

Abstract
Aims
Cathepsin C (CTSC) is necessary for the activation of several serine proteases including neutrophil elastase (NE), cathepsin G and proteinase 3. GSK2793660 is an oral, irreversible inhibitor of CTSC that is hypothesized to provide an alternative route to achieve NE inhibition and was tested in a Phase I study.
Methods
Single escalating oral doses of GSK2793660 from 0.5 to 20 mg or placebo were administered in a randomized crossover design to healthy male subjects; a separate cohort received once daily doses of 12 mg or placebo for 21 days. Data were collected on safety, pharmacokinetics, CTSC enzyme inhibition and blood biomarkers.
Results
Single, oral doses of GSK2793660 were able to dose‐dependently inhibit whole blood CTSC activity. Once daily dosing of 12 mg GSK2793660 for 21 days achieved ≥90% inhibition (95% CI: 56, 130) of CTSC within 3 h on day 1. Only modest reductions of whole blood enzyme activity of approximately 20% were observed for NE, cathepsin G and proteinase 3. Seven of 10 subjects receiving repeat doses of GSK2793660 manifested epidermal desquamation on palmar and plantar surfaces beginning 7–10 days after dosing commencement. There were no other clinically important safety findings.
Conclusions
GSK2793660 inhibited CTSC activity but not the activity of downstream neutrophil serine proteases. The palmar–plantar epidermal desquamation suggests a previously unidentified role for CTSC or one of its target proteins in the maintenance and integrity of the epidermis at these sites, with some similarities to the phenotype of CTSC‐deficient humans.
Keywords: cathepsin C, clinical pharmacology, clinical trial, serine protease
What is Already Known about this Subject
Cathepsin C is a key activator of the proenzymes of several neutrophil granule‐associated serine proteases, such as neutrophil elastase.
There is currently very limited clinical evidence on the precise role and importance of cathepsin C in human disease.
Inhibition of cathepsin C could offer an alternative approach to neutrophil elastase inhibition by reducing the production of activated proenzyme in neutrophils and other cells.
What this Study Adds
In a first‐time‐in‐human study, GSK2793660, an oral, irreversible inhibitor, inhibited whole blood cathepsin C activity in a dose‐dependent manner.
The unexpected finding of palmar–plantar epidermal desquamation on repeat dosing with GSK2793660 suggests a previously unidentified role for cathepsin C or one of its target proteins in the maintenance and integrity of the epidermis at these sites.
Introduction
Cathepsin C (CTSC) or dipeptidyl peptidase I (DPPI, EC. 3.4.4.9) has been identified as the key activator of the proenzymes of several neutrophil granule‐associated serine proteases (NSPs): neutrophil elastase (NE), cathepsin G (CTSG) and proteinase 3 (PR3) 1, 2, 3. CTSC also plays a role in the activation of other leucocyte‐derived serine proteases including granzymes A & B, mast cell chymase, and neutrophil serine protease 4 4. The important role of CTSC in the activation of the neutrophil proteases has been verified from complete or near complete loss of function mutations of the CTSC gene in humans associated with Papillon–Lefèvre syndrome (PLS) and Haim–Munk syndrome, rare autosomal recessive diseases characterized by hyperkeratosis of the palms and soles of the feet and severe periodontitis 5, 6, 7, 8, 9. It has been reported that PLS patients have a near complete loss of neutrophil‐derived NE, CTSG and PR3 activities and a moderate (~50%) reduction in both Granzyme A and B activities. Despite the lack of active NSPs, PLS patients do not manifest significant immunodeficiency, suggesting that active NSPs are not essential for neutrophil antimicrobial activity.
The biological functions of NSPs include degradation of extracellular matrix, inflammatory cell recruitment, and induction of mucus hypersecretion 10. Direct inhibition of NE has been of significant interest in respiratory diseases, including cystic fibrosis, non‐cystic fibrosis bronchiectasis and chronic obstructive pulmonary disease, as well as other inflammatory diseases, that all have a common component of an inappropriate, exaggerated inflammatory response during which leucocytes release proteases and other cytotoxic products that cause tissue injury. Unfortunately, direct inhibition of NE has proved challenging and no NE inhibitor has demonstrated significant clinical efficacy 11.
Other than the genetic association with PLS, there is currently very limited clinical evidence on the precise role and importance of CTSC in human disease. However, given the importance of CTSC in the activation of NSPs, particularly NE, inhibition of CTSC could offer an alternative approach to NE inhibition by reducing the production of activated proenzyme in neutrophils and other cells. To our knowledge, only one other CTSC inhibitor has been evaluated in humans (AZD‐7986, NCT02653872) and results are not yet reported. GSK2793660 is an irreversible, substrate competitive, potent and selective CTSC inhibitor, with an IC50 value between <0.43 and 1 nm against CTSC and a kinact/Ki value of 9.0 × 104 m −1 s−1. The effectively irreversible nature of GSK2793660 inhibition of CTSC enables a high degree of enzyme inhibition that is maintained over a long interval. We conducted a first‐time‐in‐human, Phase I study to investigate the safety, pharmacokinetics and pharmacodynamics of single and repeat doses of GSK2793660 in healthy, adult subjects.
Methods
First‐time‐in‐human clinical trial design (NCT02058407, GSK study no. 200186)
This was a two‐part study conducted in healthy, adult subjects. Part A (single dose escalation) consisted of two interlocking cohorts of nine subjects each (Supplemental Figure S1). It was planned that each subject in Cohorts 1 and 2 would take part in three single‐dose study periods and receive ascending dose levels of GSK2793660. Planned doses across the two cohorts were 0.5, 1, 3, 10, 20 and 50 mg. Placebo control was maintained by using placebo replacement for each subject in one of the treatment periods, according to the prespecified randomization schedule. In each treatment period, six subjects were assigned to GSK2793660 treatment and three subjects were assigned to placebo treatment. The decision to proceed to the next dose level of GSK2793660 was made by the study team and the Investigator based on safety, tolerability and pharmacokinetic and enzyme inhibition data obtained in at least four active subjects at the prior dose level.
For the repeat dose component of the study (Part B), up to two cohorts of 15 subjects (10 subjects assigned to GSK2793660, five subjects assigned to placebo) were to be enrolled (Supplemental Figure S1). It was planned that a single dose strength of 12 mg, dosed once daily in Cohort 3 and twice daily in Cohort 4 (total daily dose 24 mg), would be explored. Subjects in Cohort 3 also took part in a food effect assessment. On Day 1, GSK2793660/placebo was administered under fasted conditions. On Day 2 subjects received GSK2793660/placebo following the standard Food and Drug Administration high fat meal 12.
The study received favourable ethical opinion from the Brent Research Ethics Committee (London, UK) as well as regulatory approval by the Medicines and Healthcare products Regulatory Agency. The study was conducted at Hammersmith Medicines Research (London, UK) in accordance with the revised Declaration of Helsinki (2008), Good Clinical Practice and all applicable regulatory requirements. Full written informed consent was obtained from all participants before the performance of any study‐specific procedures
Safety assessments
For each subject, adverse events and serious adverse events were collected from the start of first dosing until the final follow‐up contact. Safety was monitored by the measurement of ECGs, vital signs, clinical laboratory assessments (clinical chemistry, haematology and urinalysis), and assessment of adverse events.
Pharmacokinetics and pharmacodynamics
Determination of GSK2793660 concentrations in human plasma
GSK2793660 is unstable at pH > 4 and converts to two thermodynamically more stable cyclised products; therefore, to prevent this cyclization, all plasma standards, quality control samples, validation and study samples required acidification.
Plasma concentrations of GSK2793660 free base were determined using a sensitive and selective validated liquid chromatography–tandem mass spectrometry analytical method following sample preparation by protein precipitation (details of the assay methods are in the Supplemental Material). The lower limit of quantification for the current GSK2793660 assay was defined as 0.2 ng ml–1.
CTSC, NE, CTSG and PR3 enzyme activities were measured in samples from whole blood using validated methods. Details of the assay methods are in the Supplemental Material.
Statistical analysis
The primary objective of this study was to assess the safety and tolerability of single and repeat oral doses of GSK2793660, and as such no formal hypotheses were tested. The sample size was based on feasibility and was deemed an adequate number to provide an assessment of safety, PK and PD prior to progression into longer duration studies.
Pharmacokinetic analysis
Pharmacokinetic parameters were calculated by standard noncompartmental analysis using WinNonlin Pro 5.2 or higher. All calculations of noncompartmental parameters were based on actual sampling times.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 13, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 14.
Results
Thirty‐three adult, healthy subjects were randomized into the trial; subject disposition and demographics are outlined in Table 1. All subjects completed the planned study. In Part A (single doses), the planned doses were adjusted during the study in response to emerging data; actual doses administered were 0.5 mg, 2 mg, 6 mg, 12 mg and 20 mg. In Part B (repeat dosing), the dose administered was 12 mg once‐daily for 21 days.
Table 1.
Baseline characteristics of study subjects
| Part A | Part B | ||
|---|---|---|---|
| Placebo | GSK2793660 | ||
| All subjects, n (% male) | 18 (89) | 5 (100) | 10 (100) |
| Age (years) mean (range) | 41 (21–55) | 30 (21–39) | 34 (23–46) |
| Body mass index (kg/m 2) mean (SD) | 24 (2.7) | 25 (1.31) | 24 (2.74) |
| Race , n (%) | |||
| African‐American/African Heritage | 5 (28) | 1 (20) | 1 (10) |
| White/Caucasian/European Heritage | 13 (72) | 3 (60) | 9 (90) |
| Asian/Southeast Asian Heritage | 0 | 1 (20) | 0 |
Pharmacokinetics of GSK2793660
Following single dose administration of GSK2793660, maximum plasma concentrations of GSK2793660 were attained at approximately 0.5 to 1 h post‐dose (median estimates). Thereafter, plasma concentrations of GSK2793660 declined in a mono‐exponential manner, with geometric mean terminal elimination half‐lives (t½) ranging from 0.6 to 1.7 h across all dose levels (although t½ could not be estimated in subjects at the lowest dose of 0.5 mg due to plasma levels falling below detectable limits shortly after Cmax; Figure 1).
Figure 1.
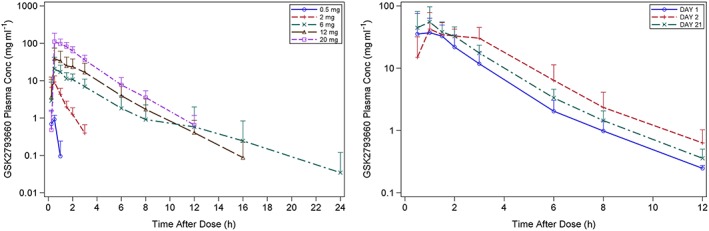
Plasma concentration time profiles of GSK2793660 following single doses (left) and repeat doses (right) of GSK2793660. Data are expressed as mean concentrations ± 95% confidence intervals on a semilogarithmic scale
Plasma concentrations following repeated once‐daily oral administration of 12 mg GSK2793660 are presented in Figure 1. On Day 1 (fasted), Day 2 (following a high‐fat meal), and Day 21 (fasted and at steady state), plasma concentrations of GSK2793660 were generally measurable up to 12 h post‐dose in most subjects; however, in all instances, concentrations fell below detectable limits within 24 h (i.e. before the next dose). Following repeated once‐daily oral dosing of 12 mg GSK2793660, there was a small degree of accumulation in plasma, with an accumulation ratio of approximately 1.47 (90% CI: 1.24, 1.73). Selected PK parameters for single and repeat dose are summarized in Table 2. Oral dosing of 12 mg GSK2793660 following a high‐fat meal, resulted in a 1.6‐fold increase in AUC (90% CI: 1.43, 1.80) with no change in Cmax (1.06‐fold, 90% CI: 0.71, 1.57).
Table 2.
Pharmacokinetic parameters for GSK2793660 following single and repeat doses
| Treatment | Cmax (ng ml–1)a | AUC(0–24) (ng h ml–1)a | tmax (h)b | t1/2 (h)a |
|---|---|---|---|---|
| GSK2793660, 0.5 mg | 1.04 (46.3) | 0.326 (72.4) | 0.5 (0.25–0.50) | ND[Link] |
| GSK2793660, 2 mg | 9.61 (31.1) | 8.460 (36.8) | 0.5 (0.25–1.00) | 0.636 (14.0) |
| GSK2793660, 6 mg | 24.53 (54.1) | 47.967 (45.0) | 1.0 (0.50–3.00) | 1.415 (40.5) |
| GSK2793660, 12 mg | 50.69 (41.5) | 95.853 (47.9) | 0.85 (0.50–3.00) | 1.732 (30.1) |
| GSK2793660, 20 mg | 126.36 (49.5) | 255.883 (28.3) | 0.75 (0.5–1.5) | 1.631 (11.2) |
| GSK2793660 12 mg x 21 days | Day 1 (fasted) | |||
| 49.00 (61.8) | 86.977 (41.9) | 0.75 (0.50–1.50) | 1.384 (24.9) | |
| Day 2 (fed) | ||||
| 51.86 (42.4) | 139.399 (27.1) | 1.000 (1.00–3.00) | 1.444 (20.9) | |
| Day 21 (fasted) | ||||
| 72.56 (39.6) | 127.591 (22.1) | 1.00 (0.50–2.00) | 1.566 (18.2) | |
Geometric mean (CV%);
Median (range);
ND: not determined
Pharmacodynamics of GSK2793660
In Part A, maximum inhibition of whole blood CTSC activity at each dose level was observed within 1–3 h of dosing (Figure 2). At the 2 mg dose level, inhibition of CTSC ranged between 22% and 74% compared with placebo across the sampling time points from 1–144 h post‐dose. The percentage inhibition for the 6 mg, 12 mg and 20 mg single doses was similar at each time point, and ranged from approximately 57 to 99% compared with placebo. At these three higher dose levels, inhibition was maintained through the 144 h time point and longer. Follow‐up sampling revealed that by Day 14, CTSC enzyme activity was back to baseline levels.
Figure 2.
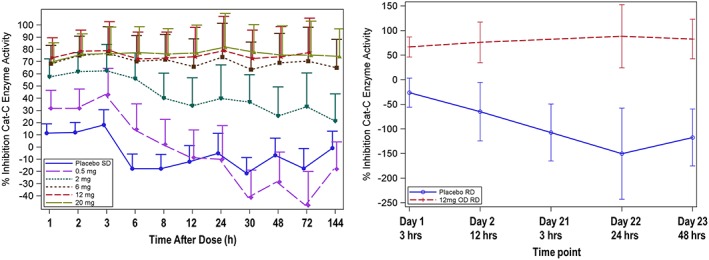
Whole blood cathepsin C inhibition after single (left) and 21 days of repeat dosing (right) with placebo or GSK2793660 12 mg once daily. Data are expressed as adjusted means ± 95% confidence intervals
Based on these data, a dose of 12 mg once daily was selected for the first repeat dose cohort to ensure a high probability of observing complete inhibition of CTSC. Following repeat dose administration of 12 mg GSK2793660, CTSC inhibition of 93% (95% CI: 56, 130) inhibition was observed within 3 h of the Day 1 dose, compared with placebo. This level of inhibition was maintained across the repeat dose sampling period (Figure 2).
The impact of GSK2793660 on whole blood NSP activity is shown in Figure 3. NE activity was reduced by 7–47% following repeat dose administration of 12 mg GSK2793660 compared with placebo. CTSG activity was reduced by up to 47% and proteinase 3 activity was reduced up to approximately 37% following repeat dose administration of 12 mg GSK2793660 compared with placebo. For all of these measurements, there was fluctuation across the time points such that no consistent relationship between time and enzyme activity levels could be determined.
Figure 3.
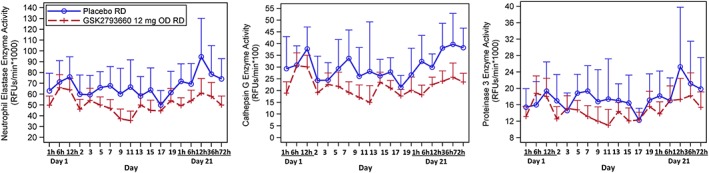
Whole blood activity of neutrophil elastase (left), cathepsin G (centre) and proteinase 3 (right) following 21 days repeat dosing with placebo or GSK2793660 12 mg once daily. Data are expressed as adjusted means ± 95% confidence intervals
Safety
A total of nine adverse events (AEs) in six subjects (33%) were reported during Part A of the study. Of these, five were reported after administration of placebo, and four following administration of GSK2793660 (Table S1). The only AEs considered drug related were two episodes of dizziness. No subjects withdrew due to an AE during the single‐dose period. In Part B, 9 (90%) of the 10 subjects who were administered repeat doses of GSK2793660 reported 20 AEs (Table S1) vs. two (40%) subjects reporting two AEs after receiving placebo.
Seven subjects receiving repeat doses of GSK2793660 experienced skin desquamation (peeling of the skin on the hands, fingers and/or feet) that was considered a drug‐related AE by the investigator (Figure 4). These AEs began to emerge approximately after 7–10 days of once‐daily dosing with 12 mg GSK2793660, lasted for several weeks and were limited to the palmar and plantar surfaces. The skin desquamation was not associated with significant visual signs of inflammation, exudates or pain with the exception of one subject who reported discomfort on his plantar surfaces during exercise. All AEs associated with skin desquamation were considered mild in intensity by the study investigator. The follow up period for the study was extended to allow the AEs to be followed to resolution. All but one of these AEs was ultimately confirmed as resolved (one subject was lost to follow‐up). Other drug‐related AEs included dry skin, pruritis and pruritic rash (reported in one subject each on GSK2793660) and skin exfoliation reported in one subject on placebo. Despite the skin findings, no subjects withdrew due to an AE.
Figure 4.
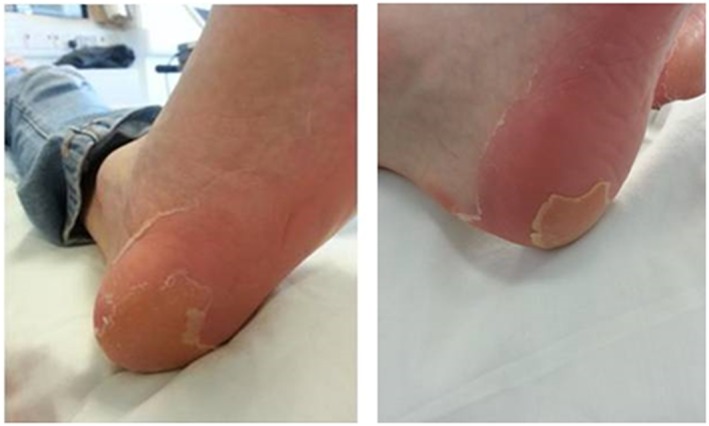
Epidermal desquamation following 21 days repeat dosing with GSK2793660 12 mg once daily
There were no clinically significant changes in vital signs, clinical laboratory assessments, or ECG readings. There were no significant differences in either the exposure to GSK2793660 or the percent CTSC inhibition between those subjects who developed skin changes and those who did not. The decision was made to stop the study based on prespecified criteria for the frequency of adverse events, i.e. two or more subjects experiencing AEs that were of moderate or severe intensity and were consistent across subjects, and considered possibly drug‐related by the study investigator. Consequently, the planned final cohort of Part B (cohort 4) was not conducted.
Discussion
GSK2793660 is one of the first CTSC inhibitors to be tested in humans. The primary objective of the study was to assess the safety and tolerability of single and repeat oral doses of GSK2793660 in healthy subjects. Secondary objectives included assessment of GSK2793660 pharmacokinetics and pharmacodynamics (inhibition of CTSC enzyme activity and the activity of downstream NSPs).
GSK2793660 exhibited a very short systemic half‐life (~1.5 h). While target mediated clearance cannot be ruled out entirely, the current observations are more likely to stem from saturable first pass extraction. This would explain the greater than dose proportional exposure with single higher doses, increased exposure with high fat meal, and some accumulation with repeat dosing. Inhibition of whole blood CTSC enzyme activity was observed following all single doses tested, reaching a maximal inhibition of approximately 80%. Although GSK2793660 was rapidly cleared from the plasma, recovery of CTSC enzyme activity in peripheral blood did not occur until 14 days following dose administration. This is consistent with the irreversible mechanism of action of GSK2793660 (data not shown) and the time needed for the maturation and release of a new population of neutrophils from the bone marrow and turnover of peripheral blood neutrophils 15.
CTSC enzyme activity is essential for the processing and activation of NSPs in both animal models and in humans 16, 17. The human and animal evidence indicates that extensive loss of CTSC activity is needed to prevent activation of NSP proenzymes 17, 18, 19. CTSC −/+ mice and the CTSC −/+ parents of PLS subjects have reduced CTSC, but either no change in NSP activity 1, no symptoms related to CTSC 7 or no loss of NSP activity / protein 17. Together, the animal and human data suggest that in vivo high levels of inhibition of CTSC are necessary to achieve significant downstream effects.
In vitro, variable levels of CTSC are needed to inhibit NE or CTSG 1, 19. Intravenous infusion of a CTSC inhibitor for 2 weeks achieved high levels of CTSC inhibition (> 90%) but resulted in only 50–80% inhibition of NSPs 1. Similarly, in work from our group, in mice, nearly complete pharmacological inhibition of CTSC (96–100%) resulted in partial inhibition of downstream NSPs 18. There are few examples where both CTSC activity and NSP activity were determined in cases of CTSC loss of activity <90%. Recent studies continue to suggest high levels of inhibition (>75%) are required, although the absolute levels are not completely consistent across studies 20, 21. We do not understand why the levels of NSP inhibition in this study are so low, as 80% inhibition of CTSC was achieved. It is possible that in humans, this level is simply insufficient, unlike cellular studies or in vivo in other species. Other possibilities are simply speculative, such as compartmentalization of CTSC and inhibitor in such a way that a CTSC compartment that is sufficient for NSP activation remains active, or that the time course of neutrophil maturation allows a sufficient window for generation of CTSC and NSP activation between each dose, due to the particular kinetics of this compound.
With once daily repeat dosing of 12 mg GSK2793660 for 21 days, only modest inhibition of whole blood NSP enzyme activity was observed for any of the three downstream NSPs evaluated. The modest inhibition of downstream NSPs observed in this study does not support the hypothesis that 80% sustained CTSC inhibition would lead to reductions in NSP activity 1, 18. One could speculate that the residual CTSC activity was sufficient to promote complete NSP activation during neutrophil maturation and that inhibition levels would need to approach 100% to achieve an effect on NSP activation. We conducted extensive assay validation to confirm substrate specificity with selective inhibitors, and included relevant controls. We cannot, however, exclude the possibility that there are assay artefacts due to unidentified enzymes in the cell lysate or sample processing.
The development of skin desquamation after 7–10 days dosing with GSK2793660 in the healthy subjects was unexpected. CTSC‐deficient mice develop normally and have no skin abnormalities, and in preclinical toxicology studies both rats and dogs dosed for up to 3 months with high doses of GSK2793660 did not manifest any skin abnormalities (unpublished observations: GSK2793660 Investigator's Brochure, GSK data on file). Patients with PLS manifest skin lesions that range from scaly skin to hyperkeratosis; these lesions typically develop between the ages of 6 months and 4 years 9. The relatively rapid occurrence of the epidermal peeling in this clinical trial suggests a previously unidentified and important role for CTSC or one or more of its substrate proteins in the development and maintenance of the integrity of plantar and palmar epidermal surfaces; it has been postulated that CTCS may be important in the processing of proteins such as keratins 22.
The physical appearance of the desquamation was reminiscent of acral skin peeling, a genetic condition resulting in palmoplantar peeling. Other observations of palmoplantar peeling without erythema or inflammation include some case studies of sirolimus treatment, some infections and certain tyrosine kinase inhibitors 23. One preliminary study of genetic acral skin peeling found altered proteolytic activity and kallikrein (KLK5) expression in skin from patients 24. The specific mechanism by which cathepsin C inhibition by GSK2793660 leads to skin peeling would require more work to elucidate, but numerous proteolytic activities contribute to maintenance of skin differentiation and integrity, perhaps including cathepsin C expressed in keratinocytes.
The results of the clinical study with GSK2793660 suggest that relatively high levels of pharmacologic CTSC inhibition can be achieved with a CTSC inhibitor in humans but this does not translate to a significant effect on downstream NSPs within 21 days. In addition, high CTSC inhibition was associated with epidermal desquamation on the palmar and plantar surfaces in most subjects who received GSK2793660 once a day for 21 days. This finding will need to be considered for subsequent development of CTSC inhibitors. Recently, another CTSC inhibitor entered clinical development (AZD7986, clinicaltrials.gov identifier NCT02303574). It will be important to see how the results with AZD7986 compare with those of GSK2793660.
Competing Interests
All authors, with the exception of M.B., were employees and shareholders of GSK at the time the study was conducted. M.B. is an employee of HMR.
The authors would like to thank the study volunteers and the staff at Hammersmith Medicines Research (London, UK). The authors thank Pragathi Kotha Venkata (GSK) for assistance with preparation of the figures.
GSK was the sponsor of the clinical trial.
Contributors
A.L.L., R.J.M., N.G., J.B., N.D., A.C., T.H., D.C. and B.E.M. made substantial contributions to the conception and design of the clinical trial. All authors participated in the analysis and interpretation of study results. A.L.L., R.J.M. and B.E.M. drafted the manuscript. All authors had full access to the data, critically reviewed the manuscript for intellectual content, and gave approval of the final version of the manuscript.
Supporting information
Figure S1 Study design
Table S1 All adverse events
Table S2 Drug‐related adverse events
Miller, B. E. , Mayer, R. J. , Goyal, N. , Bal, J. , Dallow, N. , Boyce, M. , Carpenter, D. , Churchill, A. , Heslop, T. , and Lazaar, A. L. (2017) Epithelial desquamation observed in a phase I study of an oral cathepsin C inhibitor (GSK2793660). Br J Clin Pharmacol, 83: 2813–2820. doi: 10.1111/bcp.13398.
Clinical Trial Registration: Clinical Trials.gov registration NCT02058407 (GSK Study No. 200186), date of initial registration 09 January 2014.
References
- 1. Methot N, Guay D, Rubin J, Ethier D, Ortega K, Wong S, et al In vivo inhibition of serine protease processing requires a high fractional inhibition of cathepsin C. Mol Pharmacol 2008; 73: 1857–1865. [DOI] [PubMed] [Google Scholar]
- 2. Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. Papillon–Lefèvre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J Immunol 2004; 173: 7277–7281. [DOI] [PubMed] [Google Scholar]
- 3. Hart TC, Hart PS, Michalec MD, Zhang Y, Firatli E, Van Dyke TE, et al Haim–Munk syndrome and Papillon–Lefèvre syndrome are allelic mutations in cathepsin C. J Med Genet 2000; 37: 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perera NC, Wiesmuller KH, Larsen MT, Schacher B, Eickholz P, Borregard N, et al NSP4 is stored in azurophil granules and released by activated neutrophils as active endoprotease with restricted specificity. J Immunol 2013; 191: 2700–2707. [DOI] [PubMed] [Google Scholar]
- 5. Nagy N, Valvi P, Csoma Z, Sulak A, Tripolszki K, Farkas K, et al CTSC and Papillon–Lefèvre syndrome: detection of recurrent mutations in Hungarian patients, a review of published variants and database update. Mol Genet Genomic Med 2014; 2: 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sorensen OE, Clemmensen SN, Dahl SL, Ostergaard O, Heegard NH, Glenthoj A, et al Papillon–Lefèvre syndrome patient reveals species‐specific requirements for neutrophils defenses. J Clin Invest 2014; 10: 4539–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hewitt C, McCormick D, Linden G, Turk D, Stern I, Wallace I, et al The role of cathepsin C in Papillon–Lefèvre syndrome, prepubertal periodontitis, and aggressive periodontitis. Hum Mutat 2004; 23: 222–228. [DOI] [PubMed] [Google Scholar]
- 8. Toomes C, James J, Wood AJ, Wu CL, McCormick D, Lench N, et al Loss‐of‐function mutations in the cathepsin C gene result in periodontal disease and palmoplantar keratosis. Nat Genet 1999; 23: 421–424. [DOI] [PubMed] [Google Scholar]
- 9. Haneke E. The Papillon–Lefèvre syndrome: keratosis palmoplantaris with periodontopathy. Report of a case and review of the cases in the literature. Hum Genet 1979; 51: 1–35. [DOI] [PubMed] [Google Scholar]
- 10. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010; 62: 726–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Henriksen PA. The potential of neutrophil elastase inhibitors anti‐inflammatory therapies. Curr Opin Hematol 2014; 21: 23–28. [DOI] [PubMed] [Google Scholar]
- 12. FDA . 2002. Guidance for industry, food effect bioavailability and fed bioequivalence studies, Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER), December 2002. Available at: http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126833.pdf. (last accessed 31 March 2016).
- 13. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walker RJ, Willemze R. Neutrophil kinetics and the regulation of granulopoiesis. Rev Infect Dis 1980; 2: 282–292. [DOI] [PubMed] [Google Scholar]
- 16. Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil‐derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest 2002; 109: 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Haar SF, Jansen DIC, Schoenmaker T, De Vree H, Everts V, Beersten W. Loss‐of‐function mutations in cathepsin C in two families with Papillon–Lefèvre syndrome are associated with deficiency of serine proteases in PMNs. Human Mutat 2004; 23: 524–530. [DOI] [PubMed] [Google Scholar]
- 18. Podolin PL, Bolognese BJ, Foley JP, Carpenter DC, Logan GA, Wixted WE, et al Cathepsin C inhibition: a novel approach for the treatment of chronic respiratory diseases. Am J Respir Crit Care Med 2013; 187: A1500. [Google Scholar]
- 19. Methot N, Rubin J, Guay D, Beaulieu C, Ethier D, Reddy TJ. Inhibition of the activation of multiple serine proteases with a cathepsin C inhibitor requires sustained exposure to prevent pro‐enzyme processing. J Biol Chem 2007; 282: 20836–20846. [DOI] [PubMed] [Google Scholar]
- 20. Gardiner P, Wikell C, Clifton S, Shearer J, Benjamin A, Peters SA. Neutrophil maturation rate determines the effects of dipeptidyl peptidase 1 inhibition on neutrophil serine protease activity. Brit J Pharm 2016; 173: 2390–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guarino C, Hamon Y, Croix C, Lamort A‐S, Dallet‐Choisy S, Marchand‐Adam S, et al Prolonged pharmacological inhibition of cathepsin C results in elimination of neutrophil serine proteases. Biochem Pharmacol 2017; 131: 52–67. [DOI] [PubMed] [Google Scholar]
- 22. Nuckolls GH, Slavkin HC. Paths of glorious proteases. Nat Genet 1999; 23: 378–380. [DOI] [PubMed] [Google Scholar]
- 23. Liu LS, McNiff JM, Colegio OR. Palmoplantar peeling secondary to sirolimus therapy. Am J Transplant 2014; 14: 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pampalakis G, Kiritsi D, Zingkou E, Valari MD, Bruckner‐Tuderman L. Sotiropoulou G elevated epidermal proteolysis and altered KLK5 expression in Acral peeling skin syndrome. J Invest Dermatology 2016; 9 (supp 2): S176. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study design
Table S1 All adverse events
Table S2 Drug‐related adverse events