

Abstract
Mass-spectrometric analyses of Xe released from acid-treated U ore reveal that apparent Xe fission yields significantly deviate from the normal values. The anomalous Xe structure is attributed to chemically fractionated fission (CFF), previously observed only in materials experienced neutron bursts. The least retentive CFF-Xe isotopes, 136Xe and 134Xe, typically escape in 2:1 proportion. Xe retained in the sample is complimentarily depleted in these isotopes. This nucleochemical process allows understanding of unexplained Xe isotopic structures in several geophysical environments, which include well gasses, ancient anorthosite, some mantle rocks, as well as terrestrial atmosphere. CFF is likely responsible for the isotopic difference in Xe in the Earth’s and Martian atmospheres and it is capable of explaining the relationship between two major solar system Xe carriers: the Sun and phase-Q, found in meteorites.
I. INTRODUCTION
Stable xenon isotopes have high fission yields and are rare in rocks. These properties allow for accurate resolution of isotopic differences between Xe components which originated from various fissile materials, planetary atmospheres, solar wind, as well as nucleosynthetic products of s, r, and p processes in stars, while corresponding differences in other elements are barely visible. Xe isotopic signatures of various processes are presently well established and routinely used as genetic markers of terrestrial and extraterrestrial materials.
Fission fragments of heavy nuclei are neutron rich. To normalize the neutron-to-proton ratio the immediate fission fragments undergo a series of β− decays ending with a stable isobar. Isotopes 136Xe, 134Xe, 132Xe, 131Xe, and some 129Xe are such isobars and are usually called fission Xe for short. The fission chain with M = 130 ends at nearly stable 130Te, and therefore 130Xe is effectively “shielded” from fission. Light stable Xe isotopes Xe, Xe, and Xe are proton rich and are not produced in fission. These differences allow for accurate decomposition of Xe containing both fission and nonfission isotopes.
For stable fission Xe isotopes the β− -active precursors in fission chains are the isotopes of Sn, Sb, Te, and I with half-lives ranging from seconds to 1.57 × 107 years for 129I. Therefore, the formation of different Xe isotopes is delayed from the moment when actual fission has occurred, with this delay differing for each Xe isotope. In the case of the pulsed neutron-induced fission, such as for the Oklo reactors, the diffusion of Xe precursors is much faster during the active fission cycle, due to the higher temperatures, than when the reactor is dormant. This effect causes a separation of Xe isotopes having long and short lived β-active precursors and was called chemical fractionation fission (CFF; a term proposed by Frank Podosek [1], confusingly close, but unrelated to, CCF, the acronym for carbonaceous chondrite fission). Kennett and Thode [2] were among the first who systematically studied the formation of CFF-Xe released from irradiated U3O8 powder. At low extraction temperatures the apparent fission yields of 131Xe and 132Xe showed ~ 10-fold enrichments. A modified version of this experiment included the isotopic analyses of the Xe accumulated in a neutron-irradiated quartz ampoule containing U-oxide powder [3,4]. Here, Xe in the ampoule was found to be enriched in 136Xe and 134Xe, complimentarily to the low temperature Xe releases from the irradiated powder itself which was depleted in these isotopes. In a natural environment, CFF-Xe was reproducibly observed in minerals from the Oklo reactors in Gabon [5,6] which still remain the only occurrence of natural nuclear reactors. There were several previous observations of 131Xe and 132Xe enrichments, similar to those found in the Oklo ores, but they were either not reproducible [7] or tentatively attributed to neutron capture reactions on Te [8,9].
Xenon carried by nanodiamond separated from primitive meteorites is anomalously enriched in both heavy and light isotopes, which is reflected in the acronym Xe-HL. To explain the isotopic compositions of Xe-HL Ott [10] and Richter et al. [11] suggested a neutron burst intermediate between s− and r processes which modified the products of a regular r process by the separation of stable Xe isotopes and their radioactive precursors on short time scales after supernova explosions. Clear manifestations of the CFF process was observed at sites of nuclear tests which had been simultaneously subjected to neutron bursts and temperature shocks [12–14]. Previously, CFF was considered to be a rather exotic process observed mainly in the environments with pulsed neutron flux or short neutron bursts. However, the experiment reported here demonstrates that CFF process can modify apparent fission yields not only in the reactors and at nuclear test sites but also in conventional U-bearing ores where the relatively low neutron flux is nearly constant over time and the temperature changes are slow.
II. EXPERIMENT
The sample used in this work was a fragment of uranium rich ore from Streltsovka caldera in Trans-Baikalia (STR-Nk), the world largest volcanogenic uranium field near the Russia-China border and Russia’s largest uranium resource (~280,000 tons, [15]). It represents a typical mineralogy of the Streltsovka U ores, an assemblage of various uranium minerals (pitchblende, brannerite, coffinite) closely overgrown with silicates, forming a U-Si metagel as the result of secondary alteration of coffinite. The mineralogy, geochemistry, and geology of U-ore veins from Streltsovka have been extensively studied [[16], and references therein]. The U-Pb age of the primary uranium mineralization was determined to be 133 ± 4Ma. Five Streltsovka samples analyzed prior to this study gave Xe retention ages from 44 to 118 Ma, indicating variable Xe losses which are typical for these U minerals [17]. All Xe analyses were performed on bulk samples and therefore represent a mixture of xenon released from different minerals. In contrast to previous studies, in this work the sample STR-Nk has been subjected to chemical treatments prior to Xe analyses in order to study distribution of fission Xe isotopes in the acid resistant mineral phases.
The sample was crushed and sieved through a 200-mesh stainless steel screen allowing all particles less than 74 μm to pass through. Then the powder was treated with 6M HNO3 at room temperature. Most of the visible reaction lasted for only about 10 min, and after 3 h the unsolved residue was separated from the solution, washed, dried, and weighed. Approximately 80 wt% of the original sample was dissolved by this treatment (we cannot exclude losses of the finer grains during the washing). The treated sample STR-Nk2 was then loaded into the extraction system and kept under vacuum at < 10−8 Torr for 2 weeks at 200 °C prior to Xe analyses. This rather severe processing was done to remove atmospheric contamination which was expected to be significant due to the large surface area and perhaps fresh active surfaces formed by the acid treatment [18].
The samples were subjected to stepwise pyrolysis. Chemically active gases (O, N, CO2, etc.) were adsorbed by freshly deposited Ti films and St 707 getter alloy from SAES. The remaining noble gases were cryogenically separated on activated charcoal at −90 °C. He, Ne, Ar, and partially Kr were pumped away and Xe was analyzed in the magnetic sector mass spectrometer. This built-in-house instrument has a high transmission electron impact ion source with cylindrical symmetry of electron optics and does not require an electron focusing magnet [19,20]. It was successfully used for analyses of stable Xe isotopes produced by weak decay of 128,130Te [21,22], 130,132Ba [23], and strong nuclear interactions [6]. Since then the NP10 SAES getter pump has been installed in close proximity to the ion source in order to adsorb H+ and H2+, the most significant residual peaks in the mass spectrum. This improvement decreased the contribution of protonated Xe ions interfering with 130,131,132Xe peaks and, to some extent, reduced of the space charge effects which make sensitivity pressure dependent. With ion transmission exceeding 90% and Xe sensitivity of 2 mA/Torr (~5 times higher than modern commercial noble gas mass-spectrometers) at the same 200-μA electron emission, our instrument was better suited for accurate analyses of fission Xe from mg-size samples of Streltsovka U ore than commercial Noblesse and Helix MC+ also operating in our lab.
The two lightest and least abundant isotopes, 124Xe and 126Xe, were not measured in order to improve the counting statistics of the fissiogenic isotopes which were the main interest of this study. The measured isotopic compositions were corrected for any small atmospheric contaminations by using the observed 130Xe, an isotope shielded from fission. Within experimental errors no residual 128Xe (another fission-shielded isotope) was observed, indicating that any atmospheric Xe present was not isotopically fractionated and that all of it had been effectively removed.
III. RESULTS AND ANALYSIS
A. Xenon release profiles
The concentrations and isotopic compositions of fission Xe before and after acid treatment, corrected for small amounts of atmospheric contributions, are shown in Table I. The concentration of fission Xe in the treated sample (STR-Nk2) decreased threefold compared to the original sample STR-Nk1, suggesting that the U-bearing mineral phases are more susceptible to acid etching than silicates. The temperature release profiles of both of these samples have one broad peak with maximum release slightly above 1000 °C. The width of the peak may be the result of low temperature resolution and could possibly have been improved by finer steps. However, another possible reason for the broad Xe releases is the superposition of several release profiles corresponding to the different uranium oxides. Natural U oxides are complex mixtures of UO2, U3O8, and UO3 (generally, UOx, where x varies from 2.16 to 2.96). Diffusion parameters of Xe in these oxides differ [3]. The maximum Xe release from the acid treated sample STR-Nk2 occurs ~ 100 °C higher than that from the untreated SRT-Nk1 (Fig. 1), confirming previously observed preferential leaching of the less refractory U minerals [16].
TABLE I.
Concentrations and isotopic compositions of fission Xe released from untreated U-rich sample from Streltsovka ore field and from the powdered sample treated with 6M HNO3 for 3 h. Data are corrected for small instrumental mass discrimination and atmospheric contamination. The composition of Xe lost during the treatment is calculated from the total compositions. Stated errors for are 1σ. The uncertainties of Xe concentrations are ~8%, based on the reproducibility of standards over the course of analyses.
| T,°C | 136Xe, 10−9 cm3 STP/g | 134Xe /136Xe | 132Xe /136Xe | 131Xe /136Xe | 129Xe /136Xe |
|---|---|---|---|---|---|
| STR-Nk1 - original sample from Streltsovka U ore | |||||
| 600 | 0.06 | 0.913 ± 0.049 | 0.54 ± 0.11 | 0.180 ± 0.086 | 0.025 ± 0.020 |
| 820 | 0.53 | 0.887 ± 0.012 | 0.582 ± 0.017 | 0.166 ± 0.012 | 0.018 ± 0.013 |
| 930 | 0.89 | 0.914 ± 0.010 | 0.597 ± 0.009 | 0.163 ± 0.004 | 0.019 ± 0.003 |
| 1090 | 5.57 | 0.912 ± 0.008 | 0.595 ± 0.006 | 0.161 ± 0.003 | 0.018 ± 0.002 |
| 1310 | 2.95 | 0.906 ± 0.010 | 0.593 ± 0.007 | 0.175 ± 0.002 | 0.018 ± 0.002 |
| 1750 | 0.50 | 0.906 ± 0.011 | 0.598 ± 0.017 | 0.159 ± 0.011 | 0.019 ± 0.010 |
| Total | 10.5 | 0.909 ± 0.004 | 0.594 ± 0.004 | 0.165 ± 0.002 | 0.018 ± 0.001 |
| STR-Nk2 - sample treated with 6M HNO3 for 3 h | |||||
| 450 | 0.05 | 1.1 ± 0.2 | 0.86 ± 0.07 | 0.345 ± 0.08 | 0.66 ± 0.20 |
| 600 | 0.09 | 1.16 ± 0.09 | 0.825 ± 0.04 | 0.272 ± 0.05 | 0.59 ± 0.11 |
| 700 | 0.11 | 1.05 ± 0.08 | 0.725 ± 0.055 | 0.244 ± 0.04 | 0.25 ± 0.09 |
| 800 | 0.04 | 0.8 ± 0.2 | 0.587 ± 0.078 | 0.17 ± 0.06 | 0.14 ± 0.06 |
| 900 | 0.24 | 0.902 ± 0.007 | 0.595 ± 0.009 | 0.164 ± 0.005 | 0.021 ± 0.005 |
| 1000 | 0.65 | 0.915 ± 0.006 | 0.603 ± 0.009 | 0.159 ± 0.004 | 0.017 ± 0.003 |
| 1150 | 1.62 | 0.911 ± 0.004 | 0.595 ± 0.009 | 0.162 ± 0.003 | 0.019 ± 0.003 |
| 1400 | 0.88 | 0.915 ± 0.008 | 0.596 ± 0.010 | 0.155 ± 0.003 | 0.018 ± 0.003 |
| 1750 | 0.06 | 0.91 ± 0.02 | 0.59 ± 0.02 | 0.148 ± 0.009 | 0.016 ± 0.008 |
| Total | 3.74 | 0.923 ± 0.005 | 0.609 ± 0.005 | 0.167 ± 0.003 | 0.049 ± 0.005 |
| Xenon lost during etching | |||||
| 6.76 | 0.901 ± 0.007 | 0.586 ± 0.007 | 0.164 ± 0.004 | 0.001 ± 0.003 | |
| Reference fission compositions [4] | |||||
| 238U spontaneous | 0.825 ± 0.006 | 0.579 ± 0.009 | 0.082 ± 0.003 | 0.002 ± 0.001 | |
| 235U neutron induced | 1.241 ± 0.013 | 0.672 ± 0.002 | 0.411 ± 0.001 | 0.105 ± 0.003 | |
FIG. 1.
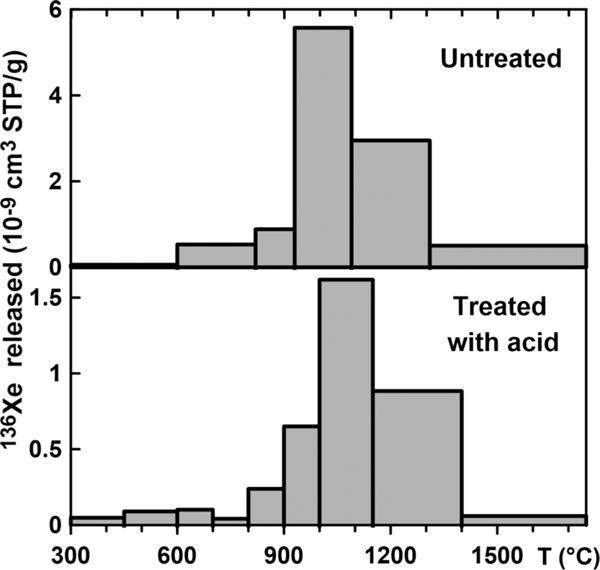
Release of radiogenic 136Xe from the original STR-Nk1 sample (top) and from the sample STR-Nk2 treated with HNO3 for 3 h (bottom). About 2/3 of fission 136Xehas been lost as a result of the treatment and the low-temperature Xe release peak became apparent in the leached STR-Nk2.
A less prominent, but more intriguing, difference in the release profiles of STR-Nk1 and STR-Nk2 is observed below 800 °C. These low temperature releases of fission Xe, unresolvable in untreated samples, become clearly visible after the acid treatment (Fig. 1). Since low-temperature gas releases are often associated with surface residence, it is possible that some of the fission Xe, initially formed within the minerals, is brought close to surfaces by crushing and leaching. Regardless of the origin of the low-temperature peaks, they are clearly not due to atmospheric Xe introduced during the sample treatment since all data presented in Fig. 1 have been corrected for minor atmospheric contributions using the 130Xe, shielded from fission.
B. Isotopic compositions of fission Xe
Fission Xe in U ores is typically a binary mixture of two major components: the products of spontaneous fission of 238U and that of neutron-induced fission of 235U. The amount of 238U-derived Xe depends only on the U content and the Xe retention age, while Xe from 235U is a function of the U content and the dose of thermal neutrons received. The neutron dose is a function of the U content, the concentration of neutron moderators, absorbers, and the geometry of the U vein. In U-bearing minerals the contribution of neutron-induced fission varies from few percentages in minerals rich in REEs (for example, monazite) to up to 40% in massive hydrothermal U veins (for the Oklo natural reactor this content is clearly much higher). In our samples from Streltsovka U ores the contribution of neutron-induced 235U fission is in the middle of this range, ~20%. This is typical not only for the Streltsovka U fields but also for many other U deposits worldwide [3].
Xe isotopes produced in spontaneous and neutron-induced fission are inseparable and indistinguishable during stepwise pyrolysis since the energy ~200 MeV released in both 235U and 238U fission is sufficient to penetrate any lattice, making similar initial fission fragment distributions. Indeed, in our original sample STR-Nk1 the isotopic composition of Xe in all temperature steps is constant within experimental uncertainties (Table I).
Fission Xe released from the treated sample STR-Nk2 also has uniform isotopic composition in all temperature steps above 800 °C. However, below 800 °C the Xe compositions are clearly anomalous (Fig. 2). All three experimental points 450°, 600°, and 700 °C lie well off of the mixing line between 238U and 235U fission, and these very same temperature steps form the distinct low-temperature Xe release peak for acid-treated sample shown in Fig. 1. How could an acid treatment cause these isotopic effects in chemically inert noble gas like Xe?
FIG. 2.
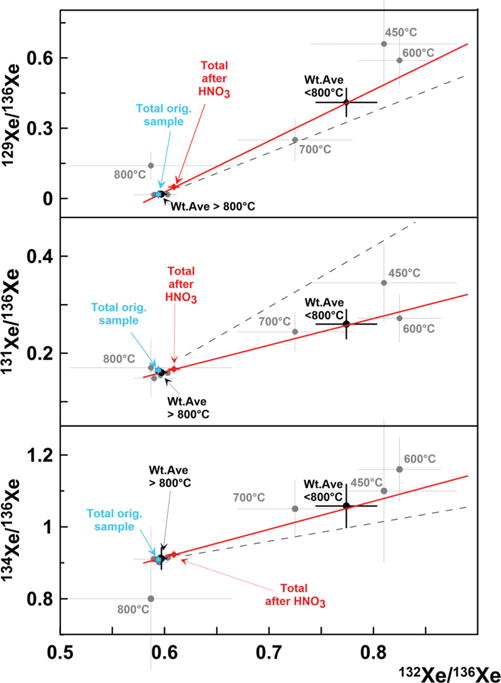
Isotopic composition of Xe released from acid treated sample STR-Nk2 during stepwise pyrolysis. All temperature fractions tend to form linear arrays (solid lines) which differ from mass-fractionation lines (dashed). Stars show the compositions of Xe from spontaneous fission of 238U and neutron-induced fission of 235U. Composition of Xe lost due to the acid treatment is calculated from the difference between the original and treated samples.
Both the low- and high-temperature points depart from the 238U −235U mixing line but in different directions (Fig. 2). However, the data point representing all temperature fractions combined together (“Total” in Table I) lies almost exactly on the 238U −235U mixing line. Experimental points from both STR-Nk1 and STR-Nk2 tend to form a linear array which differs from a mass-dependent fractionation line. This suggests that the isotopically anomalous Xe released at the low temperatures is not produced by mass fractionation but by the redistribution of fission Xe isotopes between different mineral phases with different thermal properties and/or surface areas.
The isotopic spectrum of Xe plotted in Fig. 3 represents the excesses of different isotopes in the low-temperature gas extractions from the acid-treated sample STR-Nk2: δM = 100%[(MXe /136Xe)lowT/(MXe /136Xe)total − 1)]. These isotopic excesses are traditionally normalized to 136Xe since it has the highest yield in fission. The relative isotopic excesses vary from ~15% for the 134Xe/136Xe ratio to ~740% for 129Xe /136Xe. The 132Xe /136Xe and 131Xe /136Xe ratios have intermediate excesses of ~27% and ~55%, respectively.
FIG. 3.
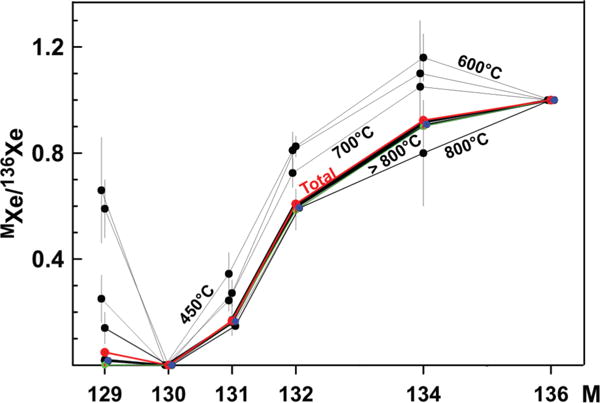
Composition of fission Xe released from the acid-treated sample STR-Nk2 at different extraction temperatures. These compositions are normalized to 136Xe. Errors are 1σ.
From the Q values of the radioiodine isotopes we can estimate maximum recoil energies of Xe from momentum conservation, but the actual values will be somewhat lower since some energy is carried away by the neutrinos. However, even the maximum recoil energies are less than 1 eV, too small to produce any significant recoil displacement of Xe within the lattice or induce significant breaking of chemical bonds. The β-active precursor remains kinetically in place and the only reasonable explanation for the observed CFF effect is chemically driven diffusion of the fission-produced radioiodine and possibly other radioactive precursors of Xe in the fission chains. Note, in Fig. 4, that there is general correlation between relative isotopic excesses (as defined above) with the half-lives of the different iodine precursor isotopes. Apparently, the longer an iodine isotope lives before decay, the greater distance it can diffuse. When the iodine decays, the resulting Xe isotope will reproduce the spatial distribution of its precursor since any recoil effect is negligible. Note here that no CFF would occur if iodine and Xe diffused identically and ended up in identical sites. However, CFF effects are clearly observed here so the properties of the different isobaric chains must clearly differ. These differences could be due to iodine which, unlike (normal) Xe, can have significant chemical activity and is presumably the more mobile of the two (chemically enhanced diffusion). It could also be due to chemical bonds which are more easily formed by the more reactive iodine precursor. These chemical bonds then could be inherited by a stable Xe end member, in the manner described by Hohenberg et al. [18].
FIG. 4.
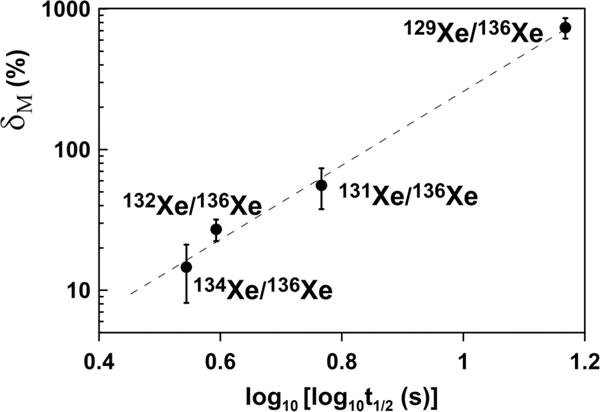
Weighted average of Xe relative (to 136Xe) isotopic excesses δM = 100% × [(MXe /136Xe)lowT/(MXe /136Xe)total − 1)] at low (<800 °C) extraction temperatures vs. half-lives of corresponding iodine isotopes, Xe precursors in fission chains. The correlation spans over 13 orders of magnitude in half-lives. Errors are 1σ.
Among all of the radioiodine isotopes produced in the spontaneous fission of 238U, 136I (1.39m), and its short-lived isobaric precursors, have the highest yield, with decay of 136I producing approximately half of the stable 136Xe. The other half is generated by direct yield of 136Xe, the highest direct yield of any Xe isotope. Thus, quite a bit of 136Xe appears almost instantaneously with fission and Xe fragment remains near the end of a fission damage track where the crystalline structure has experienced the most disruption. This could be one key to CFF because 136Xe is the least retained fission-produced Xe isotope in the acid residue. It remains in its resting place only as long as the fission track remains intact. However, as amply demonstrated in fission-track dating, acid treatment preferentially attacks the damaged regions of the fission track, liberating any Xe isotope that remains in this zone. Since Xe and its radioactive precursors are equally large atoms, the observation of CFF must be related to chemical differences.
In the fission chain A = 134 the 42-min 134Te has the highest yield. It decays to 134I (53 min). The direct yield of 134Xe is at least two orders of magnitude less than the yield of its precursors, 134I and 134Te. Therefore, the formation of stable 134Xe from decay along the isobaric chain is delayed for about an hour after the fission. During this time the more chemically active atoms of the 134Te and 134I, precursors of 134Xe, may have a greater chance to react with the environment than the precursor of 136Xe. Therefore, 134Xe may reside farther from the damaged zone and may have implicit chemical bonds and thus have a better chance than 136Xe to survive acid treatment. This observed trend continues as we go to the iodine isotopes with longer half-lives: several hours for 132I, ~8 days for 131I, and gigantic 16 Myr for 129I. Regardless of the specific details, we have observed that CFF is in play here in a manner that seems to be consistent with enhanced chemical mobility of reactive precursors.
Our experiment has demonstrated that the CFF process can be at work in conventional terrestrial rocks with uniform neutron flux. As shown in Fig. 4, it also demonstrates an apparent correlation between the magnitude of CFF at a given isotope and the iodine precursor half-life. Diffusion of intermediate fission products apparently changes the final distribution of Xe isotopes. The least retentive 136Xe and 134Xe escape first, making the remaining fission Xe complimentarily enriched in 132Xe, 131Xe, and 129Xe. This remaining Xe is released at low extraction temperatures, perhaps indicating association with near surface residences, and probably associated with the smallest grains. This implies that CFF process works only on a small scale and must be averaged on a large scale, in the bulk rock. Indeed, in the untreated sample STR-Nk1 we do not observe the CFF effects (Table I and Fig. 2).
Both aqueous alteration and weathering reduce effective grain sizes increasing the surface areas and exposing radiation-damaged zones. In this case, partial fission xenon losses could be accompanied with isotopic modification caused by the CFF effects. Finally, it should be stressed that CFF is not yet another Xe component with a unique isotopic composition but is a physiochemical process modifying nominal fission yields with continuously changing isotopic distributions due to these effects.
C. Observations of CFF-Xe in terrestrial rocks and gases
The CFF process in the natural environment should be observable in U-bearing rocks that have experienced some alterations which expose locations where hot atoms of Te and I migrated and later decayed to stable Xe. Using accelerator mass spectrometry Fabryka-Martin et al. [24] found 10-fold excess of 129I in ground water from the flow system of Stripa granite in Sweden. The authors attributed the observed excess of 129I to “subsurface production by spontaneous fission of 238U in microfractures in the granite matrix and subsequent transfer of this 129I by diffusion.” Fission iodine is highly mobile in the environment [25], especially when natural organic matter is present [26]. This iodine will eventually decay into 129Xe which will be partially dissolved in ground water and partially released into the atmosphere. In any case, it is obviously not related to primordial 129Xe.
There are several reproducibly observed occurrences of CFF-Xe enriched in 129–132Xe. The characteristic signatures of CFF-Xe were found in CO2 well gas from Harding County, New Mexico [27–30] (Fig. 5). Since there were many well gases with nearly normal Xe composition, it was thought that the Harding County gas is somewhat unique, specific to this particular location. However, Xe isotopic signature in natural gases from Gujarat (India) (Fig. 5) was found to be almost identical to that from Harding County [31].
FIG. 5.
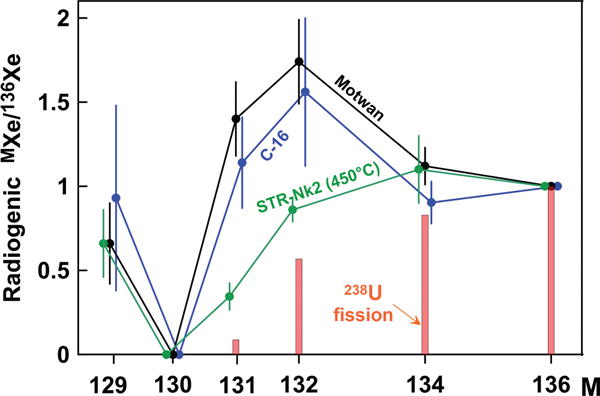
Composition of fission Xe in CO2 well gases from C-16, New Mexico [27], Motwan, Gujarat/India [31], and the lowest temperature extraction from the acid-treated sample STR-Nk2 (this work). Bars show fission yields from spontaneous fission of 238U. All compositions are normalized to 136Xe. Errors are 1σ.
129–132Xe enrichments resembling CFF-Xe are observed not only in natural gases but also in MORB glasses, diamonds, and some continental rocks. These observations were summarized in Fig. 7 from Ref. [5] and we will not discuss them here. The most extreme manifestation of the CFF process was observed in fines from Archean anorthosite from the Fiskenaesset Complex in West Greenland [7]. Analyses of coarse mineral separates from the very same anorthosite block [32] showed normal Xe corresponding to 238U fission spectrum (Fig. 6). This confirms the critical role of the effective grain size: The CFF process changes the apparent fission yields in fine-grained materials and is unlikely to be observable in whole, course-grained rocks.
FIG. 7.
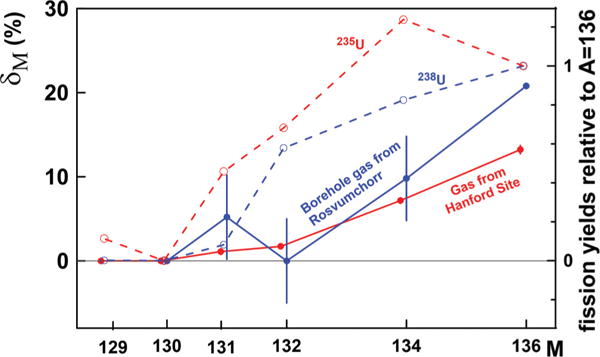
Isotope excess δM = 100%[(MXe /136Xe)sample/ (MXe /136Xe)atm. − 1)] in Xe released from boreholes in Rosvumchorr tunnel, Kola Peninsula [34], and in gas bubbles in groundwater from soil containing highly radioactive transuranic waste at DOE’s Hanford site [35] (solid lines). Fission yields of Xe isotopes from 235U and 238U (normalized to 136Xe) are shown for comparison (dashed lines). In both cases the 136Xe excess is twice as large as the 134Xe excess.
FIG. 6.
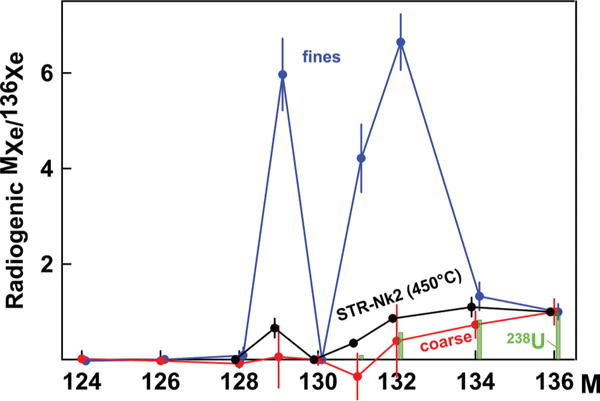
Composition of fission Xe in Archean anorthosite from Fiskenaesset Complex in West Greenland. Xe in the coarse-grained fraction [32] is indistinguishable from of fission Xe from 238U (green bars). Xe from the fine-grained fraction of the same anorthosite block [7] has an anomalous pattern similar to the low-temperature Xe extraction of the acid-treated sample STR-Nk2 (this work). Errors are 1σ.
What is rarely observed is the complimentary depletion in 129–132Xe isotopes, which should look like enrichment in 134Xe and 136Xe. As discussed in Sec. IIIC, these two heaviest fission Xe isotopes are the least retentive when subjected to the CFF process [33]. The most likely places to observe 134Xe and 136Xe enrichments are U-bearing rocks or materials which are currently subjected to aqueous alteration or weathering—the two likely processes which expose radiation-damaged zones where 134Xe and 136Xe are preferentially located. We found two examples where these conditions are apparently met. The first one is a CO2 gas from shallow boreholes in the tunnel Rosvumchorr, Kola Peninsula [34]. In spite of large error bars, the enrichment in 136Xe and 134Xe is evident (Fig. 7). The second example is Xe in gaseous bubbles dissolved in water from soil containing highly radioactive human-made transuranic waste at the DOE’s Hanford Site in southwest Washington State [35]. Both gases releasing from these very different objects have similar Xe structure: They are enriched in 136Xe and 134Xe, with the former being approximately twice higher than the latter (Fig. 7). Both isotopic patterns significantly differ from neutron-induced fission of 235U and spontaneous fission of 238U. Fission Xe remaining in these objects should be complimentary depleted in these isotopes.
IV. DISCUSSION
This section is devoted to several geo- and astrophysical implications of the CFF effect we experimentally observed in the laboratory. We will revisit previously published Xe analyses which do not have satisfactory explanations and show that the CFF effect can potentially provide these explanations. In our discussion here three parent nuclei are principal for Xe isotopic composition: (1) 238U (t1/2 = 4468 Ma), (2) 244Pu (t1/2 = 82Ma), and (3) 129I (t1/2 = 16Ma). The first two produce 136−131Xe (and almost no 129Xe) by spontaneous fission, while the 129I β decay produces 129Xe. The last two nuclei are presently extinct, but Xe produced by their decay is still observable, providing the basis for “xenology,” which utilizes Xe isotopes as genetic markers of nuclear processes in the distant past.
A. Modelling of early terrestrial evolution
Preferential losses of the least retentive CFF 136Xe and 134Xe may produce an apparent correlation between excesses of remaining 129Xe, 131Xe, and 132Xe. This is clearly visible in Fig. 2 and also suggested by Fig. 5, 6, and 8. Correlation between excesses of 129Xe and 136Xe in mantle samples is usually considered a mixture between products of extinct 129I and 244Pu. This correlation provides a foundation for modeling of the early terrestrial evolution.
FIG. 8.
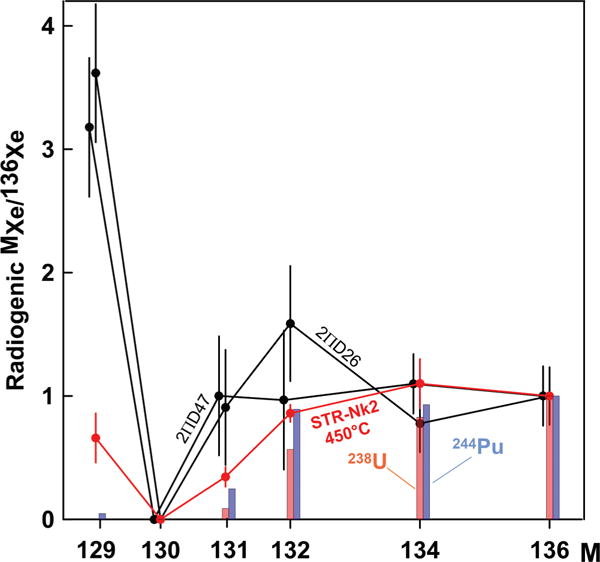
Radiogenic Xe in precisely measured midocean ridge basalt glasses 2ΠD47 and 2ΠD26 [36] are not consistent with fission spectrum of 244Pu (blue bars). This inconsistency is likely due to a CFF process which modified a 244Pu fission yield. Similar modification is observed in low-temperature Xe extraction from acid-treated sample STR-Nk2 (this work) which is due to CFF modified yields of 238U (red bars). Errors are 1σ.
Kunz et al. [36] claimed that 244Pu-derived fission Xe has been identified in the Earth’s mantle. However, as noted by Marty and Mathew [37], “their data interpretation is clearly biased, first because the trapped Xe is assumed to be a primitive (244Pu fission-free) atmospheric component and, second their data are not compatible with their inferred two-component mixture.” Indeed, Fig. 8 shows that at least two of the most precisely measured Xe compositions of midocean ridge basalt glasses are not consistent with 244Pu and/or 238U. These compositions are closer to CFF-Xe with characteristic enrichment in 129–132Xe isotopes implying that CFF process could be at work here. It also suggests that not all 129Xe in mantle derived sample may be originated from decay of primordial 129I.
B. Origin of fission Xe in the terrestrial atmosphere
Terrestrial atmospheric Xe contains only small percentages of fission products from 238U and/or extinct 244Pu. Takaoka [38] used two-dimensional (2D) correlation analysis for carbonaceous chondrules, lunar soils, and achondrites and found that in order to fit the terrestrial Xe to the correlation lines it is necessary to add some fission Xe to the atmosphere. The isotopic structure of this fission Xe was estimated to be 136Xe /134Xe /132Xe /131Xe = 1/0.9/1.68/1.12 close to 244Pu for 134Xe /136Xe, but very different for 131Xe /136Xe and 132Xe /136Xe. This unusual structure was thought to be due to fission of a superheavy element (SHE; Ref. [39]). But after the SHE hypothesis was abandoned, the isotopic structure of the Takaoka’s fission Xe was dismissed and never considered seriously. Pepin and Phinney [40] assumed that 244Pu is solely responsible for fission Xe in terrestrial atmosphere. The presence of extinct 244Pu in the Earth interior is now supported by Xe analyses of ancient zircons [41] and some mantle derived samples [42]; however, the presence of 244Pu-derived fission Xe in the atmosphere remains a reasonable, but still unconfirmed, assumption.
Igarashi [43] applied principal component analysis to the expanded database of meteorite Xe and found that terrestrial fission Xe is neither from 244Pu nor from 238U. The structure of the Igarashi’s fission Xe (136Xe /134Xe/132Xe/131Xe = 1/1.15/1.51/0.79) turned out to be very close to Takaoka’s Xe. This is remarkable considering these compositions were obtained 23 years apart using different mathematical treatment and different databases. Figure 9 shows both compositions as the isotopic shifts from 244Pu fission Xe as well as CFF-Xe releases below 800 °C from the acid treated sample STR-Nk2. In spite of large error bars, the similarity of apparent isotopic shifts shown in Fig. 9 suggest that fission Xe in a terrestrial atmosphere is likely the 244Pu fission Xe affected by the CFF process.
FIG. 9.
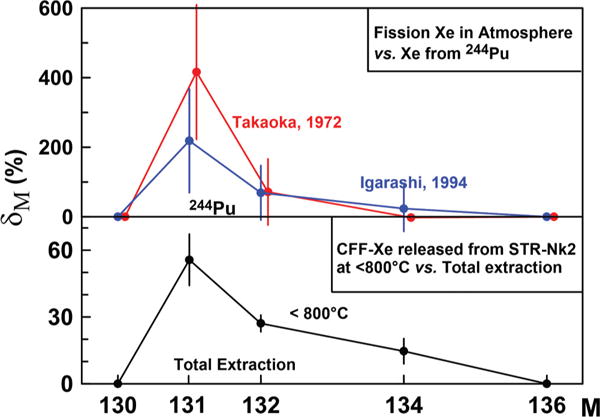
Xe isotopic excesses in terrestrial atmosphere derived by Takaoka [38], Igarashi [43] δM = 100%[(MXe /136Xe)/(MXe / 136Xe)ref, − 1)] (top) and CFF-Xe released at low temperature from acid-treated sample STR-Nk2 (bottom, this work). Terrestrial fission Xe is plotted relative to fission Xe from 244Pu, and CFF-Xe from STR-Nk2 is shown relative to the total Xe composition in this sample. The similarity of the isotopic patterns suggests that terrestrial fission Xe is also formed by the CFF process.
C. Fission Xe in the Mars and Earth atmospheres
In contrast to the Earth, the Martian atmosphere apparently does not contain measurable amount of fission Xe; its isotopic composition is well described by mass-fractionated solar Xe with the addition of 129Xe from extinct primordial 129I (t½ = 16 Ma) [44]. The 138% excess of 129Xe in the Martian atmosphere (Fig. 10) seemingly suggests that the terrestrial atmosphere is “younger” than the Martian atmosphere, supporting significant loss of the early Earth atmosphere. According to planetary sciences, this loss was caused by a catastrophic event that stripped the Earth of its primary atmosphere and ejected a large amount of terrestrial rocks which formed our moon (giant impact theory). The lost primary atmosphere was likely enriched in 136Xe and 134Xe due to CFF modification of 244Pu fission, similar to the Xe lost during the acid treatment of the sample STR-Nk2. After the primary atmosphere was lost along with the least-retentive CFF 136Xe and 134Xe, the remaining Xe became complimentary depleted in these isotopes (Fig. 10). The Earth was and is warmer than Mars. It is therefore plausible that a carrier of 244Pu-derived Xe was partially degassed on the Earth but still remains in the solid Mars. In a certain sense, Mars resembles our initial sample STR-Nk1, while the Earth is similar to our acid-treated sample STR-Nk2.
FIG. 10.
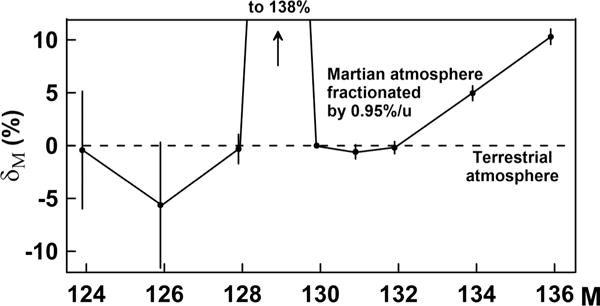
Fractionated Martian atmosphere [44] relative to the Earth’s atmosphere. δM = 100%[(MXe /136Xe)Mars/(MXe / 136Xe)Earth. − 1)].
The study of Xe in planetary atmospheres is beyond the scope of this paper. This section is only to demonstrate that CFF is seemingly capable of explaining the isotopic differences of Xe in the atmospheres of Earth and Mars. If our hypothesis is correct, then the starting Xe compositions for both Earth and Mars were similar and close to solar. It also means that that there is no need for a hypothetical progenitor of terrestrial Xe (U-Xe [40]).
D. Xenon composition in phase Q
Phase Q (Q is for quintessence) is a carbonaceous substance in primitive meteorites which contains most of the heavy noble gases [45]. Phase Q is the second-largest reservoir of Xe after the Sun. It is isolated by chemical dissolution that results in 98% loss of the meteorite mass. The bulk of Xe survives this chemical attack and remains in the acid residue. Further treatment with HNO3 causes little loss of mass but essentially complete loss of Xe. This Xe, called Xe-Q [46], has the isotopic structure which is close to mass-fractionated solar Xe with a small variable admixture of Xe-HL, a “presolar” component apparently produced by p and s and r processes in supernovae and associated with diamond-rich separates from primitive meteorites [47]. Modeling of Xe-Q composition suggested that it can be made of solar wind Xe fractionated by 8.2‰/u with addition of 1.6% of Xe-HL and 0.1% of Xe-S produced by the astrophysical s process [48]. This association of solar and “presolar” components in phase Q does not have a satisfactory explanation. The CFF process (if it was at work during the formation of phase Q) opens a possibility to explain isotopic structure of Xe-Q. Figure 11 shows isotopic excesses δM in Xe-Q relative to solar wind Xe. These excesses are remarkably similar to those found in Xe from gas bubbles releasing from the DOE Hanford site and also from the borehole gas in the Rosvumchorr tunnel (Fig. 7). Having a completely different nature and origin, these objects may have one thing in common—they selectively captured the least retentive CFF-Xe isotopes: from 235U in the Hanford site, from 238U in the tunnel, and from 244Pu in phase Q. Therefore, we can model the composition of Xe-Q as 6.5‰/u fractionated solar wind with 3% (for 136Xe) addition of CFF-Xe from 244Pu. The fractionation of solar Xe required by this model is significantly smaller than in the case where the presolar components are considered, which is comforting. This interpretation also implies that phase Q is likely of “local” rather than “presolar” origin.
FIG. 11.
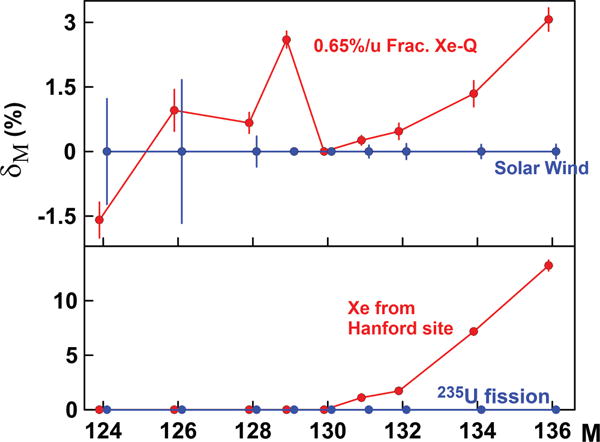
Xe-Q can be modeled as fractionated by 6.5‰ /u solar Xe [48] with ~3% addition of 136Xe, ~1.5% of 134Xe, and <0.5% of 132Xe and 131Xe (top). The isotopic structure of the required for this model addition matches the pattern of Xe found in the gas bubbles released from the Hanford site [35] (bottom). This model does not require any presolar Xe components.
V. CONCLUSION
Chemical treatment of common U-bearing ore, originally containing well-understood canonical fission products, results in Xe with the anomalous isotopic composition, clearly observable at low extraction temperatures. The magnitude of this effect is correlated with half-lives of radioiodine, a water-soluble Xe precursor in fission chains. The only reasonable explanation for this effect is CFF.
CFF-Xe is not a Xe component with fixed isotopic composition. It is a process which modifies apparent fission yields in open systems. CFF-Xe escaping the system typically consists of ~½ of 136Xe and ~¼ of 134Xe with the rest being shared among 132Xe, 131Xe, and 129Xe. CFF-Xe remaining in the system is complimentary and usually is observed at low extraction temperatures.
The isotope structure of fission Xe released from the acid-treated U-ore sample in our laboratory experiment is very close to anomalous and not-well-understood Xe composition reported in well gasses, ancient anorthosite, some mantle derived rocks, and inferred fission Xe composition in the terrestrial atmosphere. The CFF process can potentially provide a natural explanation for Xe isotopic anomalies in these objects and establish a simple relationship between two major Xe reservoirs in the solar system: the Sun and Phase Q, an enigmatic carbonaceous substance containing essentially all Xe in primitive meteorites.
The CFF concept provides us with certain flexibility in modelling of Xe isotopic composition and it may help to establish genetic links between major reservoirs of heavy noble gases.
Acknowledgments
The sample STR-Nk used in this study was kindly provided by Dr. A. A. Nikitin, Russian Academy of Sciences. This work is supported by NASA Grants No. NNX13AD14G and No. NNX14A124G.
References
- 1.FA Podosek (private communication).
- 2.Kennett TJ, Thode HG. Can J Phys. 1960;38:945. [Google Scholar]
- 3.Meshik AP. PhD thesis. Vernadsky Institute; 1988. [Google Scholar]
- 4.Shukolyukov Yu A, Jessberger EK, Meshik AP, Vu Minh D, Jordan JL. Geochim Cosmochim Acta. 1994;58:3075. [Google Scholar]
- 5.Meshik AP, Kehm K, Hohenberg CM. Geochim Cosmochim Acta. 2000;64:1651. [Google Scholar]
- 6.Meshik AP, Hohenberg CM, Pravdivtseva OV. Phys Rev Lett. 2004;93:182302. doi: 10.1103/PhysRevLett.93.182302. [DOI] [PubMed] [Google Scholar]
- 7.Jeffery PM. Nature. 1971;233:260. doi: 10.1038/233260a0. [DOI] [PubMed] [Google Scholar]
- 8.Drescher J, Kirsten T, Schäfer K. Earth Planet Sci Lett. 1998;154:247. [Google Scholar]
- 9.Pinti DL, Matsuda J, Maruyama Sh. Chem Geol. 2001;175:387. [Google Scholar]
- 10.Ott U. Astrophys J. 1996;463:344. [Google Scholar]
- 11.Richter S, Ott U, Begemann F. Nature. 1998;391:261. [Google Scholar]
- 12.Pravdivtseva OV. Proceedings of IX All-Union Symposium on Isotope Geochemistry. Moscow: Vernadsky Institute, USSR, Academy of Sciences; 1986. p. 289. [Google Scholar]
- 13.Meshik AP, Pravdivtseva OV, Hohenberg CM. Geochim Cosmochim Acta. 2004;68(11 Suppl):A504. [Google Scholar]
- 14.Cassata WS, Prussin SG, Knight KB, Hutcheon ID, Isselhardt BH, Renne P. J Environ Radioact. 2014;137:88. doi: 10.1016/j.jenvrad.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 15.Chabiron A, Cuney M, Poty B. Mineralium Deposita. 2003;38:127. [Google Scholar]
- 16.Aleshin A. PhD thesis. de l’Université Henry Poincaré, Nancy-I; 2008. [Google Scholar]
- 17.Shukolyukov Yu A, Meshik AP. Chem Geol (Isot Geosci Sect) 1987;66:123. [Google Scholar]
- 18.Hohenberg CM, Thonnard N, Meshik A. Meteorit Planet Sci. 2002;37:257. [Google Scholar]
- 19.Baur H. PhD thesis. Swiss Federal Institute of Technology; Zurich: 1980. [Google Scholar]
- 20.Hohenberg CM. Rev Sci Instrum. 1980;51:1075. [Google Scholar]
- 21.Bernatowicz T, Brannon J, Brazzle RH, Cowsik R, Hohenberg CM, Podosek F. Phys Rev C. 1993;47:806. doi: 10.1103/physrevc.47.806. [DOI] [PubMed] [Google Scholar]
- 22.Meshik AP, Hohenberg CM, Pravdivtseva OV, Bernatowicz TJ, Kapusta YS. Nucl Phys A. 2008;809:275. [Google Scholar]
- 23.Meshik AP, Hohenberg CM, Pravdivtseva OV, Kapusta YS. Phys Rev C. 2001;64:035205. [Google Scholar]
- 24.Fabryka-Martin JT, Davis SN, Elmore D, Kubik PW. Geochim Cosmochim Acta. 1989;53:1817. [Google Scholar]
- 25.Kahn M, Kleinberg J. Radiochemistry of Iodine. National Academy of Sciences–National Research Council; Oak Ridge, TN: 1997. (Nuclear Science Series). NAS-NS-3062. [Google Scholar]
- 26.Steinberg SM, Schmett GT, Kimble G, Emerson DW, Turner MF, Rudin M. J Radioanalyt Nucl Chem. 2008;277:175. [Google Scholar]
- 27.Boulos MS, Manuel OK. Science. 1971;174:1334. doi: 10.1126/science.174.4016.1334. [DOI] [PubMed] [Google Scholar]
- 28.Hennecke EW, Manuel OK. Earth Planet Sci Lett. 1975;27:346. [Google Scholar]
- 29.Phinney D, Tennyson J, Frick U. J Geophys Res. 1978;83:2313. [Google Scholar]
- 30.Smith SP, Reynolds JH. Earth Planet Sci Lett. 1981;54:236. [Google Scholar]
- 31.Murty SVS. Chem Geol (Isot Geosci Sect) 1992;94:229. [Google Scholar]
- 32.Azuma Sh, Ozima M, Hiyagon H. Earth Planet Sci Lett. 1993;114:341. [Google Scholar]
- 33.Meshik AP, Pravdivtseva OV, Hohenberg CM. Meteorit Planet Sci. 2015;50(Suppl. S1) 5371.pdf. [Google Scholar]
- 34.Shukolyukov Yu A. The Decay of Uranium Nuclei in Nature. Atomizdat; Moscow: 1970. [Google Scholar]
- 35.Dresel PE, Olsen KB, Hayes JC, McIntyre SR, Waichier SR, Kennedy BM. J Radioanal Nucl Chem. 2008;276:763. [Google Scholar]
- 36.Kunz J, Staudacher Th, Allègre CJ. Science. 1998;280:877. doi: 10.1126/science.280.5365.877. [DOI] [PubMed] [Google Scholar]
- 37.Marti K, Mathew KJ. J Earth Syst Sci. 1998;107:425. [Google Scholar]
- 38.Takaoka N. J Mass Spectrom Soc Jpn. 1972;20:287. [Google Scholar]
- 39.Anders E, Heymann P. Science. 1969;164:821. doi: 10.1126/science.164.3881.821. [DOI] [PubMed] [Google Scholar]
- 40.R. O. Pepin and D. Phinney (unpublished).
- 41.Turner G, Harrison TM, Holland G, Mojzsis SJ, Gilmour J. Science. 2004;306:89. doi: 10.1126/science.1101014. [DOI] [PubMed] [Google Scholar]
- 42.Mukhopadhyay S. Nature. 2012;486:101. doi: 10.1038/nature11141. [DOI] [PubMed] [Google Scholar]
- 43.Igarashi G. In: Farley KA, editor. Volatiles in the Earth and Solar System; AIP Conf Proc No 341; Pasadena, California. New York: American Institute of Physics Press; 1995. [Google Scholar]
- 44.Swindle TD, Caffee MW, Hohenberg CM. Geochim Cosmochim Acta. 1986;50:1001. [Google Scholar]
- 45.Lewis RS, Srinivasan B, Anders E. Science. 1975;190:1251. doi: 10.1126/science.195.4274.209. [DOI] [PubMed] [Google Scholar]
- 46.Tang M, Anders E. Geochem Cosmochem Acta. 1988;52:1235. [Google Scholar]
- 47.Lewis RS, Tang M, Wacker Anders JF, Steel E. Nature. 1987;326:160. [Google Scholar]
- 48.Meshik AP, Pravdivtseva OV, Hohenberg CM, Burnett DS. Geochim Cosmochim Acta. 2014;127:326. doi: 10.1016/j.gca.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]