

Abstract
Aims
Human cytomegalovirus constitutes a prevalent and serious threat to immunocompromised individuals and requires new treatments. Letermovir is a novel viral‐terminase inhibitor that has demonstrated prophylactic/pre‐emptive activity against human cytomegalovirus in Phase 2 and 3 transplant trials. As unchanged letermovir is primarily excreted via the liver by bile, this trial aimed to assess the effect of hepatic impairment on letermovir pharmacokinetics.
Methods
Phase 1, open‐label, parallel‐group pharmacokinetic and safety comparison of multiple once‐daily oral letermovir in female subjects with hepatic impairment and healthy matched controls. For 8 days, subjects with moderate hepatic impairment (n = 8) and their matched healthy controls (n = 9) received 60 mg letermovir/day and those with severe hepatic impairment (n = 8) and their matched healthy controls (n = 8) received 30 mg letermovir/day. Pharmacokinetic parameters were determined from blood samples.
Results
For subjects with moderate hepatic impairment, maximal observed concentration at steady state (Css,max) and the area under the concentration vs. time curve over a dosing interval at steady state (AUCτ,ss) for total letermovir were 1.37‐fold (90% confidence interval: 0.87, 2.17) and 1.59‐fold (0.98, 2.57) higher, respectively, than in healthy subjects. For subjects with severe hepatic impairment, Css,max and AUCτ,ss values of total letermovir were 2.34‐fold (1.91, 2.88) and 3.82‐fold (2.94, 4.97) higher, respectively, compared with healthy subjects.
Conclusions
Moderate hepatic impairment increased exposure to letermovir <2‐fold, while severe hepatic impairment increased letermovir exposure approximately 4‐fold as compared with healthy subjects. Letermovir 60/30 mg/day was generally well‐tolerated in subjects with hepatic impairment.
Keywords: antivirals, cytomegalovirus, hepatic insufficiency, letermovir, pharmacokinetics, terminase inhibitor
What is Already Known about this Subject
Phase 2 and 3 trials have demonstrated the safety and efficacy of letermovir as human cytomegalovirus prophylaxis or pre‐emptive therapy in adult transplant recipients.
Unchanged biliary excretion is the major route of letermovir elimination.
This trial evaluated the influence of moderate‐to‐severe hepatic impairment on the pharmacokinetics, safety and tolerability of letermovir.
What this Study Adds
Increases in letermovir plasma concentrations were observed following multiple oral doses of once‐daily letermovir 60 mg in subjects with moderate hepatic impairment and letermovir 30 mg in subjects with severe hepatic impairment.
These doses were generally well tolerated in all subjects with hepatic impairment.
Introduction
Human cytomegalovirus (HCMV) infection or reactivation can be life‐threatening to vulnerable recipients of transplants and is considered a major limitation to the success of such treatments. Disease manifestations are dependent on the type of transplant, but commonly include fever, myelosuppression, pneumonia, gastrointestinal disease and retinitis 1, 2.
Strategies to prevent HCMV infection of transplant recipients include prophylaxis, where all patients at risk receive an antiviral agent for a defined period of time, or pre‐emptive therapy, where treatment is initiated once a viral threshold is reached in an individual patient. Using antivirals in this way has reduced the risk of HCMV complications post transplantation; however, current antivirals used against HCMV are associated with significant adverse events, including bone marrow suppression and renal toxicity 3, 4, 5.
Letermovir (AIC246/MK‐8228), like its predecessor compounds 6, 7, 8, targets the viral terminase complex of HCMV, thereby inhibiting the formation and release of infectious virus particles. Unlike nucleoside analogues, which are prodrugs and require intracellular phosphorylation by the virus for activation 9, terminase inhibitors such as letermovir do not require an activation step, thereby offering protection to cells that are not yet infected. Terminase inhibitors are also active against viruses that have become resistant to currently approved anti‐HCMV drugs that target the viral polymerase.
In Phase 2 and 3 trials, letermovir demonstrated prophylactic and pre‐emptive activity against HCMV in post‐transplant patients 10, 11, 12. In these studies, letermovir was generally well tolerated, with a safety profile comparable with placebo and with no indication of haematological toxicity or nephrotoxicity 10, 11. In addition, letermovir treatment of a single patient, who had demonstrated multidrug‐resistant HCMV led to the powerful suppression of HCMV and was associated with clinical improvement 13. Recently, the Phase 3 study using HCMV prophylaxis with letermovir in stem cell transplant recipients met its primary endpoint 12.
The effect of hepatic impairment on letermovir pharmacokinetics (PK) has yet to be determined. An extensive body of nonclinical information has been obtained on the metabolism and PK of letermovir and this information was, in addition to toxicological studies, used to obtain regulatory approval for this clinical trial to study hepatic impairment. Letermovir is protein unbound with the percentage of drug binding in plasma ranging from 0.73% (dog) to 2.38% (mouse); in humans, the value was 1.33%. Letermovir was also found to be primarily excreted via the biliary route in the rat. None of the human cytochrome P450 (CYP450) isoenzymes were found to mediate metabolism of letermovir. There was evidence for letermovir inhibiting UDP‐glucuronosyltransferase 1A1, although there was no clinical evidence for this impacting on bilirubin clearance. In a human mass balance trial, 8 healthy male subjects received a [14C]‐letermovir tracer dose together with the last dose of a twice‐daily 80 mg letermovir treatment; excretion in urine was negligible (1.43% of radioactivity) while 93.3% of radioactivity was found in faeces, 70.5% being identified as unchanged letermovir and 6.0% as O‐glucuronide metabolite (data on file). Thus, biliary excretion of primarily unchanged letermovir via organic anion‐transporting polypeptide (OATP)1B1 and OATP1B3 transporters provides the major route of elimination; and so, impaired hepatic function might influence letermovir plasma concentrations and subsequent systemic exposure.
The current trial evaluated the PK, safety and tolerability of letermovir in subjects with moderate or severe hepatic impairment. The formation of glucuronides from letermovir was also investigated in the form of unbound (deglucuronidated) letermovir.
Methods
Trial design and objectives
This was a Phase 1, open‐label, parallel‐group comparison trial designed primarily to investigate the influence of moderate‐to‐severe hepatic impairment on letermovir PK following multiple, oral daily doses (AiCuris protocol: AIC246‐01‐I‐10, Merck protocol: MK‐8228‐P015). Secondary objectives were to investigate the safety and tolerability of letermovir and the formation of glucuronides. This two‐centre trial was performed at GOUVPO Russian Peoples' Friendship University, Center of Applied Clinical Pharmacology, Clinical Hospital #3 and City Clinical Hospital #64, Moscow, Russia.
Overall, subjects were enrolled into the following four groups: women with moderate hepatic impairment; healthy women who were individually matched to the women with moderate hepatic impairment; women with severe hepatic impairment; healthy women who were individually matched to the women with severe hepatic impairment.
Eligible subjects were hospitalised from the day of arrival (Day –1) to Day 11 (72 h postdosing). Included subjects were assigned a four‐digit subject number according to the order of arrival during the evening before trial medication intake on Day –1.
The trial design was chosen in accordance with the current European Medicines Agency guidance Guideline on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Hepatic Function 14. Each participant was required to provide written informed consent before starting the trial or any trial procedure. The study was conducted in accordance with principles of good clinical practice and was approved by the appropriate institutional review boards and regulatory agencies.
Trial population
Female subjects of any ethnic origin, aged between 18 and 65 years (inclusive), with a body mass index (BMI) between ≥18 and ≤34 kg m−2 and body weight >50 kg were eligible.
Hepatic impairment was defined according to Child–Pugh classification: subjects with moderate hepatic impairment were defined as Class B, and those with severe hepatic impairment defined as Class C.
Healthy subjects were individually matched to those with hepatic impairment with respect to age (± 10%), BMI (± 10%) and ethnic origin (i.e. healthy controls matched with moderate hepatic impairment and with severe hepatic impairment). Subjects were considered healthy based on a screening examination including medical history, physical examination, blood pressure, pulse rate, electrocardiogram (ECG) assessment and clinical laboratory results, and shown to have no clinically relevant malignant, cardiovascular, renal, gastrointestinal, hepatic, metabolic, endocrine, neurological or psychiatric abnormalities.
All women of childbearing age were required to use adequate contraception. Women of nonchildbearing potential could be included if surgically sterile (i.e. documented complete hysterectomy or bitubal ligations; partial hysterectomy was not sufficient) or postmenopausal for ≥2 years (i.e. history of no menses for at least 24 months).
Use of any agent known to inhibit or induce xenobiotic metabolising enzymes or transporters, which may have modulated letermovir exposure, within 14 days prior to the first dose was not allowed. In addition, any food or beverage known to inhibit or induce enzyme activity was also prohibited from 1 week prior to first drug administration until discharge, and alcohol, methylxanthine‐containing beverages or food and tobacco products were not permitted from 48 h prior to entry until discharge. Concomitant medication needed for the treatment of underlying liver disease and single intake of a drug could be accepted if judged by the investigators to have no clinical relevance and no relevance for the trial objectives. Limited amounts of paracetamol were allowed to treat adverse events (AEs). During the trial, patients with moderate hepatic impairment reported the use of spironolactone, ursodeoxycholic acid, lactulose and paracetamol. Subjects with severe hepatic impairment reported the use of spironolactone, propranolol, furosemide, ursodeoxycholic acid, amoxi‐clavulanico, lactulose, ornithine, folic acid, menadione and kendural C.
Subjects were excluded if they had any diseases or surgery of the gastrointestinal tract that may interfere with drug absorption and/or elimination, or had participated in a drug trial within 60 days prior to first administration of the trial medication.
Trial treatment
Subjects with hepatic impairment were treated in a stepwise manner to protect their safety. Subjects with moderate hepatic impairment and their matched controls received oral letermovir 60 mg once daily for 8 days. This initial dose was selected as the anticipated increase of plasma concentration and prolongation of terminal elimination half‐life of letermovir in subjects with moderate hepatic impairment at the 60‐mg dose were considered to be covered by plasma concentrations observed in earlier clinical trials. After reviewing safety and PK data for the subjects with moderate hepatic impairment, it was decided that the subjects with severe hepatic impairment and their matched controls should receive oral letermovir 30 mg once daily for 8 days. The decision to use a lower dose for these groups was also based on the concomitant medications expected for severe hepatic impairment that may influence PK and exposure to letermovir above levels previously studied.
Letermovir was provided as an immediate‐release tablet formulation containing 30 mg of letermovir per tablet. All subjects received letermovir once daily for 8 days (Days 1–8) in the morning.
Sampling and assessments
Blood sampling for determination of trough letermovir concentrations was carried out predose in the morning of Days 1–7. On Day 8, blood samples were taken predose and 0.25, 0.50, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24, 36, 48, 60 and 72 h after last drug administration. Additional samples were taken for subjects with hepatic impairment at 96 h and 120 h after the last administration of letermovir on Day 8 because a decreased clearance may lead to a prolonged half‐life of letermovir. The samples obtained on Day 8 at pr‐dose and at 1.5 h and 6 h post‐dose were also used for determination of letermovir glucuronides after hydrolysis in plasma. Blood sampling for determination of unbound letermovir fraction concentrations was carried out 1.5 h post‐dose on Day 8.
Letermovir plasma concentrations were determined by a validated liquid chromatography–tandem mass spectrometry method with a lower limit of quantification of 1 ng ml−1. The analytical assay of the plasma samples was carried out at A&M Labor für Analytik und Metabolismusforschung Service GmbH, Bergheim, Germany. The bioanalysis was conducted according to good laboratory practice regulations.
Data analysis, PK evaluations and statistics
No formal estimation of the sample size was performed. All subjects with bioanalytical data were included in the PK analysis. For the statistical analysis, only paired data (i.e. from subjects with moderate/severe hepatic impairment and their individually matched healthy subject) were included. Therefore, in the event that it was not possible to calculate either maximal observed concentration at steady state (Css,max) or the area under the concentration vs. time curve over a dosing interval at steady state (AUCτ,ss) for a subject, both this subject and her individual matched subject were excluded from the statistical analysis for the applicable PK parameter. All subjects who received at least one dose of trial medication were considered valid for the safety evaluation.
PK and statistical analyses were done by Kinesis Pharma B.V., Breda, The Netherlands, using the validated computer program WinNonlin Professional (version 4.1; Pharsight Corporation, Mountain View, California, USA). Noncompartmental analysis model 200 (extravascular input, plasma data) was applied for the PK analysis. Microsoft Excel (version 2007; Microsoft, Redmond, Washington, USA) and SAS (version 9.1.3; SAS Institute Inc., Cary, North Carolina, USA) were also used. In some cases, letermovir concentration after hydrolysis (representing free nonconjugated letermovir in plasma and letermovir originating from letermovir glucuronides) was lower than the free nonconjugated letermovir concentration. In these cases, the calculated letermovir concentrations originating from glucuronides were set to ‘0’. In case of ‘0’ values, no glucuronide vs. free letermovir ratios were calculated.
The primary PK variables were exposure, as measured by AUCτ,ss and Css,max, based on total and unbound plasma concentrations.
Secondary PK variables included minimal observed concentration, average steady‐state concentration over the dosing interval, time to reach the maximal observed concentration, apparent elimination rate constant, apparent elimination half‐life, apparent oral clearance and apparent volume of distribution at steady state based on total and unbound plasma concentrations.
Least square (LS) means were calculated for the groups with moderate and severe hepatic impairment as a percentage of the reference to their matched controls, for AUCτ,ss and Css,max of letermovir using an analysis of variance model with the logarithm of PK parameters as a dependent variable and hepatic status as a fixed effect (based on total and unbound plasma concentrations). Only paired data (i.e. data pairs of subjects with moderate or severe hepatic impairment and their individually matched healthy subject) were included in the statistical analysis. Using the LS means and the intersubject variance, the point estimate and the 90% confidence intervals for the difference in means on a log scale between hepatic impaired group (test) and normal hepatic function group (reference) were conducted and transformed using antilogarithms. The P value associated with this assessment was extracted in the treatment comparison. A linear mixed‐effects model was also used, controlling for hepatic function as a fixed effect and subject as a random effect. No formal hypothesis testing was done; all analyses were exploratory in nature. Differences among LS means were calculated with 90% confidence intervals and compared with predefined bioequivalence limits of 80–125%.
Safety assessments
AEs were monitored and recorded 30 min before letermovir administration on Days 1–8, throughout the day postdose on Days 1 and 8 and at the post‐trial examination. AEs were also monitored in subjects with hepatic impairment on their additional visits on Days 12 and 13.
Vital signs (heart rate and blood pressure), physical examination, 12‐lead ECG and standard clinical laboratory assessments were recorded throughout the trial.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 15, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 16, 17.
Results
Trial population
Between 20 July 2010 and 4 August 2011, 33 subjects were enrolled in the trial. There were eight subjects enrolled in each group except for the moderate hepatic impairment control group which included nine subjects. All subjects received the trial drug and completed the trial; however, one subject in the moderate hepatic impairment control group was replaced due to elevated cholesterol constituting dyslipidaemia of clinical relevance. Therefore, the data for the original subject were excluded from the PK results, but not from the safety data set.
Demographic characteristics and Child–Pugh classifications were balanced across the trial groups (Table 1). All subjects with hepatic impairment had liver cirrhosis. Of those subjects with moderate hepatic impairment, 4/8 (50%) subjects tested positive for hepatitis C and the remaining subjects had a history of alcohol abuse. Of those subjects with severe hepatic impairment, all had a history of alcohol abuse and 3/8 (38%) tested positive for hepatitis C.
Table 1.
Baseline demographic characteristics and Child–Pugh classifications for the groups with hepatic impairment and matched healthy subjects
| Moderate hepatic impairment (n = 8) | Healthy (moderate impairment matched) controls (n = 9) | Severe hepatic impairment (n = 8) | Healthy (severe impairment matched) controls (n = 8) | |
|---|---|---|---|---|
| Female, n (%) | 8 (100) | 9 (100) | 8 (100) | 8 (100) |
| Age, years, mean ± SD | 52.5 ± 7.0 | 52.4 ± 7.6 | 52.9 ± 9.8 | 52.4 ± 8.9 |
| BMI, kg m −2, mean ± SD | 24.8 ± 4.6 | 26.0 ± 3.8 | 27.6 ± 3.5 | 27.6 ± 2.4 |
| Child–Pugh classification a | ||||
| Total score, mean ± SD | 7.3 ± 0.5 | – | 10.4 ± 0.7 | – |
| Liver disease, n (%) | ||||
| Class B | 8 (100) | – | – | – |
| Class C | – | – | 8 (100) | – |
BMI, body mass index; INR, international normalised ratio; SD, standard deviation.
Assignment to Child–Pugh class is based on the sum of points based on the following criteria:
• Hepatic encephalopathy: 1 = Absent / 2 = Grade 1 or 2 / 3 = Grade 3 or 4
• Ascites: 1 = Absent / 2 = Mild / 3 = Moderate
• Bilirubin: 1 = <34 / 2 = 34 to 51 / 3 = >51 μmol l−1
• Albumin: 1 = >3.5 / 2 = 2.8 to 3.5 / 3 = <2.8 g dl−1
• Prothrombin time: 1 = <1.7 / 2 = 1.7 to 2.3 / 3 = >2.3 INR
→ Child–Pugh A (5–6 points); Child–Pugh B (7–9 points); Child–Pugh C (≥10 points)
Plasma PK of letermovir
Steady‐state conditions were generally achieved in all groups prior to full PK blood sampling on Day 8.
In all four groups, mean total letermovir plasma concentrations started to increase immediately after dosing, reaching a maximal plasma concentration about 1.5–2.5 h after dosing and decreasing thereafter (Figure 1). Exposure was higher in subjects with hepatic impairment than in healthy subjects at most time points, with the difference most pronounced in subjects with severe hepatic impairment.
Figure 1.
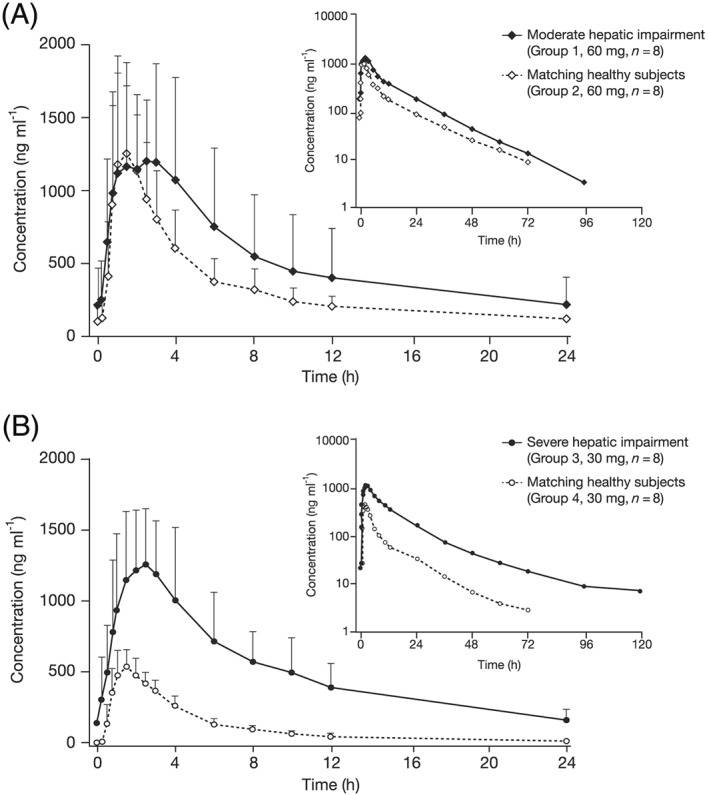
Mean letermovir plasma concentration‐time profiles at steady state for (a) subjects with moderate hepatic impairment versus matched healthy subjects (letermovir 60 mg ± 90% confidence intervals, inset: semi‐logarithmic scale) and (b) subjects with severe hepatic impairment versus matched healthy subjects (letermovir 30 mg ± 90% confidence intervals, inset: semi‐logarithmic scale)
As a proportion of the total letermovir plasma concentration, the mean unbound fraction was 1.09% vs. 0.95% in the moderate impairment vs. matched control group, and 1.41% vs. 0.99% in the severe impairment vs. matched control group, indicating somewhat higher unbound fractions with an increasing degree of hepatic impairment.
PK parameters for letermovir (total and unbound) are shown in Tables 2 and 3. Css,max and AUCτ,ss for total letermovir were 1.37‐fold (90% confidence interval: 0.87, 2.17) and 1.59‐fold higher (0.98, 2.57), respectively, in subjects with moderate hepatic impairment compared with matched healthy controls (Table 2). Css,max and AUCτ,ss for total letermovir were 2.34‐fold (1.91, 2.88) and 3.82‐fold (2.94, 4.97) higher, respectively, in subjects with severe hepatic impairment compared with matched healthy controls (Table 2); this was a statistically significant increase.
Table 2.
Plasma PK parameters of total letermovir at steady state (Day 8) in subjects with moderate and severe hepatic impairment and matched healthy control subjects
| Moderate hepatic impairment (n = 8) | Healthy (moderate impairment matched) controls (n = 8) | Severe hepatic impairment (n = 8a) | Healthy (severe impairment matched) controls (n = 8a) | |||||
|---|---|---|---|---|---|---|---|---|
| AUC τ,ss, ng.h ml−1 | 11 960 ± 8164 | 7121 ± 3310 | 10 863 ± 3986 | 2732 ± 525 | ||||
| C ss,max, ng ml −1 | 1687.0 ± 592.4 | 1361.0 ± 643.3 | 1206.0 ± 326.8 | 511.5 ± 111.7 | ||||
| C min, ng ml −1 | 140.60 ± 198.30 | 67.44 ± 47.55 | 135.3 ± 55.97 | 19.03 ± 8.44 | ||||
| t max, h | 2.00 (0.75–4.00) | 1.50 (1.00–2.50) | 2.00 (1.00–4.00) | 1.50 (1.00–2.50) | ||||
| t 1/2z, h | 13.23 ± 2.97 | 14.71 ± 5.10 | 19.56 ± 7.04 | 13.84 ± 5.79 | ||||
| C L/F, l h −1 | 6.695 ± 3.21 | 10.56 ± 5.99 | 3.094 ± 1.07 | 11.43 ± 2.75 | ||||
| V ss/F, l | 127.20 ± 69.09 | 241.30 ± 221.10 | 74.79 ± 28.08 | 233.40 ± 95.57 | ||||
| LS means | LS means | |||||||
|---|---|---|---|---|---|---|---|---|
| Moderate hepatic impairment | Matched healthy subjects | LS means ratio % (90% CI) | P value | Severe hepatic impairment | Matched healthy subjects | LS means ratio % (90% CI) | P valueb | |
| AUC τ,ss, ng.h ml −1 | 10 178 | 6411 | 158.8 (98.21–256.7) | 0.112 | 10 251 | 2682 | 382.2 (294.0–496.9) | <0.0001 |
| C ss,max, ng ml −1 | 1608 | 1174 | 137.0 (86.58–216.6) | 0.247 | 1173 | 500.8 | 382.2 (294.0–496.9) | <0.0001 |
All data are mean ± standard deviation except tmax, which is median (range); λz, apparent elimination rate constant; AUCτ,ss, area under the concentration vs. time curve over a dosing interval at steady state; CL/F, apparent oral clearance; Cmin, minimal observed concentration; Css,av, average steady‐state concentration over the dosing interval; Css,max, maximal observed concentration at steady state; CI, confidence interval; LS, least square; PK, pharmacokinetic; SD, standard deviation; t½,z, apparent elimination half‐life; tmax, time to reach the maximal observed concentration; Vss/F, apparent volume of distribution at steady state.
n = 6 for λz, t1/2z and Vss/F.
P values were calculated using an analysis of variance model with the logarithm of PK parameters as dependent variable and hepatic status as fixed effect.
Table 3.
Plasma PK parameters of unbound letermovir at steady state in subjects with moderate and severe hepatic impairment and matched healthy control subjects
| Moderate hepatic impairment (n = 8) | Healthy (moderate impairment matched) controls (n = 8) | Severe hepatic impairment (n = 8a) | Healthy (severe impairment matched) controls (n = 8a) | |||||
|---|---|---|---|---|---|---|---|---|
| AUC τ,ss, ng.h ml −1 | 141.30 ± 125.30 | 66.93 ± 31.91 | 155.40 ± 94.91 | 26.58 ± 6.61 | ||||
| C ss,av, ng ml −1 | 5.885 ± 5.217 | 2.79 ± 1.33 | 6.47 ± 3.95 | 1.11 ± 0.28 | ||||
| C ss,max, ng ml −1 | 18.86 ± 9.548 | 12.93 ± 6.64 | 17.67 ± 10.67 | 5.05 ± 1.62 | ||||
| C min, ng ml −1 | 1.79 ± 2.83 | 0.63 ± 0.43 | 1.84 ± 0.79 | 0.19 ± 0.11 | ||||
| LS means | LS means | |||||||
|---|---|---|---|---|---|---|---|---|
| Moderate hepatic impairment | Matched healthy subjects | LS means ratio % (90% CI) | P valueb | Severe hepatic impairment | Matched healthy subjects | LS means ratio % (90% CI) | P valueb | |
| AUC τ,ss, ng.h ml −1 | 108.3 | 59.72 | 181.4 (102.8–320.0) | 0.0860 | 138.8 | 25.88 | 536.2 (386.3–744.4) | <0.0001 |
| C ss,max, ng ml −1 | 17.11 | 10.94 | 156.4 (92.96–263.3) | 0.1521 | 15.88 | 4.831 | 328.7 (233.2–463.2) | <0.0001 |
AUCτ,ss, area under the concentration vs. time curve over a dosing interval at steady state; Cmin, minimal observed concentration; Css,av, average steady‐state concentration over the dosing interval; Css,max, maximal observed concentration at steady state; CI, confidence interval; LS, least square; PK, pharmacokinetic
Dosing interval (τ) = 24 h. All data are mean ± standard deviation.
n = 6 for λz, t1/2z and Vss/F.
P values were calculated using an analysis of variance model with the logarithm of PK parameters as dependent variable and hepatic status as fixed effect.
For unbound letermovir, Css,max and AUCτ,ss were 1.57‐ and 1.81‐fold higher, respectively, in subjects with moderate hepatic impairment compared with controls (Table 3). Css,max and AUCτ,ss for unbound letermovir were 3.29‐ and 5.36‐fold higher, respectively, in subjects with severe hepatic impairment compared with controls (Table 3).
Subjects with moderate hepatic impairment demonstrated geometric mean letermovir glucuronide/letermovir ratios at 1.5 h and 6 h post‐dose of 6.3% and 8.7%, respectively, vs. 7.7% and 6.7%, respectively, in matched healthy subjects.
Severe hepatic impairment geometric mean letermovir glucuronide/letermovir ratios at 1.5 h and 6 h post‐dose were 10.1% and 14.8%, respectively, vs. 3.5% and 3.2%, respectively, in the matched healthy individuals.
In some cases, the letermovir concentration after hydrolysis was lower than the free letermovir concentration (more frequently observed for the groups with severe impairment and their matched controls than for the groups with moderate impairment and their matched controls); then the calculated letermovir concentrations originating from glucuronides were set to ‘0’ and no letermovir glucuronide/letermovir ratios were calculated.
Safety
Treatment‐emergent AEs occurred in two subjects with moderate hepatic impairment, two healthy subjects in the matched control group, three subjects with severe hepatic impairment and one healthy subject in the matched control group. There were no deaths, serious AEs or discontinuations due to AEs during the trial. Three cases of headache (moderate) in the moderate impairment and the matched control group and one case of skin rash (mild) in the moderate impairment group were considered by investigators to be related to trial treatment. There were no treatment‐related AEs in subjects with severe hepatic impairment. Any clinically relevant changes in laboratory parameters, vital signs, ECG or physical examination observations were determined to be related to the underlying conditions and not deemed to be treatment related.
Discussion
Letermovir is an anti‐HCMV drug with a novel mechanism of action (terminase inhibition) that is primarily eliminated unchanged via biliary excretion into the faeces. Hepatic impairment appeared to increase exposure to total and unbound letermovir following multiple oral daily dosing in this Phase 1 trial in subjects with moderate and severe hepatic impairment, compared with matching healthy subjects. The effect was more pronounced in subjects with severe hepatic impairment who received a lower 30‐mg daily letermovir dose; increases in total letermovir concentrations by up to 3.82‐fold for AUC and 2.34‐fold for maximum observed concentration were found in subjects with severe hepatic impairment as compared with matched healthy controls. This effect was further pronounced for unbound letermovir in subjects with severe hepatic impairment compared with healthy individuals. Increased exposure may be linked with compromised biliary efflux transporters in subjects with hepatic impairment, but this hypothesis requires further investigation. Although higher exposure to letermovir was reported for subjects with hepatic impairment, this was not accompanied by clinical findings (e.g. any increased AE rates) potentially due to the decreased dose applied in the severely impaired subjects.
A somewhat higher (approximately 40%) mean unbound fraction has been found in patients with severe hepatic impairment compared with healthy subjects. Since the pharmacologically active fraction is increased, this may have an influence on efficacy and safety in patients. However, these data were generated from a relatively small sample size and it has, thus, to be further evaluated in future patient trials.
This trial supports biliary excretion being the primary route of letermovir clearance as identified in previous studies (data on file). It is known that letermovir is not cleared by the CYP450 isoenzymic system (data on file) and thus it may be the case that decreased glucuronidation would be expected to lead to increased systemic exposure to letermovir as CYP‐mediated clearance would not occur. The trial indicates that glucuronidation is not a major route of elimination for letermovir and as such diminished glucuronidation capacity in subjects with hepatic impairment will have a minimal impact on letermovir overall clearance. Even in patients with severe hepatic impairment, the capacity for glucuronidation of letermovir was maintained. The geometric means of the letermovir glucuronide/letermovir ratios were comparable for subjects with moderate hepatic impairment and their healthy controls. Furthermore, letermovir glucuronide/letermovir geometric means ratios were in a similar range in subjects with severe impairment to the values for moderate impairment and the moderate impairment matched healthy controls. However, the same geometric means ratios were lower in the severe impairment matched healthy controls compared with the subjects with severe hepatic impairment. Regardless, the presence of glucuronides of letermovir shows that glucuronidation is a functional route of clearance in subjects with both moderate and severe hepatic impairment, but it is not a major route of letermovir elimination.
In some cases, the letermovir concentration after hydrolysis of the O‐acylglucuronide was lower than the free letermovir concentration. This was more frequently observed for the groups with severe impairment and their matched controls than for the groups with moderate impairment and their matched controls. The reason for this discrepancy was not apparent. However, it cannot be discounted that letermovir glucuronide is a composite of isomeric ester O‐glucuronides, which may be demonstrating differing stabilities under the hydrolysis conditions. As such, the concentrations of letermovir posthydrolysis must be interpreted with caution.
When interpreting the results of this trial in future clinical settings, consideration should also be given to the limitations of the small sample sizes involved and the need to confirm findings in transplant recipients on concomitant immunosuppressive drugs. In addition, the doses administered in this trial were lower than the therapeutic doses (240/480 mg) that have been evaluated in the Phase 3 trial of letermovir vs. placebo for the prevention of HCMV following haematopoietic stem cell transplant (Clinicaltrials.gov: NCT02137772). Further investigations of differing doses of letermovir in subjects with hepatic impairment are warranted before drawing definitive conclusions about the need for potential dose reductions in these patients.
Conclusions
Increases in total letermovir AUC of approximately 1.6‐fold and Css,max of approximately 1.4‐fold compared with matched healthy individuals were observed in subjects with moderate hepatic impairment after administration of multiple doses of letermovir 60 mg once daily for 8 days. Letermovir AUC and Css,max were increased approximately 3.8‐ and 2.4‐fold in patients with severe hepatic impairment receiving letermovir 30 mg once daily for 8 days compared with matched healthy individuals. Letermovir was generally well‐tolerated in subjects with hepatic impairment, although the doses used in this study were lower than evaluated therapeutic doses. Further investigations are required to generate definitive conclusions about letermovir dose adjustments in patients with hepatic impairment.
Competing Interests
Funding for this research was provided by AiCuris Anti‐infective Cures GmBH. D.K., D.M., K.E.‐Z., H.‐P.S., H.Z. and H.R.‐S. are current or former employees of AiCuris Anti‐infective Cures GmBH. V.S.M. and Z.D.K. have nothing to disclose.
Medical writing assistance was provided by Nicole Jones for Complete Medical Communications, Hackensack, NJ, USA. This assistance was funded by Merck & Co., Inc., Kenilworth, NJ, USA.
Contributors
D.K. contributed to study design, medical responsibility during conduct, data evaluation and interpretation, writing and reviewing the manuscript. D.M. contributed to study design, method validation, writing and reviewing the manuscript. K.E.‐Z. contributed to study design, data analysis, data interpretation, writing and review of the manuscript. V.M. contributed to data acquisition and interpretation, writing and reviewing the manuscript. Z.K. contributed to data acquisition and interpretation, writing and reviewing the manuscript. H.‐P.S. contributed to study design, data analysis, data interpretation, writing and review of the manuscript. H.Z. contributed to study design, data analysis, data interpretation, writing and review of the manuscript. H.R.‐S. contributed to study design, data analysis, data interpretation, writing and review of the manuscript.
Kropeit, D. , McCormick, D. , Erb‐Zohar, K. , Moiseev, V. S. , Kobalava, Z. D. , Stobernack, H.‐P. , Zimmermann, H. , and Rübsamen‐Schaeff, H. (2017) Pharmacokinetics and safety of the anti‐human cytomegalovirus drug letermovir in subjects with hepatic impairment. Br J Clin Pharmacol, 83: 2678–2686. doi: 10.1111/bcp.13376.
References
- 1. Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep 2012; 14: 633–641. [DOI] [PubMed] [Google Scholar]
- 2. Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Infect Dis Clin North Am 2010; 24: 319–337. [DOI] [PubMed] [Google Scholar]
- 3. Melendez DP, Razonable RR. Letermovir and inhibitors of the terminase complex: a promising new class of investigational antiviral drugs against human cytomegalovirus. Infect Drug Resist 2015; 8: 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jacobsen T, Sifontis N. Drug interactions and toxicities associated with the antiviral management of cytomegalovirus infection. Am J Health Syst Pharm 2010; 67: 1417–1425. [DOI] [PubMed] [Google Scholar]
- 5. Salzberger B, Bowden RA, Hackman RC, Davis C, Boeckh M. Neutropenia in allogeneic marrow transplant recipients receiving ganciclovir for prevention of cytomegalovirus disease: risk factors and outcome. Blood 1997; 90: 2502–2508. [PubMed] [Google Scholar]
- 6. Buerger I, Reefschlaeger J, Bender W, Eckenberg P, Popp A, Weber O, et al A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J Virol 2001; 75: 9077–9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reefschlaeger J, Bender W, Hallenberger S, Weber O, Eckenberg P, Goldmann S, et al Novel non‐nucleoside inhibitors of cytomegaloviruses (BAY 38‐4766): in vitro and in vivo antiviral activity and mechanism of action. J Antimicrob Chemother 2001; 48: 757–767. [DOI] [PubMed] [Google Scholar]
- 8. Zimmermann H, Lischka P, Ruebsamen‐Schaeff H. Letermovir (AIC246) a novel drug under development for prevention and treatment of cytomegalovirus infections acting via a novel mechanism of action. Eur Infect Dis 2011; 5: 112–114. [Google Scholar]
- 9. Sullivan V, Talarico CL, Stanat SC, Davis M, Coen DM, Biron KK. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus‐infected cells. Nature 1992; 359: 85. [DOI] [PubMed] [Google Scholar]
- 10. Chemaly RF, Ullmann AJ, Stoelben S, Richard MP, Bornhäuser M, Groth C, et al Letermovir for cytomegalovirus prophylaxis in hematopoietic‐cell transplantation. N Engl J Med 2014; 370: 1781–1789. [DOI] [PubMed] [Google Scholar]
- 11. Stoelben S, Arns W, Renders L, Hummel J, Muhlfeld A, Stangl M, et al Preemptive treatment of cytomegalovirus infection in kidney transplant recipients with letermovir: results of a phase 2a study. Transpl Int 2014; 27: 77–86. [DOI] [PubMed] [Google Scholar]
- 12. Marty FM, Ljungman PT, Chemaly RF, Maertens JA, Snydman DR, Duarte RF, et al A phase III randomized, double‐blind, placebo‐controlled trial of letermovir (LET) for prevention of cytomegalovirus (CMV) infection in adult CMV‐seropositive recipients of allogeneic hematopoietic cell transplantation (HCT). Paper presented at the BMT Tandem Meetings 2017, 2017.
- 13. Kaul DR, Stoelben S, Cober E, Ojo T, Sandusky E, Lischka P, et al First report of successful treatment of multidrug‐resistant cytomegalovirus disease with the novel anti‐CMV compound AIC246. Am J Transplant 2011; 11: 1079–1084. [DOI] [PubMed] [Google Scholar]
- 14. European Medicines Agency . Evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000370.jsp. Updated 2005 (last accessed 22 May 2017).
- 15. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 2015; 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]