PCR products were excised from a 1.5% agarose gel and purified by AxyPred Gel Extraction Kit (Axygen, Scientific Inc., Union City, CA, United States).
Sequence screening and analysis
To identify the population of lactase genes, Qiime (v1.8.0, http://qiime.org/) software[15] was used to align the sequencing results and perform cluster analysis, principal component analysis (PCA) and ACE abundance indexing, and Simpson diversity indexing of the analysis results. The principal coordinates analysis (PCoA), nonmetric multidimensional scaling (NMDS), and heatmap were carried out in R for diversity and similarity. In addition, USEARCH (v5.2.236, http://www.drive5.com/usearch/) was used to exclude chimeric sequences. According to the software, fragments with sequence similarity over 97% were considered as one operational taxonomic unit (OTU)[16]. Evolution and abundance of bacterial lactase genes were analyzed by MEGAN (http://ab.inf.uni-tuebingen.de/software/megan5/) software[17].
Statistical analysis
SPSS21.0 software (IBM Corp, Armonk, NY, United States) was used for statistical analysis. Results are expressed as means ± SE. To compare the significance of differences, the pairwise t-test was used with P values < 0.05.
RESULTS
Comparison of operational taxonomic units
To investigate the variety and abundance of the expression of lactase genes, the OTUs were measured. We found that there were 871 and 963 OTUs expressed in mice in the model and control groups, respectively. Of these, 690 were identical in the two groups (Figure 1). These findings indicated that antibiotics-induced diarrhea reduced the expression of certain lactase genes and triggered different expression responses to symptoms.
Figure 1.
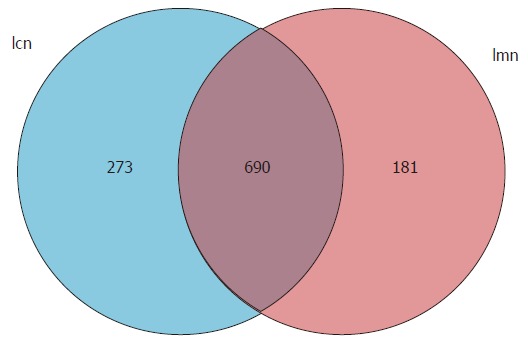
Venn diagram of operational taxonomic units based on the sequences with over 97% similarity under a similar level of clustering. lcn: Control group; lmn: Model group.
Alpha diversity analysis
Alpha diversity and abundance analysis were estimated by four indices from the Simpson or Shannon diversity analysis, and Chao1 or ACE. According to the definition, a higher value of the Chao1 or ACE index indicated greater abundance of a bacterial population. Higher Shannon and Simpson indices indicated a more diverse bacterial population. Chao1 and ACE indices in the control group were significantly higher than those in the model group (P < 0.05; Table 1). This suggested that antibiotics reduced the abundance of bacterial lactase genes in the intestinal contents. As the difference in the Simpson and Shannon indices between the two groups was small, the difference in diversity was insignificant.
Table 1.
Alpha diversity index
| Group | Chao1 | ACE | Simpson | Shannon |
| Control group | 503.00 ± 20.07 | 585.25 ± 40.84 | 0.89 ± 0.02 | 4.81 ± 0.28 |
| Model group | 446.67 ± 18.04a | 500.20 ± 5.10 a | 0.91 ± 0.02 | 4.92 ± 0.27 |
P < 0.05 vs control.
Beta diversity analysis
PCA analysis was used to measure the variation among individuals within the same group. It can be seen that the distribution was more concentrated in the control group compared with the model group, and the distance between the two groups was relatively great. The percentage contributed to variation of PC1 and PC2 was 91.3% and 8.11%, respectively (Figure 2). These findings suggest that the response to antibiotics-induced diarrhea differed among individuals, and antibiotic modeling changed the structure of the bacterial lactase genes.
Figure 2.
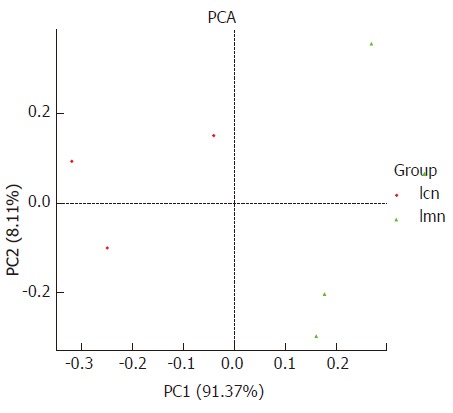
Principal component analysis diagram of bacterial lactase gene similarity at genus level based on DNA sequence data. Each point in the figure represents a sample. Points with the same color belong to the same group. The closer the distance between two points, the smaller the difference in the microbial community. lcn: Control group; lmn: Model group;PCA: principal component analysis.
To further investigate the homogenous bacterial lactase genes, PCoA was used. In particular, the distances between samples within each group sample were measured. The results showed that the distances between samples from the control group were significantly smaller than those from the model group (Figure 3). NMDS analysis was carried out to compare the similarity of lactase genes expressed within groups. Each point represented one sample, and the points with different colors were the various samples. The closer the distance between two points was, the higher the similarity between two samples, and the smaller the difference was. The distribution of samples from the control group was tightly concentrated compared to the samples from the model group (Figure 4). This indicated that the variations in lactase gene expression in response to antibiotics-induced diarrhea were significantly different from those in the control group.
Figure 3.
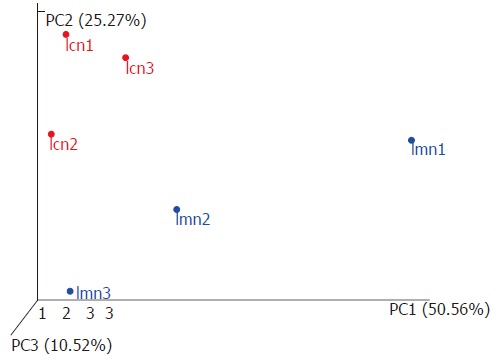
Three-dimensional sorting graph of samples based on weighted UniFrac principal coordinates analysis. Each point in the figure represents a sample. Points with the same color belong to the same group. The closer the distance between two points, the smaller the difference in the microbial community. lcn1-3 were control groups 1-3, and lmn1-3 were model groups 1-3.
Figure 4.
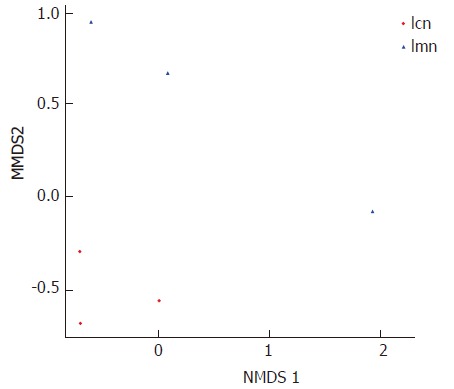
Two-dimensional map of samples based on weighted UniFrac nonmetric multidimensional scaling analysis. Each point in the figure represents a sample. Points with the same color belong to the same group. The closer the distance between two points, the smaller the difference in the microbial community. lcn: Control group; lmn:Model group.
Analysis of bacterial lactase gene source and abundance
The pie chart of each branch point of the classification tree shows the abundance of the classification unit in each sample. The larger the fan area, the higher the corresponding abundance of the taxon became. Based on the origins of lactase genes, the phylogenetic tree results showed that the majority of them were members of families, such as Actinobacteria, Firmicutes and Proteobacteria. Interestingly, some of the lactase genes were not from the above species, and there was no clear clue to show which types of bacteria they belonged to (Figure 5).
Figure 5.
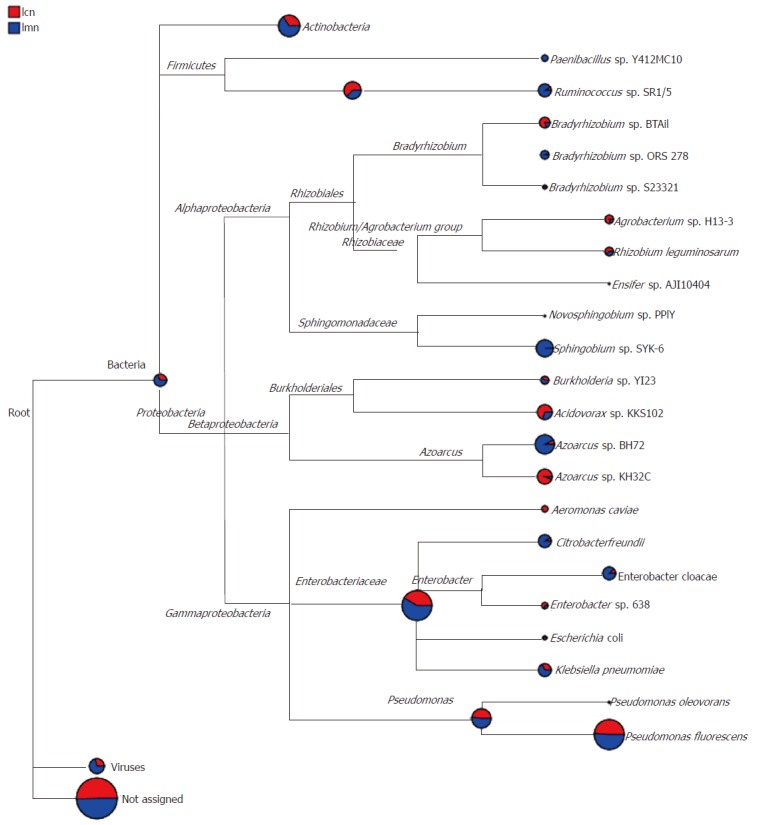
Species evolution and abundance information. The pie chart of each branch point of the classification tree shows the abundance of the classification unit in each sample. The larger the fan area, the higher the corresponding abundance of the taxon became. lcn: Control group; lmn: Model group.
The data strongly indicated that a mixture of antibiotics induced diarrhea and stimulated the growth of diverse bacteria. Some of the sensitive bacterial lactase genes were inhibited or killed, and insensitive genes were reproduced. In addition, the population sizes, such as Bradyrhizobium sp. BTAi1, Agrobacterium sp. H13-3, Acidovorax sp. KKS102, Azoarcus sp. KH32C and Aeromonas caviae, were hugely different in the intestinal contents of mice from the model group and control group.
At the genus level, the difference was more apparent. For example, Gordonia, Mycobacterium, Frankia, Microbacterium, Novosphingobium and Aeromonas were commonly seen in the intestinal contents of control mice. However, the lactase genes from these bacteria were not detectable in model mice. In contrast, Micromonospora, Paenibacillus and Ensifer were only found in the model group (Table 2). With regard to quantity, there was no significant difference in bacterial lactase genes in terms of genus in the intestinal contents of mice with antibiotics-induced diarrhea; however, the number of unclassified genes was increased (P < 0.05) and the abundance of no blast hit genes was reduced (P < 0.01; Table 2).
Table 2.
Effects of antibiotics-induced diarrhea on species abundance of bacterial lactase genes at genus level in intestinal contents
| Genus | Control group | Model group |
| Corynebacterium | 0.000942 ± 0.000365 | 0.001943 ± 0.000637 |
| Gordonia | 0.000114 ± 0.00197 | 0 |
| Mycobacterium | 0.000282 ± 0.000286 | 0 |
| Frankia | 0.000017 ± 0.000029 | 0 |
| Modestobacter | 0.000013 ± 0.000002 | 0.000098 ± 0.000155 |
| Microbacterium | 0.000005 ± 0.000009 | 0 |
| Arthrobacter | 0.000559 ± 0.000233 | 0.000658 ± 0.000308 |
| Micromonospora | 0 | 0.000016 ± 0.000017 |
| Streptomyces | 0.000081 ± 0.000031 | 0.000886 ± 0.000877 |
| Eggerthella | 0.000030 ± 0.000027 | 0.000074 ± 0.000039 |
| Paenibacillus | 0 | 0.000086 ± 0.000150 |
| Ruminococcus | 0.000039 ± 0.000055 | 0.000455 ± 0.000788 |
| Bradyrhizobium | 0.000244 ± 0.000171 | 0.000202 ± 0.000134 |
| Agrobacterium | 0.000140 ± 0.00146 | 0.000023 ± 0.000026 |
| Ensifer | 0 | 0.000026 ± 0.000045 |
| Rhizobium | 0.000100 ± 0.000173 | 0.000074 ± 0.000128 |
| Novosphingobium | 0.000011 ± 0.000020 | 0 |
| Sphingobium | 0.000019 ± 0.000033 | 0.002638 ± 0.004569 |
| Burkholderia | 0.000046 ± 0.000014 | 0.000079 ± 0.000012 |
| Acidovorax | 0.000515 ± 0.000258 | 0.000251 ± 0.000250 |
| Azoarcus | 0.001446 ± 0.002341 | 0.002971 ± 0.004730 |
| Aeromonas | 0.000075 ± 0.000130 | 0 |
| Citrobacter | 0.000051 ± 0.0000089 | 0.0005557 ± 0.000691 |
| Enterobacter | 0.000122 ± 0.000047 | 0.000380 ± 0.000303 |
| Escherichia | 0.000022 ± 0.000007 | 0.000017 ± 0.000017 |
| Klebsiella | 0.000211 ± 0.000040 | 0.000421 ± 0.000219 |
| Pseudomonas | 0.040033 ± 0.008991 | 0.045158 ± 0.031454 |
| Unclassified | 0.729202 ± 0.059025 | 0.866470 ± 0.048913a |
| No blast hit | 0.184757 ± 0.050216 | 0.024207 ± 0.004006a |
P < 0.05 vs control.
To confirm our findings, heatmap analysis provided by the R package was used, and both the diversity and abundance of bacterial species were clearly shown (Figure 6). The colors in the heatmap images are from high (red) to low abundance (blue). In addition, the images also show the distinction/differences in individuals within the same group. From the image, the lactase genes of Gordonia, Frankia, Novosphingobium, Mycobacterium, Agrobacterium, Aeromonas and Microbacterium were highly present in the contents from the model mice compared with those from the control mice. However, the difference in these bacteria was consistent from mouse to mouse in the control group.
Figure 6.
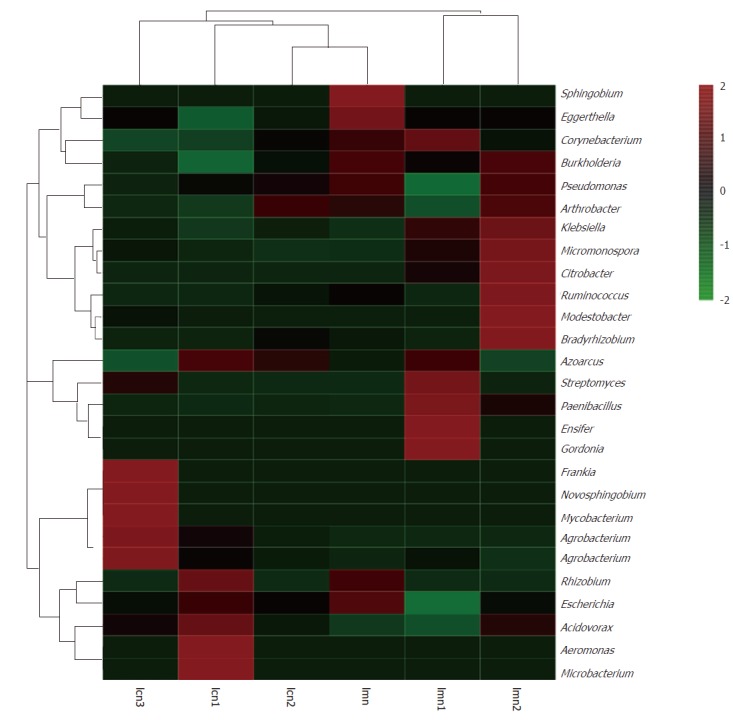
Heatmap analysis at genus level combined with cluster analysis. lcn1-3 were control groups 1-3, and lmn1-3 were model groups 1-3.
DISCUSSION
It is well known that the function or activity of a protein is closely related to its structure and modifications. Maintaining routine biological functions and stresses, such as antibiotics-induced diarrhea, requires highly regulated protein expression and its modifications. As with proteins, enzymes require the same[18-21]. In our current research, mice with diarrhea triggered by a mixture of antibiotics showed less diverse bacterial growth than control mice[22-24]. The mechanism of antibiotics-induced diarrhea has been studied in a wide range of diverse microbes[25-27], but there has been less research carried out on functional enzymes.
The current study aimed to determine the mechanism of lactase activity from the viewpoint of genetic diversity and provide a basis for antibiotics-induced diarrhea. The results showed that the number of OTUs, Chao1 index and ACE index of bacterial lactase genes in the intestinal contents were lower in mice with antibiotics-induced diarrhea than in control mice (P < 0.05). There were also significant differences between these mice shown by PCA, PCoA and NMDS analysis, which indicated changes in community structure and decreased diversity of lactase genes in antibiotics-induced diarrhea. A possible reason for this could be the diversity of bacterial species, or known species with inhibitory activity. Unknown types of lactase will contribute to the functions triggered by stresses, such as diarrhea.
Besides the unknown species living in the intestinal contents of model mice, bacteria such as Actinobacteria, Proteobacteria and Firmicutes were more abundant in these mice than in control mice. There were 79.33 ± 0.58 and 76.00 ± 4.58 different genera in the control group and model group (Table 3), respectively, which reflected the diversity of bacterial lactase genes in the intestinal contents. However, bacteria such as Gordonia, Mycobacterium, Frankia, Microbacterium, Novosphingobium and Aeromonas were exclusively detected in the control group. The differences in species and abundance between the two groups indicated that the intestinal environment in model mice treated with antibiotics may be significantly different from that in control mice.
Table 3.
Effects of antibiotic-induced diarrhea on species number of bacterial lactase genes at genus level in intestinal contents
| Species number at genus level | |
| Control group | 79.33 ± 0.58 |
| Model group | 76.00 ± 4.58 |
To maintain intestinal health in mice, diverse bacteria may be critical, and our observations also suggested this. For example, the abundance of lactase genes which originated from bacterial species Bradyrhizobium sp. BTAi1, Agrobacterium sp. H13-3, Acidovorax sp. KKS102, Azoarcus sp. KH32C and Aeromonas caviae was lower in the model group than in the control group. The same pattern was observed at the bacterial genus level. In addition to Gordonia, Frankia, Novosphingobium, Mycobacterium, Agrobacterium, Aeromonas and Microbacterium, the abundance of other lactase genes was higher in the model group. All these changes in bacteria indicated that antibiotics reduced or increased the number of certain lactase-producing strains, and thereby affected the level of intestinal lactase activity.
In summary, lactase genes are good indicators for determining the diversity of bacteria in intestinal contents, and may be used to monitor the health of mice by comparing the abundance and diversity of bacterial lactase gene expression. The results of this study show that antibiotics-induced diarrhea reduced the diversity of bacterial lactase genes in the intestinal contents and decreased lactase activity by altering the number of lactase-producing strains or reducing the number of key lactase strains, leading to diarrhea. Our current investigation provides strong support for the potential application of this strategy in the clinic.
ARTICLE HIGHLIGHTS
Research background
Studies have shown that drug- or antibiotics-induced diarrhea is associated with intestinal lactase dysfunction due to loss of activity. Thus, treatment with lactase supplements is a good option for most types of diarrhea due to the importance of lactase activity in the control of intestinal function. Various isoforms of the lactase gene have been identified and are widely expressed in the intestinal tract, with diversity enzyme activities. The expression, protein modification and isoforms can change in different micro-environments. Antibiotics-associated diarrhea is not only associated with dysbacteriosis but also intestinal lactase activity damage, leading to diarrhea. In the present study, we found that the activity of lactase in intestinal contents was significantly reduced in mice with antibiotics-induced diarrhea.
Research motivation
The mechanism of antibiotics-induced diarrhea has been studied in a wide range of diverse microbes. However, less research has been carried out on functional enzymes. In our preliminary study, we found that the activity of lactase in intestinal contents was significantly reduced in mice with antibiotics-induced diarrhea. The present study was conducted in order to determine the mechanism of lactase activity from the viewpoint of genetic diversity and provide a basis for antibiotics-induced diarrhea.
Research objectives
This study was carried out in order to provide a basis for the mechanism of antibiotics-induced diarrhea and to determine whether the alterations in activity were caused by its expression. We compared the diversity of bacterial lactase genes expressed in model mice with antibiotics-induced diarrhea and in control mice.
Research methods
Twelve mature specific pathogen-free Kunming mice were randomly allocated to the control and model groups, with six mice in each group. The mouse model of antibiotics-induced diarrhea was created by gastric perfusion with mixed antibiotics (23.33 mL·kg-1·d-1) composed of gentamicin sulfate and cephradine capsules administered for 5 days; the control group received an equal amount of sterile water. The contents of the jejunum and the ileum were then collected and metagenomic DNA was extracted, followed by analysis of bacterial lactase genes using operational taxonomic units after amplification and sequencing. Qiime software was used to align the sequencing results and carry out cluster analysis, principal component analysis (PCA), ACE abundance indexing and Simpson diversity indexing analysis. Principal coordinates analysis (PCoA), nonmetric multidimensional scaling (NMDS) and heatmap analysis were carried out in the R for diversity and similarity. SPSS21.0 software was used for statistical analysis and the results are expressed as means ± SE.
Research results
The results showed that there were significant differences in Chao1 and ACE indices between the two groups (P < 0.05). As shown by PCA, PCoA and NMDS analysis, sample distribution in the control group was relatively intensive and differences among individuals were small, while in the model group, they were dispersed and more diversified. The bacterial lactase genes in the intestinal contents from the control mice were related to Proteobacteria, Actinobacteria, Firmicutes and unclassified bacteria. Of these, Proteobacteria showed the greatest abundance. In contrast, the bacterial population was less diversified and abundant in model mice, as the abundance of Bradyrhizobium sp. BTAi1, Agrobacterium sp. H13-3, Acidovorax sp. KKS102, Azoarcus sp. KH32C and Aeromonas caviae was lower than that in the control group. In addition, of the known species, the control group and model group had their own unique genera, respectively. For example, Gordonia, Mycobacterium, Frankia, Microbacterium, Novosphingobium and Aeromonas were only seen in the control group. However, Micromonospora, Paenibacillus and Ensifer were only found in the model group. To confirm our findings, the diversity and abundance of bacterial species were clearly shown using heatmap analysis. The lactase genes of Gordonia, Frankia, Novosphingobium, Mycobacterium, Agrobacterium, Aeromonas and Microbacterium were highly present in the intestinal contents from the model group compared with the control group.
Research conclusions
Antibiotics mainly changed the number of the lactase-producing strains or reduced the number of key lactase strains. Antibiotics reduce the diversity of the intestinal bacterial flora, change the lactase gene strains, and transform their structures.
Antibiotics-induced diarrhea reduced the diversity of bacterial lactase genes in the intestinal contents and decreased lactase activity by altering the number of lactase-producing strains or reducing the number of key lactase strains.
The activity of lactase in intestinal contents was significantly reduced in mice with antibiotics-induced diarrhea. The new hypotheses that this study proposed involves how to screen and identify certain key lactase-producing strains in intestinal contents.
The bacterial lactase gene primers were designed to analyze the diversity of bacterial lactase genes in intestinal contents of mice with antibiotics-induced diarrhea by PCR, gene diversity analysis and bioinformatics techniques.
There were significant differences between control group and model group mice shown by PCR, PCoA and NMDS analysis. Lactase from different bacterial sources has different nature and activity. The diversity of lactase-producing bacteria leads to diversity of lactase genes and their activities.
Antibiotics-induced diarrhea reduced the diversity of bacterial lactase genes in the intestinal contents and decreased lactase activity by altering the number of lactase-producing strains or reducing the number of key lactase strains, leading to diarrhea.
Research perspectives
Lactase genes are good indicators for determining the diversity of bacteria in intestinal contents, and may be used to monitor the health of mice by comparing the abundance and diversity of bacterial lactase gene expression. The lactase genes will help to explore the regulation mechanism of traditional Chinese medicine on intestinal lactase activity based on the relationship between intestinal lactase diversity and antibiotics-induced diarrhea. The best methods for future research are enzyme technology and gene diversity analysis technology.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was approved by the Institutional Review Board of Hunan University of Chinese Medicine, Changsha, China.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Animal Ethics and Welfare Committee of Hunan University of Chinese Medicine, Animal License SCXK (Xiang) 2013-0004.
Conflict-of-interest statement: The authors declare no conflict of interest related to this article.
Data sharing statement: No additional data are available.
Peer-review started: July 18, 2017
First decision: August 30, 2017
Article in press: September 26, 2017
P- Reviewer: Fujino Y, Teramoto-Matsubara OT, Zouiten-Mekki L S- Editor: Gong ZM L- Editor: Filipodia E- Editor: Ma YJ
References
- 1.Hickson M. Probiotics in the prevention of antibiotic-associated diarrhoea and Clostridium difficile infection. Therap Adv Gastroenterol. 2011;4:185–197. doi: 10.1177/1756283X11399115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rogawski ET, Westreich DJ, Becker-Dreps S, Adair LS, Sandler RS, Sarkar R, Kattula D, Ward HD, Meshnick SR, Kang G. Antibiotic treatment of diarrhoea is associated with decreased time to the next diarrhoea episode among young children in Vellore, India. Int J Epidemiol. 2015;44:978–987. doi: 10.1093/ije/dyv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin ML, Ma XL, Zhou WJ. Advances in research of antibiotic-associated diarrhea. Linchuang Jianyan Zazhi. 2012;30:456–457. [Google Scholar]
- 4.Peng HZ, Ren LH. Relationship between Antibiotic Associated Diarrhea and Lactose Intolerance. Zhongguo Quanke Yixue. 2011;14:2999–3006. [Google Scholar]
- 5.Wang H, Zhang L, Wang XL, Huang T, Wang LJ. Study on the relation between infantile diarrhea and lactose intolerance. Zhongguo Weishengtaixue Zazhi. 2007;19:222–224. [Google Scholar]
- 6.Luo WX, Xie M, Gao LW. Observation on the therapeutic effect of lactase on lactose intolerance in infants with diarrhea. Haixia Yaoxue. 2016;28:153–154. [Google Scholar]
- 7.Yang HX, Du HT, Tian YM. Research progress of lactase and its gene. Anhui Nongye Kexue. 2014;42:5497–5499. [Google Scholar]
- 8.Wang ZY, Sun JQ, Xie H, Li Z. Effects of lactose supplementation on improving tolerance of lactose. Zhongguo Xiaohua Zazhi. 2010;30:632–635. [Google Scholar]
- 9.Rhimi M, Aghajari N, Jaouadi B, Juy M, Boudebbouze S, Maguin E, Haser R, Bejar S. Exploring the acidotolerance of beta-galactosidase from Lactobacillus delbrueckii subsp. bulgaricus: an attractive enzyme for lactose bioconversion. Res Microbiol. 2009;160:775–784. doi: 10.1016/j.resmic.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Juajun O, Nguyen TH, Maischberger T, Iqbal S, Haltrich D, Yamabhai M. Cloning, purification, and characterization of β-galactosidase from Bacillus licheniformis DSM 13. Appl Microbiol Biotechnol. 2011;89:645–654. doi: 10.1007/s00253-010-2862-2. [DOI] [PubMed] [Google Scholar]
- 11.Zhong WQ, Song QF, Cheng T, Li J. Research on lactase and its gene engineering. Zhongguo Rupin Gongye. 2008;36:47–50. [Google Scholar]
- 12.Zeng A, Zhang HL, Tan ZJ, Cai Y, Cai GX, Zhou SN. The Construction of mice diarrhea model due to dysbacteriosis and curative effect of ultra-micro Qiweibaizhusan. Weishengwuxue Tongbao. 2012;39:1341–1348. [Google Scholar]
- 13.Wu H, Zhou SN, Guo C, Tan ZJ, Cai GX, Zeng A, Zhang HL. A metagenome DNA extracting method of intestinal flora in mice for molecular diversity analysis based on PCR technology. Zhongguo Weishengtaixue Zazhi. 2012;24:648–651. [Google Scholar]
- 14.Long CX, He L, Liu YJ, Hui HY, Tan ZJ, Li DD. Universal primer for analysis of the diversity of intestinal bacterial lactase gene. Yingyong Yu Huanjing Shengwu Xuebao. 2017;23:758–763. [Google Scholar]
- 15.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaxter M, Mann J, Chapman T, Thomas F, Whitton C, Floyd R, Abebe E. Defining operational taxonomic units using DNA barcode data. Philos Trans R Soc Lond B Biol Sci. 2005;360:1935–1943. doi: 10.1098/rstb.2005.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan CC, Shi L. Diversity of helicobacter pylori clinical isolates by the method of polymerase chain reaction-restriction fragment length polymorphism. Xiandai Xiaohua Ji Jieru Zhiliao. 2008;13:18–21. [Google Scholar]
- 19.Tan ZY, Peng GX, Xu PZ, Ai SY, Tang SH, Zhang GX, Zeng FY. Diversity and high nitrogenase activity of endophytic diazotrophs isolated from Oryza rufipogon Griff. Kexue Tongbao. 2009;54:1885–1893. [Google Scholar]
- 20.Wang JZ, Liang JR, Qiu HY, Duan R, Xiao YC, Wang X, Jing HQ. Analysis on polymorphism of Yersinia enterocolitica urease gene and urease activity. Zhongguo Renshou Gonghuanbing Xuebao. 2014;30:140–145. [Google Scholar]
- 21.Jiang XD, Guo GG, Zhang J. Association of genetic diversity for Amy6-4 gene with α-amylase activity in germplasm of barley. Zuowu Xuebao. 2014;40:205–213. [Google Scholar]
- 22.Tan ZJ, Wu H, Liu FL, Cai Y, Cai GX, Zhang HL, Zeng A. Effect of ultra-micro powder qiweibaizhusan on the intestinal microbiota and enzyme activities in mice. Shengtai Xuebao. 2012;32:6856–6863. [Google Scholar]
- 23.Zhang HL, Cai Y, Tan ZJ, Zhou SN, Guo KX, She Y, Cai GX. Effects of ultra-micro powder Qiweibaizhusan on metabolism diversity of intestinal microflora in diarrhea mice with dysbacteriosis. Yingyong Yu Huanjing Shengwu Xuebao. 2014;20:93–100. [Google Scholar]
- 24.Guo KX, Xiao XY, Liu YJ, Long CX, Tan, ZJ Effect of Qiweibaizhusan on the intestinal lactobacilli diversity in dysbacteria diarrhea mice. Yingyong Yu Huanjing Shengwu Xuebao. 2015;21:1071–1075. [Google Scholar]
- 25.Russell SL, Gold MJ, Hartmann M, Willing BP, Thorson L, Wlodarska M, Gill N, Blanchet MR, Mohn WW, McNagny KM, et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012;13:440–447. doi: 10.1038/embor.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenwood C, Morrow AL, Lagomarcino AJ, Altaye M, Taft DH, Yu Z, Newburg DS, Ward DV, Schibler KR. Early empiric antibiotic use in preterm infants is associated with lower bacterial diversity and higher relative abundance of Enterobacter. J Pediatr. 2014;165:23–29. doi: 10.1016/j.jpeds.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu DP, Du LZ, Yu JL, Ai Q, Xiao S, Cheng C, Zhang YH, He Y, PanY, Song SJ. Effect of initial empirical antibiotic treatment on the intestinal microbiota of preterm infants. Zhongguo Xunzheng Erke Zazhi. 2016;11:26–29. [Google Scholar]