

Abstract
Aims
Based on in vitro data, there is evidence to suggest that cytochrome P450 (CYP) 2C8 is involved in the metabolism of selexipag and its active metabolite, ACT‐333679. The present study evaluated the possible pharmacokinetic interactions of selexipag with gemfibrozil, a strong CYP2C8 inhibitor, and rifampicin, an inducer of CYP2C8.
Methods
The study consisted of two independent parts, each conducted according to an open‐label, randomized, crossover design. The pharmacokinetics and safety of selexipag and ACT‐333679 were studied following single‐dose administration either alone or in the presence of multiple‐dose gemfibrozil (part I) or rifampicin (part II) in healthy male subjects.
Results
Gemfibrozil had comparatively small effects on selexipag (less than 2‐fold difference in any pharmacokinetic variable) but, with respect to ACT‐333679, increased the maximum plasma concentration (Cmax) 3.6‐fold [90% confidence interval (CI) 3.1, 4.3] and the area under the plasma concentration–time curve from zero to infinity (AUC0–∞) 11.1‐fold (90% CI 9.2, 13.4). The marked increased exposure to ACT‐333679, which mediates the majority of the pharmacological activity of selexipag, was accompanied by significantly more adverse events such as headache, nausea and vomiting. Coadministration of rifampicin increased the Cmax of selexipag 1.8‐fold (90% CI 1.4, 2.2) and its AUC0–∞ 1.3‐fold (90% CI 1.1, 1.4); its effects on ACT‐333679 were to increase its Cmax 1.3‐fold (90% CI 1.1, 1.6), shorten its half‐life by 63% and reduce its AUC0–∞ by half (90% CI 0.45, 0.59).
Conclusion
Concomitant administration of selexipag and strong inhibitors of CYP2C8 must be avoided, whereas when coadministered with inducers of CYP2C8, dose adjustments of selexipag should be envisaged.
Keywords: CYP2C8, drug interactions, gemfibrozil, pharmacokinetics, rifampicin, selexipag
What is Already Known about this Subject
Selexipag, a prostacyclin receptor agonist marketed for the treatment of pulmonary arterial hypertension, is enzymatically hydrolysed to form the active metabolite, ACT‐333679. Based on in vitro data, both compounds have been found to be substrates of cytochrome P450 (CYP) 2C8 and CYP3A4.
Selexipag and ACT‐333679 are substrates of organic anion‐transporting polypeptide (OATP) 1B1 and OATP1B3. In addition, selexipag is a substrate of P‐glycoprotein, and its active metabolite is a substrate of breast cancer resistance protein.
What this Study Adds
Concomitant administration of selexipag and gemfibrozil resulted in a marked increase in the exposure to ACT‐333679, associated with a significant increase in the incidence and/or intensity of typical prostacyclin‐related adverse events. Concomitant administration of selexipag and strong inhibitors of CYP2C8 must be avoided.
Concomitant administration of rifampicin decreased exposure to ACT‐333679. The clinical efficacy of selexipag is mediated mainly by ACT‐333679 and, therefore, an increase in the dose of selexipag should be considered when administered concomitantly with CYP2C8 inducers.
Introduction
Pulmonary arterial hypertension (PAH) is a disease characterized by a progressive increase in pulmonary artery pressure and pulmonary vascular resistance, ultimately leading to right heart failure and death 1, 2. Epoprostenol, a prostacyclin receptor agonist, was the first drug to be approved for the treatment of PAH. Therapy with epoprostenol requires a chronic indwelling catheter, reconstitution of the drug and operation of an infusion pump, and carries the risk of serious bacteraemia 3, 4. Therefore, other prostanoids have been developed, such as treprostinil for continuous subcutaneous infusion 5, inhalation 6 and oral administration 7; iloprost for inhalation 8 and beraprost for oral administration 9. The short half‐life and poor oral bioavailability of the prostanoids led to the search for other compounds acting on the prostacyclin receptor. Selexipag (NS‐304, ACT‐293987) was the first orally active, selective, long‐acting, nonprostanoid prostacyclin receptor agonist approved for the treatment of PAH 10, 11, 12, 13, 14.
Selexipag is hydrolysed by carboxylesterases to its active metabolite, ACT‐333679. Both selexipag and ACT‐333679 bind selectively and with high affinity to the prostacyclin receptor 10. In vitro, ACT‐333679 is approximately 37‐fold more potent than selexipag in activating the human prostacyclin receptor and is present at 3‐ to 4‐fold higher levels than the parent drug at steady state in humans after oral selexipag administration 15. Therefore, ACT‐333679 is the major contributor to the drug effect in humans. The structures of both compounds have been published previously 16. An absolute bioavailability study in healthy subjects showed that the clearance of selexipag is 17.9 l h–1, the volume of distribution 11.7 l and the absolute bioavailability 49.4% 17. Following single‐ and multiple‐dose oral administration of selexipag to healthy subjects, the pharmacokinetics (PK) of selexipag and ACT‐333679 were dose proportional 16, 18. The time to maximum concentration was within 2.5 h and 4 h for selexipag and ACT‐333679, respectively, and the apparent terminal half‐life (t½) varied from 0.8 h to 2.5 h for selexipag and from 6.2 h to 13.5 h for ACT‐333679 15. Steady‐state conditions are reached within 3 days and no relevant accumulation in plasma, either of parent compound or ACT‐333679, occurred at steady state 18.
In vitro experiments have shown that: (i) selexipag and ACT‐333679 undergo oxidative metabolism by the cytochrome P450 (CYP) enzymes, CYP2C8 and CYP3A4; (ii) UDP‐glucuronosyltransferase (UGT) enzymes, UGT1A3 and UGT2B7, are involved in the metabolism of ACT‐333679; (iii) both selexipag and ACT‐333679 are substrates of the organic anion‐transporting polypeptide (OATP) 1B1 and OATP1B3; and (iv) selexipag is a substrate of P‐glycoprotein (P‐gp), and its active metabolite is a substrate of breast cancer resistance protein 15. The present study was designed to investigate the effects of gemfibrozil, a strong inhibitor of CYP2C8 19, 20 and rifampicin, an inducer of CYP2C8 21, 22, on the PK of selexipag and ACT‐333679 in healthy male subjects.
Methods
Subject eligibility
Subjects were eligible to participate in the study if they were male, between 18 and 55 years of age and with a body mass index (BMI) between 18.0 kg m−2 and 28.0 kg m−2. All subjects were healthy, based on examinations performed during the screening visit, with no history of significant disease and not taking any medications, including over‐the‐counter drugs and herbal medicines such as St John's wort. Further, subjects were not enrolled if they were allergic/hypersensitive to study drugs or were otherwise judged to be unsuitable to participate in the study. Prior to any study‐related procedures, all subjects signed the informed consent form after receiving a full explanation of the study.
Clinical study design
The clinical study was approved by the Ethikkommission II, Bismarckallee 8–12, 23795 Bad Segeberg, Germany, on 10 May 2016, reference number 055/16 II, and by the Federal Institute for Drugs and Medical Devices, the German health authorities. The study was conducted in accordance with the Declaration of Helsinki principles, International Council for Harmonization and Good Clinical Practice guidelines, and applicable regulations and laws.
The study consisted of two parts that were conducted according to an open‐label, randomized, two‐treatment, two‐way crossover design. In parts I and II, subjects received a single dose of 400 μg selexipag (UPTRAVI®, 200 μg film‐coated tablet, Actelion Pharmaceuticals Ltd) at least 30 min before a light breakfast either alone (treatment A) or concomitantly with gemfibrozil (GEVILON®, 600 mg film‐coated tablet, Pfizer Ltd) (treatment B, part I) or rifampicin (EREMFAT®, 600 mg film‐coated tablet, RIEMSER Pharma GmbH) (treatment B, part II). During treatment B in part I, subjects received 600 mg gemfibrozil twice daily on days 1 to 9 in the morning and evening, at least 30 min prior to breakfast and dinner. On the morning of day 4, selexipag 400 μg was administered concomitantly. In part II, once‐daily 600 mg rifampicin was administered at least 30 min before a light breakfast on days 1 to 9, with concomitant selexipag 400 μg on the morning of day 7.
Serial blood samples for the analysis of selexipag and ACT‐333679 were collected in lithium–heparin tubes for 72 h or 144 h (treatment B, part I only) after administration of selexipag. In order to prevent degradation of selexipag and ACT‐333679 in the plasma, exposure to light was minimized and sample preparation was conducted under yellow light.
Based on standard deviations (SDs) observed for area under the plasma concentration–time curve from zero to infinity (AUC0–∞) and maximum observed plasma concentration (Cmax) in a previous study 23, a sample size of 16, and assuming a geometric mean ratio of 1.00, it was estimated that the 90% confidence interval (CI) of the geometric mean ratio (treatment B/treatment A) would be 0.64 to 1.56 for the AUC0–∞ and 0.62 to 1.62 for the Cmax of selexipag, and 0.68 to 1.47 and 0.73 to 1.36 for the AUC0–∞ and Cmax, respectively, of ACT‐333679. In order to compensate for any potential dropouts, in each part of the study 20 subjects were enrolled.
Analysis of selexipag and ACT‐333679 concentrations in plasma
The concentrations of selexipag and its active metabolite in the plasma were measured using a previously described validated liquid chromatography method with tandem mass spectrometry (LC–MS/MS) 23 with slight modifications. To 50 μl of plasma, 200 μl of internal standard (stable isotope‐labelled selexipag and ACT‐333679) solution was added. After vortex mixing and centrifugation, the supernatant was evaporated under nitrogen and the residue reconstituted in 100 μl acetonitrile/water (1:1 v/v), vortex mixed, and 20 μl of the supernatant was injected onto the LC–MS/MS. The chromatographic system consisted of a pump, autosampler, degasser and analytical column (Kinetex C18, 50 × 3.0 mm ID, 2.6 μm (Phenomenex, Brechbühler, Schlieren, Switzerland). Mobile phases consisted of water containing 0.1% formic acid (v/v) and acetonitrile containing 0.1% formic acid (v/v). Mass spectrometric detection (API 5000, AB Sciex, Brugg, Switzerland) was performed with a turbo ion spray operating in positive‐ion mode at 600°C.
For both analytes, the lower limit of quantification (LLOQ) was 0.01 ng ml−1 and the method was linear from 0.01 ng ml−1 up to 20.0 ng ml−1. The concentrations of the analytes were calculated by the internal standardization method – i.e. using the peak area ratio of analyte to internal standard.
The performance of the method was monitored using quality control samples of known concentrations. Precision [percentage coefficient of variation (% CV)] of the assay was ≤5.6% for selexipag and ≤6.7% for ACT‐333679. The interassay accuracy ranged from −3.2% to 1.8% for selexipag and from −2.1% to 3.8% for ACT‐333679.
PK calculations
Noncompartmental analysis of selexipag and ACT‐333679 plasma concentrations was performed using Phoenix WinNonlin (version 6.4, Pharsight Corporation, Mountain View, CA, USA). The measured individual plasma concentrations of selexipag and ACT‐333679 were used to obtain Cmax and the time to reach Cmax (tmax) directly. AUC from zero until the last quantifiable concentration (AUC0–t) was calculated according to the linear trapezoidal rule, using the measured concentration–time values above the LLOQ. AUC0–∞ was calculated by combining AUC0–t and AUCextra. AUCextra represents an extrapolated value obtained by Ct/λz, where Ct is the last plasma concentration measured above the LLOQ and λz represents the terminal elimination rate constant determined by log‐linear regression analysis of the measured plasma concentrations of the terminal elimination phase. The t½ of selexipag and its metabolite was calculated as: ln 2/λz.
Statistical analysis of PK variables
PK variables were analysed, providing geometric means and corresponding 95% CIs for AUC, Cmax and t½, whereas the median and range are shown for tmax.
The effects of gemfibrozil and rifampicin on the AUC, Cmax and t½ of selexipag and ACT‐333679 were explored using the ratio of the geometric means and 90% CI of selexipag plus gemfibrozil or rifampicin (treatment B) as the test treatment vs. selexipag alone (treatment A) as the reference treatment. The log‐transformed values were analysed by mixed‐effects models, including treatment, sequence and period as fixed effects and subject as random effect. Differences between treatments for tmax were explored using the Wilcoxon signed‐rank test, providing the median differences and corresponding 90% CIs.
Safety
The safety of the subjects was monitored by the recording of vital signs [supine systolic and diastolic blood pressure (SBP, DBP) and pulse rate] and 12‐lead electrocardiogram (ECG), physical examination, clinical chemistry and haematology tests and adverse events (AEs) reporting. Safety results were analysed descriptively by treatment. For this, in each part, treatment B was divided into treatment B1, corresponding to gemfibrozil or rifampicin alone – i.e. day 1 to the morning of day 4 (gemfibrozil) or day 7 (rifampicin), and treatment B2, corresponding to gemfibrozil or rifampicin plus selexipag.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 24, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 25, 26.
Results
Gemfibrozil
All 20 enrolled subjects completed part I of the study as per protocol and were evaluable for PK. Of the enrolled subjects, 18 were Caucasian and two Asian. The mean age was 28.8 years (range 18–54 years) and mean BMI was 23.82 kg m−2 (range 18.1–27.9 kg m−2).
Single‐dose mean selexipag plasma concentrations increased when coadministered with gemfibrozil (Figure 1). Based on geometric mean ratios, the increase was 1.4‐fold (90% CI 1.1, 1.7) and 2.0‐fold (90% CI 1.7, 2.3) for Cmax and AUC0–∞, respectively. No effect on tmax was observed but t½ was 50% longer in the presence of gemfibrozil (Table 1). When compared with selexipag, the gemfibrozil‐induced increase in the plasma concentrations of the active metabolite, ACT‐333679, was more pronounced (Figure 1). Together, a 3.6‐fold (90% CI 3.1, 4.3) increase in Cmax and a doubling of t½ resulted in an 11.1‐fold (90% CI 9.2, 13.4) increase in the AUC0–∞ for ACT‐333679. Coadministration of gemfibrozil also delayed attainment of peak ACT‐333679 concentrations by 3.5 h (Table 1). A graph showing individual changes in the AUC0–∞ for selexipag and ACT‐333679 for the two treatments is presented in Figure 2.
Figure 1.
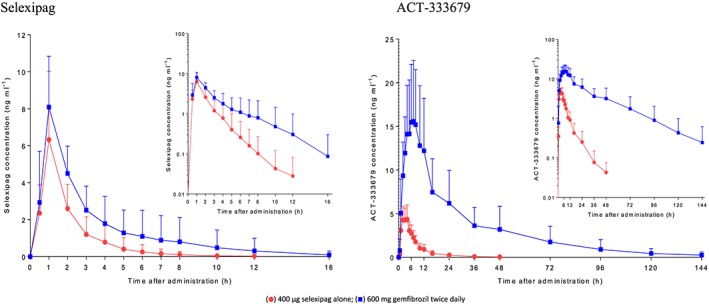
Arithmetic mean (± standard deviation) plasma concentration–time profile of selexipag and ACT‐333679 after administration of 400 μg selexipag alone and with 600 mg gemfibrozil twice daily; n = 20
Table 1.
Summary of the pharmacokinetic (PK) variables of selexipag and its active metabolite, ACT‐333679, after administration of 400 μg selexipag alone (treatment A) and with 600 mg gemfibrozil twice daily (treatment B), and geometric mean ratios for the comparison of treatments (n = 20)
| Treatment A | Treatment B | Geometric mean ratio Treatment B/A | |
|---|---|---|---|
| Selexipag | |||
| C max [ng ml −1 ] | 5.57 | 7.71 | 1.39 |
| 4.40, 7.04 | 6.49, 9.17 | 1.12, 1.72 | |
| t max [h] | 1.00 | 1.00 | 0.00 |
| 0.50, 1.00 | 0.50, 2.00 | 0.00, 0.01 | |
| AUC 0–∞ [ng*h ml −1 ] | 10.47 | 20.47 | 1.96 |
| 8.40, 13.05 | 16.17, 25.92 | 1.65, 2.32 | |
| t ½ [h] | 1.66 | 2.50 | 1.51 |
| 1.34, 2.05 | 2.12, 2.96 | 1.21, 1.87 | |
| ACT‐333679 | |||
| C max [ng ml −1 ] | 4.74 | 17.21 | 3.63 |
| 4.07, 5.51 | 14.18, 20.89 | 3.06, 4.31 | |
| t max [h] | 3.02 | 7.02 | 3.54 |
| 1.00, 4.02 | 3.00, 12.00 | 2.56, 4.60 | |
| AUC 0‐∞ [ng*h ml −1 ] | 35.93 | 398.44 | 11.090 |
| 30.71, 42.04 | 303.72, 522.71 | 9.20, 13.36 | |
| t ½ [h] | 11.04 | 24.03 | 2.18 |
| 8.87, 13.73 | 21.12, 27.35 | 1.75, 2.72 | |
Treatment A: selexipag (400 μg single dose); treatment B: selexipag (400 μg single dose) + gemfibrozil (600 mg twice daily). Data for PK variables are presented as the geometric mean [95% confidence interval (CI)], and for tmax the median (range). Data for the ratio are presented as the geometric means and 90% CI, except for tmax, for which the median difference and 90% CI are presented. AUC0–∞, area under the plasma concentration–time curve from zero to infinity; Cmax, maximum plasma concentration; t½, apparent terminal half‐life; tmax, time to reach Cmax
Figure 2.
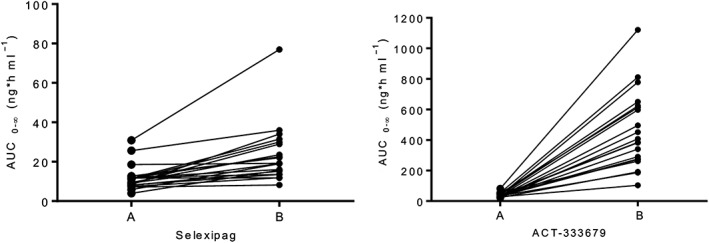
Individual area under the plasma concentration–time curve from zero to infinity (AUC0–∞) values (ng*h ml−1) of selexipag and ACT‐333679, after administration of 400 μg selexipag alone (A) and with 600 mg gemfibrozil twice daily (B); n = 20
Rifampicin
Nineteen out of 20 enrolled subjects completed part II of the study as per protocol and were included in the PK analysis. One subject withdrew informed consent during treatment B1 (rifamipicin treatment alone) after completing treatment A. At the time of withdrawing consent, the subject had ongoing AEs of a moderate hypoesthesia and a mild ear discomfort. Of the enrolled subjects, 19 were Caucasian and one African American. The mean age at screening was 35.7 years (range 18–54 years) and the mean BMI was 24.41 kg m−2 (range 18.4–27.9 kg m−2).
Single‐dose mean selexipag plasma concentrations increased when coadministered with rifampicin (Figure 3). Based on geometric mean ratios, the increase was 1.76‐fold (90% CI 1.44, 2.15) for Cmax and 1.25‐fold for AUC0–∞ (90% CI 1.11, 1.41). No effect on median tmax was observed but t½ was 34% shorter in the presence of rifampicin (Table 2). Figure 3 illustrates that, when compared with selexipag alone, the mean plasma concentrations of ACT‐333679 were initially slightly higher but from 2 h after administration onwards, the mean plasma concentrations of ACT‐333679 were lower and decreased below the LLOQ earlier. Although a 1.3‐fold (90% CI 1.1, 1.6) increase in the Cmax of ACT‐333679 was observed when compared with selexipag alone, total exposure to the metabolite (AUC0–∞) decreased by half (90% CI 0.45, 0.59). In the presence of rifampicin, the Cmax of the metabolite was attained 2 h earlier and the t½ was 63% shorter (Table 2). A graph with individual changes in the AUC0–∞ for selexipag and ACT‐333679 for the two treatments is shown in Figure 4.
Figure 3.
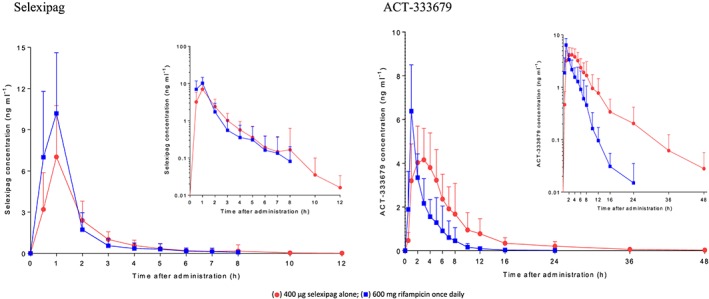
Arithmetic mean (± standard deviation) plasma concentration–time profile of selexipag and ACT‐333679 after administration of 400 μg selexipag alone and with 600 mg rifampicin once daily; n = 19
Table 2.
Summary of pharmacokinetic (PK) variables of selexipag and its active metabolite, ACT‐333679, after administration of 400 μg selexipag alone (treatment A) and with 600 mg rifampicin once daily (treatment B), and geometric mean ratios for the comparison of treatments (n = 19)
| Treatment A | Treatment B | Geometric mean ratio Treatment B/A | |
|---|---|---|---|
| Selexipag | |||
| C max [ng ml −1 ] | 6.30 | 11.14 | 1.76 |
| 5.04, 7.87 | 9.49, 13.08 | 1.44, 2.15 | |
| t max [h] | 1.00 | 1.00 | −0.01 |
| 0.95, 1.02 | 0.50, 1.00 | −0.24, 0.00 | |
| AUC 0–∞ [ng*h ml −1 ] | 10.98 | 13.83 | 1.25 |
| 9.05, 13.34 | 11.78, 16.24 | 1.11, 1.41 | |
| t ½ [h] | 1.93 | 1.27 | 0.66 |
| 1.46, 2.55 | 1.04, 1.54 | 0.52, 0.83 | |
| ACT‐333679 | |||
| C max [ng ml −1 ] | 4.74 | 6.18 | 1.30 |
| 3.99, 5.64 | 5.20, 7.35 | 1.07, 1.57 | |
| t max [h] | 3.00 | 1.00 | −2.00 |
| 0.98, 8.00 | 1.00, 6.00 | −2.50, −1.49 | |
| AUC 0‐∞ [ng*h ml −1 ] | 31.50 | 16.26 | 0.52 |
| 25.50, 38.92 | 13.65, 19.38 | 0.45, 0.59 | |
| t ½ [h] | 11.87 | 4.39 | 0.37 |
| 9.70, 14.54 | 3.54, 5.44 | 0.29, 0.469 | |
Treatment A: selexipag (400 μg single dose); treatment B selexipag (400 μg single dose) + rifampicin (600 mg once daily). Data for PK variables are presented as the geometric mean [95% confidence interval (CI)], and for tmax the median (range). Data for the ratio are presented as the geometric means and 90% CI, except for tmax, for which the median difference and 90% CI are presented. AUC0–∞, area under the plasma concentration–time curve from zero to infinity; Cmax, maximum plasma concentration; t½, apparent terminal half‐life; tmax, time to reach Cmax
Figure 4.
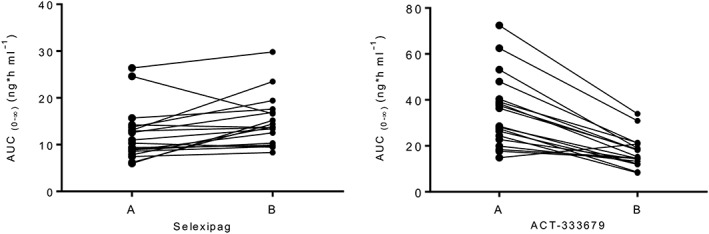
Individual area under the plasma concentration–time curve from zero to infinity (AUC0–∞) values (ng*h ml−1) of selexipag and ACT‐333679, after administration of 400 μg selexipag alone (A) and with 600 mg rifampicin once daily (B); n = 19
Safety
No serious AEs or AEs leading to study drug discontinuation were reported.
In part I, following administration of selexipag on day 1 in treatment A, the only frequently reported AE was headache (14 subjects, 70.0%). One subject reported headache during the days when gemfibrozil was administered alone (treatment B1). Following concomitant administration of selexipag and gemfibrozil on day 4 in treatment B2, headache was reported in 18/20 (90%) subjects. In treatment B2 (selexipag and gemfibrozil), all subjects experienced at least one AE, compared with 75% in treatment A (selexipag) and 15% in treatment B1 (gemfibrozil). No AEs of nausea and vomiting were reported following administration of selexipag or gemfibrozil alone. However, the majority of subjects treated with the combination of selexipag and gemfibrozil reported nausea (15/20 subjects, 75%) and vomiting (12/20 subjects, 60%) (Table 3). Most reported AEs were of mild or moderate intensity. Two subjects had severe intensity AEs (headache) during treatment B2. All but two AEs (dyspepsia and nasopharyngitis in treatment B2) were considered by the investigator to be treatment related. All reported AEs resolved by the end of the study.
Table 3.
Summary of adverse events (AEs) reported during part I by frequency
| Treatment A (n = 20) | Treatment B1 (n = 20) | Treatment B2 (n = 20) | Overall (n = 20) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Preferred term | AEs | n | % | AEs | n | % | AEs | n | % | AEs | n | % |
| Number of subjects with at least one AE | 15 | 75 | 3 | 15 | 20 | 100 | 20 | 100 | ||||
| Number of different AEs | 3 | 2 | 11 | 12 | ||||||||
| Total number of AEs | 17 | 3 | 74 | 94 | ||||||||
| Headache | 15 | 14 | 70 | 1 | 1 | 5 | 20 | 18 | 90 | 36 | 18 | 90 |
| Nausea | 18 | 15 | 75 | 18 | 15 | 75 | ||||||
| Vomiting | 23 | 12 | 60 | 23 | 12 | 60 | ||||||
| Pain in extremity | 1 | 1 | 5 | 4 | 3 | 15 | 5 | 3 | 15 | |||
| Diarrhoea | 2 | 2 | 10 | 1 | 1 | 5 | 3 | 3 | 15 | |||
| Myalgia | 2 | 2 | 10 | 2 | 2 | 10 | ||||||
| Pain in jaw | 2 | 2 | 10 | 2 | 2 | 10 | ||||||
| Presyncope | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Dyspepsia | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Arthralgia | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Neck pain | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Nasopharyngitis | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
Treatments: A = selexipag; B1 = gemfibrozil; B2 = gemfibrozil + selexipag. Data are expressed as AE (number of adverse events), n (number of subjects experiencing at least one AE) and % (percentage of subjects with at least one AE)
In part II, following administration of selexipag on day 1 in treatment A, the most frequently reported AE was headache (eight subjects, 40.0%). Other AEs included nausea and limb discomfort (two subjects each, 10.0%). Following concomitant administration of selexipag and rifampicin on day 7 in treatment B2, headache was the most frequently reported AE (five subjects, 26.3%) followed by nausea (two subjects, 10.5%). There was a tendency for fewer subjects reporting AEs after coadministration of rifampicin vs. selexipag alone (Table 4). All reported AEs were of mild or moderate intensity. All but two AEs (in treatment B1) were considered by the investigator to be treatment related.
Table 4.
Summary of adverse events (AEs) reported during part II by frequency
| Treatment A (n = 20) | Treatment B1 (n = 20) | Treatment B2 (n = 19) | Overall (n = 20) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Preferred term | AEs | n | % | AEs | n | % | AEs | n | % | AEs | n | % |
| Number of subjects with at least one AE | 8 | 40 | 4 | 20 | 6 | 31 | 12 | 60 | ||||
| Number of different AEs | 5 | 8 | 3 | 13 | ||||||||
| Total number of AEs | 16 | 8 | 9 | 33 | ||||||||
| Headache | 8 | 8 | 40 | 5 | 5 | 26.3 | 13 | 9 | 45 | |||
| Nausea | 2 | 2 | 10 | 2 | 2 | 10.5 | 4 | 4 | 20 | |||
| Vomiting | 3 | 1 | 5 | 2 | 1 | 5.3 | 5 | 2 | 10 | |||
| Limb discomfort | 2 | 2 | 10 | 2 | 2 | 10 | ||||||
| Hypoesthesia | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Somnolence | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Flatulence | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Back pain | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Pain in extremity | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Fatigue | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Peripheral oedema | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Ear discomfort | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
| Generalized pruritus | 1 | 1 | 5 | 1 | 1 | 5 | ||||||
Treatments: A = selexipag; B1 = rifampicin; B2 = rifampicin + selexipag. Data are expressed as AE (number of adverse events), n (number of subjects experiencing at least one AE) and % (percentage of subjects with at least one AE)
No clinically relevant mean changes from baseline in clinical laboratory variables, vital signs or ECG parameters were observed.
One subject randomized to treatment sequence B–A in part I had a vasovagal reaction on day 4 in treatment B2 (gemfibrozil + selexipag). The subject had an ongoing AE of headache at the time of the vasovagal reaction. The vital signs measured at the time of this reaction showed an SBP of 85 mmHg (predose on day 4: 122 mmHg), DBP of 59 mmHg (predose on day 4: 86 mmHg) and pulse rate of 46 bpm (predose on day 4: 56 bpm). The AE was mild in intensity and considered by the investigator to be treatment related. The AE resolved in 10 min without sequelae.
Discussion
Based on in vitro studies, the metabolism of selexipag is driven by initial hydrolysis to its active metabolite, ACT‐333679. In addition, both CYP2C8 and CYP3A4 are involved in the metabolism of selexipag and ACT‐333679. The metabolism of ACT‐333679 also involves UGT1A3 and ‐2B7 15.
The aim of the present study was to investigate the effect of gemfibrozil, a strong inhibitor of CYP2C8, (part I) and the effect of rifampicin, an inducer of CYP2C8, (part II) on the PK of selexipag and ACT‐333679. The study results provide important information on the contribution of the CYP2C8 pathway to the disposition of selexipag and its active metabolite, ACT‐333679.
The results of part I showed that gemfibrozil had a comparatively small effect on the exposure to selexipag (less than 2‐fold on any PK variable) but increased the AUC0–∞ of ACT‐333679 by 11‐fold.
The results of the drug–drug interaction (DDI) study with selexipag and lopinavir/ritonavir, a strong inhibitor of OATP1B1 and OATP1B3, CYP3A4 and P‐gp, showed a 2‐fold increase in exposure to selexipag in the presence of lopinavir/ritonavir 23. The nature of this increase (increase in Cmax and AUC but no significant change in t½) suggests that the interaction was the result of inhibition of OATP1B1, OATP1B3 or P‐gp rather than of CYP3A4. The increase in selexipag exposure (2.0‐fold increase in AUC, 1.4‐fold increase in Cmax, and 1.5‐fold increase in t½) in the presence of gemfibrozil could be explained by the inhibitory effect of gemfibrozil on CYP2C8 and also by its effect on OATP1B1, as gemfibrozil is also a moderate inhibitor of the latter 27.
The results of the DDI study with selexipag and lopinavir/ritonavir 23 showed only a minor change in Cmax and no change in the AUC for ACT‐333679, indicating the low dependency of ACT‐333679 on CYP3A4 enzymes and OATP transporters. Therefore, the pronounced effect of gemfibrozil on AUC, Cmax and t½ of ACT‐333679 is most likely due to the inhibition of CYP2C8 and indicates the importance of the CYP2C8 pathway in the disposition of ACT‐333679.
Strong in vivo interactions of gemfibrozil with CYP2C8 substrates are due to mechanism‐based irreversible inactivation of CYP2C8 by the gemfibrozil 1‐O‐β‐glucuronide metabolite. CYP2C8 activity can only be regained by de novo synthesis of the enzyme, and the full CYP2C8 activity recovers gradually within 3–4 days after cessation of gemfibrozil at the clinically used therapeutic dose of 600 mg twice daily 22, 28.
In part II of the study, differential effects of rifampicin were observed, with a small increase in total exposure (AUC) to selexipag and a decrease in the AUC of ACT‐333679. Inhibitory and inductive effects of rifampicin on the PK of the compounds have been also observed for other drugs such as bosentan 29 and repaglinide 30, 31.
Rifampicin is an inducer of CYP2C8 and UGT enzymes, a major inducer of CYP3A4 and a strong inhibitor of OATP1B1 and OATP1B3 32, 33. The 1.8‐fold increase in the Cmax of selexipag in the presence of rifampicin is likely to be mediated via inhibition of OATP1B1 and OATP1B3 by rifampicin. The shorter t½ of selexipag in the presence of rifampicin is indicative of CYP enzyme induction but the inhibitory effects of rifampicin outweighed the inductive effects and led to a small increase in the AUC for selexipag. In the presence of rifampicin, the t½ of ACT‐333679 was significantly shortened (by 63%), resulting in a 50% reduction in exposure (AUC) to ACT‐333679, which is likely to have been due to the inductive effect of rifampicin on CYP2C8. The initial small increase in the plasma concentrations of ACT‐333679 (Cmax), which was also observed after coadministration of lopinavir/ritonavir, is most likely mediated via inhibition of OATP1B1 and OATP1B3 by rifampicin. After cessation of rifampicin treatment, full CYP2C8 activity recovers gradually within 3–4 days 28.
In the present study, selexipag and rifampicin doses were taken simultaneously. The t½ of rifampicin at a dose of 600 mg is 2.5 h 34, whereas the t½ of CYP2C8 is 22–23 h in vitro 35, and in vivo 28, indicating that rifampicin‐induced inhibition dissipates more rapidly than rifampicin‐mediated CYP2C8 induction after discontinuation of rifampicin. Therefore, if rifampicin and selexipag are not taken simultaneously, the full effect of CYP2C8 induction on the PK of selexipag will most likely be larger than that observed in the present study. Taking into consideration the results of the DDI study with lopinavir/ritonavir 23, which showed a minor effect of OATP1B1 and OATP1B3 inhibition on the PK of ACT‐333679, the full inductive effect of rifampicin on the PK of ACT‐333679 (responsible for the majority of the drug effect) in such situations probably will not be markedly different from that observed in the present study.
In both parts I and II, the AEs reported in treatment A (selexipag) and treatment B2 (gemfibrozil or rifampicin + selexipag) were consistent with the known adverse drug reactions reported for selexipag (i.e. headache, diarrhoea, nausea, vomiting, jaw pain, myalgia, pain in the extremities, arthralgia and flushing) 15. However, the incidence and/or intensity of AEs reported after concomitant administration of selexipag and gemfibrozil were significantly higher than those following the administration of selexipag alone, which is in line with the observed increase in exposure to the active metabolite ACT‐333679, the major contributor to the effect of selexipag in humans.
In the pivotal phase 3 Prostacyclin (PG12) Receptor agonist In Pulmonary arterial HypertensiON (GRIPHON) study, selexipag was initiated in PAH patients at a dose of 200 μg twice daily, and the dose was increased weekly in twice‐daily increments of 200 μg until the individual highest tolerated dose (HTD) was attained 12. The maximum allowed dose of selexipag was 1600 μg twice daily 12, 36. In this study, no statistically significant PK/pharmacodynamic relationship with important safety parameters such as vital signs could be established 37. The occurrence of typical prostacyclin‐related AEs, such as headache, nausea and vomiting, was lower in placebo‐ than in selexipag‐treated patients. However, consistent with the concept of up‐titration to the individual HTD, no relevant difference could be determined between low and high exposures to selexipag and ACT‐333679 37. In the present DDI study, the 11‐fold increase in exposure to ACT‐333679 in the presence of gemfibrozil was associated with a significant increase in the incidence and/or intensity of prostacyclin‐related AEs. Concomitant treatment with gemfibrozil and selexipag in PAH patients would increase the exposure to ACT‐333679 to levels higher than those at the highest allowed dose of 1600 μg twice daily, and up‐titration of selexipag in these patients would probably not be possible. Therefore, concomitant treatment of selexipag and strong inhibitors of CYP2C8 should be avoided.
The effect of moderate inhibitors of CYP2C8 (e.g. clopidogrel, deferasirox, teriflunomide) 38, 39, 40, 41 on the exposure to selexipag and ACT‐333679 has not yet been studied but, based on the observed effect of gemfibrozil on the PK of ACT‐333679 in the present study, a PK interaction between moderate inhibitors of CYPC28 and selexipag cannot be excluded. Therefore, caution is required when using these drugs concomitantly with selexipag.
In conclusion, there was an increased incidence and/or intensity of typical prostacyclin‐related AEs when selexipag was administered concomitantly with gemfibrozil. Based on the observed interactions with gemfibrozil and rifampicin, concomitant administration of selexipag with strong inhibitors of CYP2C8 (e.g. gemfibrozil) must be avoided and dose adjustments should be considered when selexipag is administered with CYP2C8 inducers.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare no support from any third party organization for the submitted work and no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years. S.B., M.B., T.R. and J.D. are employees of Actelion Pharmaceuticals Ltd, the sponsor of the study, and possess stock options/shares. Selexipag (UPTRAVI®) is a product marketed by Actelion Pharmaceuticals Ltd. M.P.S. and A.H. had support from Clinical Research Services Kiel GmbH for the submitted work and were employed by Clinical Research Services Kiel GmbH in the previous 3 years.
This study was sponsored by Actelion Pharmaceuticals Ltd, Allschwil, Switzerland. The authors thank Mariya Antonova (Aprova s.r.o., Brno, Czech Republic) for statistical analysis of clinical and pharmacokinetic data, Susanne Globig (Department of Preclinical Pharmacokinetics and Metabolism, Actelion Pharmaceuticals Ltd) for conducting the bioanalysis, and Paul van Giersbergen (Van Giersbergen Consulting, Wuenheim, France) for editorial assistance.
Bruderer, S. , Petersen‐Sylla, M. , Boehler, M. , Remeňová, T. , Halabi, A. , and Dingemanse, J. (2017) Effect of gemfibrozil and rifampicin on the pharmacokinetics of selexipag and its active metabolite in healthy subjects. Br J Clin Pharmacol, 83: 2778–2788. doi: 10.1111/bcp.13379.
Marc Petersen‐Sylla was the principal investigator and Atef Halabi was the sub‐investigator of the study.
ClinicalTrial.gov registration number: NCT02770222.
References
- 1. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 2011; 8: 443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res 2014; 115: 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Badesch DB, McLaughlin VV, Delcroix M, Vizza CD, Olschewski H, Sitbon O, et al Prostanoid therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43 (12 Suppl. S): 56S–61S. [DOI] [PubMed] [Google Scholar]
- 4. Sitbon O, Vonk Noordegraaf A. Epoprostenol and pulmonary arterial hypertension: 20 years of clinical experience. Eur Respir Rev 2017; 26: pii: 160055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Skoro‐Sajer N, Lang I, Naeije R. Treprostinil for pulmonary hypertension. Vasc Health Risk Manag 2008; 4: 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Channick RN, Voswinckel R, Rubin LJ. Inhaled treprostinil: a therapeutic review. Drug Des Devel Ther 2012; 6: 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pugliese SC, Bull TM. Clinical use of extended‐release oral treprostinil in the treatment of pulmonary arterial hypertension. Integr Blood Press Control 2016; 9: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olschewski H. Inhaled iloprost for the treatment of pulmonary hypertension. Eur Respir Rev 2009; 18: 29–34. [DOI] [PubMed] [Google Scholar]
- 9. Melian EB, Goa KL. Beraprost: a review of its pharmacology and therapeutic efficacy in the treatment of peripheral arterial disease and pulmonary arterial hypertension. Drugs 2002; 62: 107–133. [DOI] [PubMed] [Google Scholar]
- 10. Kuwano K, Hashino A, Asaki T, Hamamoto T, Yamada T, Okubo K, et al 2‐[4‐[(5,6‐diphenylpyrazin‐2‐yl)(isopropyl)amino]butoxy]‐N‐(methylsulfonyl)acetamide (NS‐304), an orally available and long‐acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007; 322: 1181–1188. [DOI] [PubMed] [Google Scholar]
- 11. Asaki T, Kuwano K, Morrison K, Gatfield J, Hamamoto T, Clozel M. Selexipag: an oral and selective IP prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. J Med Chem 2015; 58: 7128–7137. [DOI] [PubMed] [Google Scholar]
- 12. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, et al Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 2015; 373: 2522–2533. [DOI] [PubMed] [Google Scholar]
- 13. Scott LJ. Selexipag: first global approval. Drugs 2016; 76: 413–418. [DOI] [PubMed] [Google Scholar]
- 14. Hardin EA, Chin KM. Selexipag in the treatment of pulmonary arterial hypertension: design, development, and therapy. Drug Des Devel Ther 2016; 10: 3747–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. US Department of Health and Human Services, Food and Drug Administration US prescribing information: Uptravi (Selexipag). December 2015. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207947s000lbl.pdf (last accessed 28 January 2017).
- 16. Kaufmann P, Okubo K, Bruderer S, Mant T, Yamada T, Dingemanse J, et al Pharmacokinetics and tolerability of the novel oral prostacyclin IP receptor agonist selexipag. Am J Cardiovasc Drugs 2015; 15: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaufmann P, Hurst N, Astruc B, Dingemanse J. Absolute oral bioavailability of selexipag, a novel oral prostacyclin IP receptor agonist. Eur J Clin Pharmacol 2017; 73: 151–156. [DOI] [PubMed] [Google Scholar]
- 18. Bruderer S, Hurst N, Kaufmann P, Dingemanse J. Multiple‐dose up‐titration study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of selexipag, an orally available selective prostacyclin receptor agonist, in healthy subjects. Pharmacology 2014; 94: 148–156. [DOI] [PubMed] [Google Scholar]
- 19. Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Gemfibrozil is a strong inactivator of CYP2C8 in very small multiple doses. Clin Pharmacol Ther 2012; 91: 846–855. [DOI] [PubMed] [Google Scholar]
- 20. Wang JS, Neuvonen M, Wen X, Backman JT, Neuvonen PJ. Gemfibrozil inhibits CYP2C8‐mediated cerivastatin metabolism in human liver microsomes. Drug Metab Dispos 2002; 30: 1352–1356. [DOI] [PubMed] [Google Scholar]
- 21. Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivistö KT. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet 2003; 42: 819–850. [DOI] [PubMed] [Google Scholar]
- 22. Backman JT, Filppula AM, Niemi M, Neuvonen PJ. Role of cytochrome P450 2C8 in drug metabolism and interactions. Pharmacol Rev 2016; 68: 168–241. [DOI] [PubMed] [Google Scholar]
- 23. Kaufmann P, Niglis S, Bruderer S, Segrestaa J, Äänismaa P, Halabi A, et al Effect of lopinavir/ritonavir on the pharmacokinetics of selexipag, an oral prostacyclin receptor agonist and its active metabolite in healthy subjects. Br J Clin Pharmacol 2015; 80: 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, et al The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tornio A, Neuvonen PJ, Niemi M, Backman JT. Role of gemfibrozil as an inhibitor of CYP2C8 and membrane transporters. Expert Opin Drug Metab Toxicol 2017; 13: 83–95. [DOI] [PubMed] [Google Scholar]
- 28. Backman JT, Honkalammi J, Neuvonen M, Kurkinen KJ, Tornio A, Niemi M, et al CYP2C8 activity recovers within 96 hours after gemfibrozil dosing: estimation of CYP2C8 half‐life using repaglinide as an in vivo probe. Drug Metab Dispos 2009; 37: 2359–2366. [DOI] [PubMed] [Google Scholar]
- 29. van Giersbergen PL, Treiber A, Schneiter R, Dietrich H, Dingemanse J. Inhibitory and inductive effects of rifampin on the pharmacokinetics of bosentan in healthy subjects. Clin Pharmacol Ther 2007; 81: 414–419. [DOI] [PubMed] [Google Scholar]
- 30. Bidstrup TB, Stilling N, Damkier P, Scharling B, Thomsen MS, Brøsen K. Rifampicin seems to act as both an inducer and an inhibitor of the metabolism of repaglinide. Eur J Clin Pharmacol 2004; 60: 109–114. [DOI] [PubMed] [Google Scholar]
- 31. Varma MV, Lin J, Bi YA, Rotter CJ, Fahmi OA, Lam JL, et al Quantitative prediction of repaglinide–rifampicin complex drug interactions using dynamic and static mechanistic models: delineating differential CYP3A4 induction and OATP1B1 inhibition potential of rifampicin. Drug Metab Dispos 2013; 41: 966–974. [DOI] [PubMed] [Google Scholar]
- 32. Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology 2002; 36: 164–172. [DOI] [PubMed] [Google Scholar]
- 33. Baciewicz AM, Chrisman CR, Finch CK, Self TH. Update on rifampin, rifabutin, and rifapentine drug interactions. Curr Med Res Opin 2013; 29: 1–12. [DOI] [PubMed] [Google Scholar]
- 34. Acocella G. Clinical pharmacokinetics of rifampicin. Clin Pharmacokinet 1978; 3: 108–127. [DOI] [PubMed] [Google Scholar]
- 35. Renwick AB, Watts PS, Edwards RJ, Barton PT, Guyonnet I, Price RJ, et al Differential maintenance of cytochrome P450 enzymes in cultured precision‐cut human liver slices. Drug Metab Dispos 2000; 28: 1202–1209. [PubMed] [Google Scholar]
- 36. Bruderer S, Hurst N, Remenova T, Dingemanse J. Clinical pharmacology, efficacy, and safety of selexipag for the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf 2017; 16: 743–751. [DOI] [PubMed] [Google Scholar]
- 37. Krause A, Machacek M, Lott D, Hurst N, Bruderer S, Dingemanse J. Population pharmacokinetics and pharmacodynamics of selexipag. CPT Pharmacometrics Syst Pharmacol 2017; 6: 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tornio A, Filppula AM, Kailari O, Neuvonen M, Nyrönen TH, Tapaninen T, et al Glucuronidation converts clopidogrel to a strong time‐dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug–drug interactions. Clin Pharmacol Ther 2014; 96: 498–507. [DOI] [PubMed] [Google Scholar]
- 39. Itkonen MK, Tornio A, Neuvonen M, Neuvonen PJ, Niemi M, Backman JT. Clopidogrel markedly increases plasma concentrations of CYP2C8 substrate pioglitazone. Drug Metab Dispos 2016; 44: 1364–1371. [DOI] [PubMed] [Google Scholar]
- 40. Skerjanec A, Wang J, Maren K, Rojkjaer L. Investigation of the pharmacokinetic interactions of deferasirox, a once‐daily oral iron chelator, with midazolam, rifampin, and repaglinide in healthy volunteers. J Clin Pharmacol 2010; 50: 205–213. [DOI] [PubMed] [Google Scholar]
- 41. Aubagio® film‐coated tablets (teriflunomide), summary of product characteristics, August 2013. Sanofi‐Aventis. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002514/WC500148682.pdf (last accessed 21 March 2017).