

Abstract
The small intestine (SI) is the main site for food absorption and glutamine utilization hence critical in metabolic disorders that involve energy balance such as diabetes. This study investigates the effects of oleanolic acid (OA) on SI morphology and some enzymes of glutamine metabolism in male Sprague-Dawley diabetic rats. High dose STZ-induced diabetes (HDD) and low dose STZ-induced diabetes (LDD) were induced by intraperitoneal injection of 60 and 40 mg streptozotocin/kg body weight respectively. Non-diabetic and diabetic rats were treated for two weeks with OA, insulin or OA + insulin in HDD study while animals in the in LDD study were treated with OA. There was significant (P<0.05) increase in the weight of the SI of diabetic animals and of villus height (VH) in the jejunum and duodenum of HDD animals. OA and insulin treatment significantly decreased VH in duodenum of HDD animals while OA treatment profoundly increased VH in normal rats. Jejunal of phosphate-dependent glutaminase (PDG) activity was unaffected by diabetes however alanine aminotransferase, aspartate aminotransferase and glutamate dehydrogenase activities were significantly (P<0.05) elevated by diabetes and treatments decreased these elevated aminotransferase activities. It is suggested that the intestine meets the energy demand in diabetes by modulating the activities of aminotransferases without change in PDG activity.
Keywords: Diabetes, rat small intestine, oleanolic acid, glutaminase, aminotransferases
Introduction
Diabetes mellitus, a metabolic disorder characterized by hyperglycaemia, currently affects 382 million people worldwide, a number estimated to rise to 592 million by the year 2035 [1]. The condition is exacerbated by post-prandial hyperglycaemia hence the importance of developing anti-diabetic drugs that limit carbohydrate uptake from the gut and which also have less side-effects than current drugs in use, particularly those for managing type II diabetes [2,3,4].
Oleanolic acid (3β-hydroxyolean-12-en-28-oic acid) (OA) is an example of such a plant-derived natural compound that has potential for managing diabetes without serious side effects. It is a ubiquitous pentacyclic triterpenoid of wide occurrence in plants [5]. OA has been shown to have therapeutic potential in the management of diabetes mellitus [6] and amongst its many effects, are the ability to increase response to exogenous insulin in type 1 diabetes [7] and to improve organ sensitivity to insulin [8]. Treatment with OA also inhibits uptake of glucose by the small intestine with a concomitant increase in glycogen biosynthesis in the intestinal wall which may important in decreasing postprandial glycaemia [9].
Additional to effects on other tissues, uncontrolled diabetes also leads to both morphological and biochemical remodeling of the gut, especially the small intestine [10]. The diabetes-induced intestinal and colonic structural and biomechanical remodeling which occurs in both animals and humans may be mediated by advanced glycation end of products (AGEs) and AGE receptors (RAGE) since their increased occurrence and expression has been demonstrated in the intestinal and colonic wall [11]. The small intestine (SI) is viewed as an important component of glucose homeostasis especially in a diabetic state [12] and diabetes induced-intestinal hyperplasia is thought to contribute to postprandial hyperglycaemia through increased total activity of disaccharidases such as sucrase and isomaltase in the entire small intestine [13]. The intestine is, however, known to use glutamine as a major source of energy in contrast with glucose whose major role appears to be the provision of carbon skeleton for the deposition of glutamate nitrogen through synthesis of alanine [14]. Thus, intestinal remodeling may derive its energy from increased glutamine oxidation.
The main enzyme that initiates intestinal glutamine oxidation is phosphate-dependent glutaminase [15]. In Type I animal diabetic states, the changes in the activity of intestinal phosphate-dependent glutaminase (PDG) have not been without contradiction. While Nagy and Kretchmer [16] have reported a decrease in PDG activity due to diabetes or fasting associated with a switch to ketone bodies as sources of energy, Watford and colleagues [17] reported an increase which was, however, not associated with increased glutamine utilization. Further, the activity of 3-oxoacid CoA-transferase a key enzyme in ketone body utilization was not increased in diabetes [17]. The question of how the intestine meets its energy and nitrogen requirements for intestinal hyperplasia as well as transport processes during the diabetic state is of importance.
In this paper, we investigate the effect of OA and insulin treatment on changes in intestinal morphology and activities of key enzymes of glutamine metabolism that result from diabetes. We expect this investigation to provide further information on how the intestine might meet its energy requirements in diabetes.
Materials and methods
Chemicals
Oleanolic acid (OA) was obtained from Shaanxi King Stone Enterprise, Xian, China. Chemicals and enzymes used in the study were purchased mainly from Sigma-Aldrich (St Loius, MO, USA) through Capital Lab Supplies, New Germany, South Africa while other chemicals were sourced from Merck (Halfway House, South Africa).
Animals
Male Sprague-Dawley rats (200-250 g) were procured from the Biomedical Resource Unit (BRU) at Westville Campus of the University of KwaZulu-Natal, South Africa. Animals received humane care according to the guidelines of Laboratory Animal Care of the National Society of Medical Research and the National Institutes of Health Guide for the Care and Use of Laboratory Animals of the National Academy of Sciences (National Institutes of Health publication no. 8023, revised 1978). This study was approved by the Animal Ethics Committee of the University of KwaZulu-Natal (Ethics approval reference: 001/14/Animal). Animals were maintained in standard environmental conditions with 12 h light/12 h dark cycle and fed a commercially available rat chow diet (Meadows, Pietermaritzburg, South Africa) with drinking water being provided ad libitum for the duration of the experiment.
Induction of diabetes
Diabetes was induced in rats fasted overnight by a single intraperitoneal injection of 60 or 40 mg streptozotocin/kg body weight dissolved in freshly prepared 0.1 M citrate buffer pH 4.5 for high dose STZ-induced (HDD) and low dose STZ-induced (LDD) diabetes (insulin insufficiency model), respectively [7,18]. These two doses were selected because they have been used previously in studies of Type I and Type II diabetes involving Sprague-Dawley rats. The dose of 40 mg streptozotocin/kg bw has been used for partial destruction of the pancreatic β cell population leading to reduced insulin secretion resembling Type II diabetes but without insulin resistance which was ensured by a fructose diet in other groups [19]. On the other hand, a dose of 60 mg streptozotocin/kg bw has been used for extensive destruction of pancreatic β cells resulting in Type I diabetes characterized by very low insulin levels [20] and very high fasting blood glucose levels [7,20]. Animals having a fasting blood glucose concentration of ≥ 18 and 8-16 mM after one week were considered as having stable diabetes in the two models respectively. However only animals with a fasting blood glucose level of 10-14 mM were used for the LDD study to avoid borderline cases.
Experimental design
In the HDD study normal/non-diabetic (N) and diabetic (D) animals were each divided into 2 and 4 groups, respectively, (n = 6). Non-diabetic control (NC) and diabetic control (DC) groups were treated orally with the vehicle (50% DMSO in physiological saline). A 50% solution of DMSO in physiological saline was used to prepare OA which was then administered orally while insulin was administered by subcutaneous injection. Other treatment groups were designated: NO and DO, which were treated with 80 mg/kg OA; DI, which was treated with 4 IU/kg insulin; and DOI, which was treated with a combination of OA, 80 mg/kg OA + insulin, 4 IU/kg. In the LDD study normal/non-diabetic (N) and diabetic (D) animals were each divided into 2 groups (n = 6). Non-diabetic control (NC) and diabetic control (DC) groups were treated with the vehicle (50% DMSO in physiological saline) whereas other groups designated NO and DO were treated with 80 mg/kg OA. All treatments were administered daily for a period of 14 days.
After fourteen days of treatment the animals were sacrificed by an overdose gas inhalant, Isofor (Safeline Pharmaceuticals, Johannesburg, South Africa) and then bled by cardiac puncture. The small intestine was immediately excised from the abdominal cavity. The duodenum was obtained as the first 10 cm beyond the pylorus and the middle 15 cm of the remaining segment was obtained as the jejunum while the ileum was taken as the 3.5 cm from the bottom [21]. The small intestine was then rinsed using physiological saline solution. Before the duodenum, jejunum and ileum (representing the small intestine) were separated, the length and weight was recorded. A piece of 1 cm from the duodenum, jejunum and ileum was preserved in 10% formalin for histological studies. The rest of the jejunum was snap-frozen in liquid nitrogen and stored at -20°C until used for enzyme assays.
Histopathological studies
Histopathological studies were done using a standard laboratory protocol for paraffin embedding. The intestinal segments fixed in 10% formalin were dehydrated in increasing concentrations (50%-90%) of ethanol and finally xylene. The tissues were then oriented longitudinally and cross-sectionally and embedded in paraffin wax and 4 µm of sections were cut using a microtome. The slides were allowed to dry overnight and then deparaffinized in p-xylene and rehydrated in gradients of ethanol (100%, 80%, 70%, and 50%) and finally rinsed in water. The slides were immediately stained for 5 min in hematoxylin stain and rinsed with water followed by counterstaining in eosin for 3 min. The slides were then mounted in distyrene plasticizer xylene (DPX), cover-slipped and allowed to settle and dry for two days before scanning using Leica SCN400, Germany (Software version: SlidePath Gateway Client Viewer 2.0).
Enzyme assays
Prior to enzyme assays, the frozen jejunum sections of the small intestine were thawed and homogenized (5 mL/mg tissue) in ice-cold buffer (50 mM Tris and 120 mM KCl, 0.5 mM PMSF and 5 mM benzamidine, pH 7.4) using Omni homogenizer (Kennesaw, GA, USA) operated at 35000 rpm.
Phosphate dependent glutaminase (PDG) was assayed at 37°C as described by Curthoys and Weiss [22] with modifications by Masola and Zvinavashe [23]. The two-step procedure involves determination of the amount of glutamate generated by PDG. The glutamate was determined from the amount of NADH produced when glutamate is oxidized in a reaction catalysed by glutamate dehydrogenase glutamate dehydrogenase (GDH). The activity of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in the SI was determined at 25°C using the method of Volmann-Mitchell and Parsons [24]. The assays involve coupled reactions catalyzed by ALT and AST which generate pyruvate and oxaloacetate, respectively. The respective products are then reduced by NADH in reactions catalyzed by lactate dehydrogenase (LDH) and malic dehydrogenase (MDH). The amount of NADH consumed by the LDH and MDH catalyzed reactions is measured spectrophotometrically and is directly proportional to the activity of ALT and AST. The activity of GDH was estimated at 25°C in the direction of glutamate formation by following the oxidation of NADH spectrophotometrically at 340 nm as described by Schmidt [25].
Statistical analysis
All data were expressed as means ± standard deviation (SD). Graphical plots and statistical analysis was done using GraphPad Prism (version 5) and InStat Software (version 3, Graph Pad Software, San Diego, California, USA). Statistical comparison among groups was done using one-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparison test. Value of P<0.05 was considered to be statistically significant.
Results
Change relative weight of the small intestine and villus morphology
The weight of the small intestine expressed per gram body weight or per unit length in both HDD and LDD was increased significantly (P<0.05) in all the diabetic groups compared to non-diabetic groups (Tables 1 and 2). There were no significant changes (P>0.05) in these parameters within non-diabetic groups or within the diabetic groups.
Table 1.
Relative small intestine weight change in High dose STZ-induced diabetes (HDD) rats
| NC | NO | DC | DO | DI | DOI | |
|---|---|---|---|---|---|---|
| SI weight/Body weight (g/g) | 0.028 ± 0.003a | 0.034 ± 0.003a | 0.063 ± 0.006b | 0.074 ± 0.013b | 0.069 ± 0.010b | 0.066 ± 0.019b |
| SI weight/length (g/cm) | 0.075 ± 0.006a | 0.086 ± 0.008a | 0.127 ± 0.017b | 0.126 ± 0.008b | 0.124 ± 0.012b | 0.124 ± 0.016b |
SI = small intestine; NC = non-diabetic control; NO = non-diabetic OA treated; DC = diabetic control; DO = diabetic OA treated; DI = diabetic insulin treated; DOI = diabetic OA + insulin treated. Results are expressed as mean ± SD (n = 6).
Indicate significant (P<0.05) difference from other groups.
Indicate significant (P<0.05) difference from other groups.
Table 2.
Relative small intestine weight change in Low dose STZ-induced diabetes (LDD) rats
| NC | NO | DC | DO | |
|---|---|---|---|---|
| SI weight/Body weight (g/g) | 0.029 ± 0.004a | 0.034 ± 0.003a,c | 0.050 ± 0.011b | 0.049 ± 0.007b,c |
| SI weight/length (g/cm) | 0.075 ± 0.006a | 0.086 ± 0.008a,c | 0.104 ± 0.009b,c | 0.116 ± 0.018b |
SI = small intestine; NC = non-diabetic control; NO = non-diabetic OA treated; DC = diabetic control; DO = diabetic OA treated. Results are expressed as mean ± SD (n = 6).
Indicate significant (P<0.05) difference from other groups.
Indicate significant (P<0.05) difference from other groups.
Indicate significant (P<0.05) difference from other groups.
HDD, LDD and OA treatment of non-diabetic animals resulted in significantly (P<0.05) elongated villi in the jejunum (Figure 1A and 1B) and this elongation was quite profound in non-diabetic animals treated with OA this being ≥ 90% higher in this group compared to the NC group. All treatments of HDD and LDD rats did not prevent this increase in jejunum villus height except OA treatment of LDD animals. HDD also increased duodenal villus height significantly (P<0.05) and this increase was lowered by all treatments. The average crypt depth was largely unchanged in all SI segments among the groups (data not shown), hence changes in mucosal height were due to changes in villi. Our results (not shown) also showed that the surface area of the jejunal villi was increased by almost 300% in NO groups compared to the respective NC groups in the HDD and LDD studies; that the duodenum and jejunum villus surface area of all diabetic groups in the HDD study was significantly higher compared to the NC group with exception of the DOI group in the duodenum; and that the villus surface area in the ileum of all groups was not significantly altered in all the groups.
Figure 1.
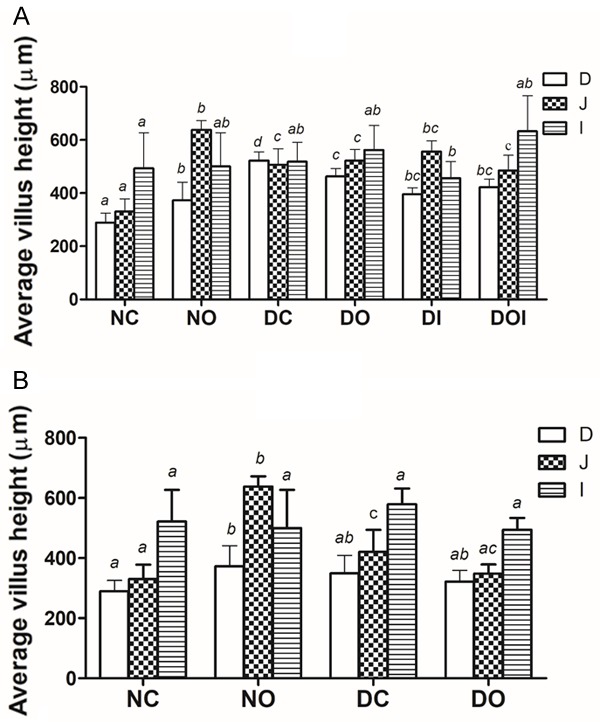
Effects of diabetes and different treatments on average villus height in the rat duodenum (D), jejunum (J) and Ileum (I) in high dose STZ-induced diabetes, (A) and low dose STZ-induced diabetes, (B) NC = non-diabetic control; NO = non-diabetic OA treated; DC = diabetic control; DO = diabetic OA treated; DI = diabetic insulin treated; DOI = diabetic OA + insulin treated. Results are expressed as means ± SD (n = 6). Different alphabets (a, b, c, d) on bars of the same shading indicates significant (P<0.05) difference from other groups.
Effect of diabetes OA and insulin on enzymes of glutamine metabolism
In both the HDD and LDD studies (Figure 2A and 2B) there was a small non-significant decrease (P>0.05) of PDG activity in the DC groups compared to their respective NC groups. There were also no significant differences in PDG activity in the treated diabetic groups compared to respective DC groups nor were there any significant differences in activity amongst the diabetic groups in each study. OA significantly reduced PDG activity (P<0.05) in non-diabetic animals of both studies.
Figure 2.
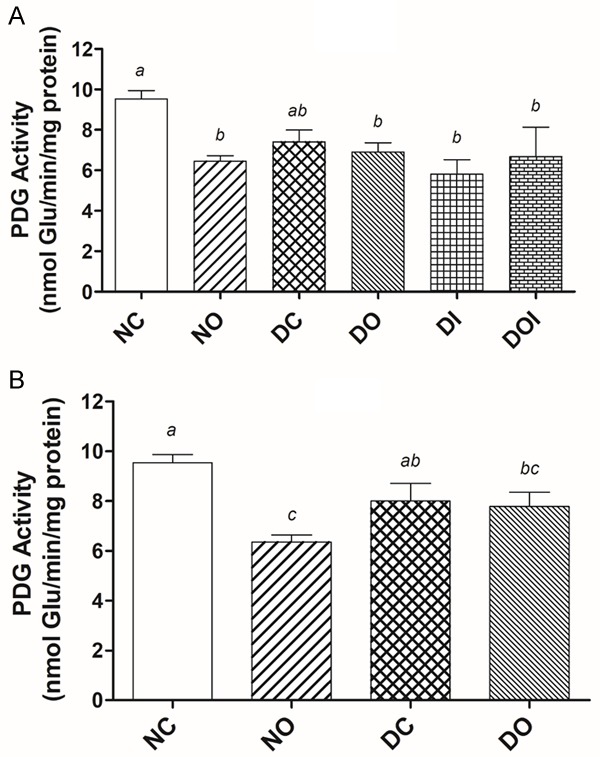
The effect of diabetes and different treatments on the activity of phosphate dependent glutaminase (PDG) in the rat jejunum in high dose STZ-induced diabetes, (A) and low dose STZ-induced diabetes, (B) Glu = glutamate; NC = non-diabetic control; NO = non-diabetic OA treated; DC = diabetic control; DO = diabetic OA treated; DI = diabetic insulin treated; DOI = diabetic OA + insulin treated. Results are expressed as means ± SD (n = 6). Different alphabets (a, b, c) on bars indicates significant (P<0.05) difference from other groups.
On the other hand, the SI activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in the HDD study were both significantly elevated in the DC group compared to the NC group (Figure 3A). Treatments of HDD diabetic groups decreased the activities of ALT and AST and this decrease was significant (P<0.05) in the DI and DOI groups compared to the DC group. Low dose STZ-induced diabetes caused a significant increase in ALT and AST activity and this increase was reduced by treatment with OA particularly with respect to ALT of which levels were reduced to those found in the normal/non-diabetic animals (Figure 3B). The changes in glutamate dehydrogenase (GDH) activity in the HDD are shown in Figure 4A. GDH was elevated by more than 100% in all HDD groups compared to the NC group and this increase was most profound in the DO group. GDH activity was also significantly (P<0.05) increased by low dose STZ and this increase was reduced by treatment with OA (Figure 4B).
Figure 3.
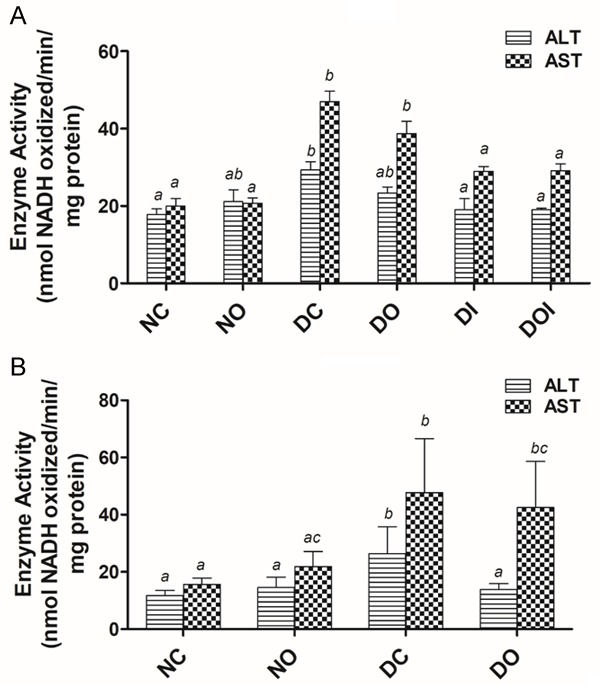
The effect of diabetes and different treatments on the activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in the rat jejunum in high dose STZ-induced diabetes, (A) and low dose STZ-induced diabetes, (B) NC = nondiabetic control; NO = non-diabetic OA treated; DC = diabetic control; DO = diabetic OA treated; DI = diabetic insulin treated; DOI = diabetic OA + insulin treated. Results are expressed as means ± SD (n = 6). Different alphabets (a, b, c) on bars of the same shading indicates significant (P<0.05) difference from other groups.
Figure 4.
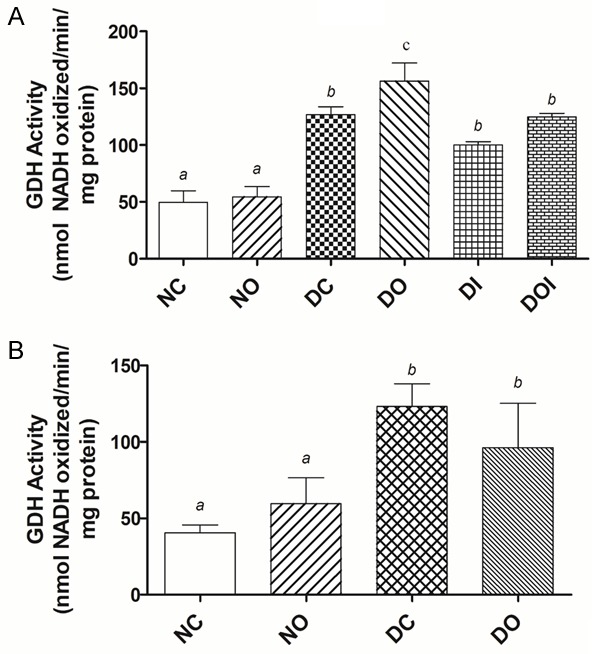
The effect of diabetes and different treatments on the activity of glutamate dehydrogenase (GDH) in the rat jejunum in high dose STZ-induced diabetes, (A) and low dose STZ-induced diabetes, (B) NC = non-diabetic control; NO = non-diabetic OA treated; DC = diabetic control; DO = diabetic OA treated; DI = diabetic insulin treated; DOI = diabetic OA + insulin treated. Results are expressed as means ± SD (n = 6). Different alphabets (a, b, c) on bars indicates significant (P<0.05) difference from other groups.
Discussion
The diabetes-induced increase in weight and length of the small intestine shown by our study has been observed in other studies of streptozotocin and alloxan induced diabetic rats and occurs as early as 8 days post-induction [26]. It is a result of both hyperplasia and hypertrophy of the SI [27] possibly mediated by glucagon like peptide-2 (GLP-2), an intestinal growth factor that is elevated in diabetic rats [28]. It is also worth noting that chronically elevated glucose levels in diabetic obese individuals, evident from elevated HbA1c, are associated with increased small intestinal enterocyte mass and increased enterocyte loss as shown by elevated levels of citrulline and intestinal fatty acid binding protein (I-FABP), respectively [29]. However, these changes were not due to increased GLP-2 secretion suggested by Fischer and colleagues [28]. Impairment of anti-proliferative genes such as p53 and transforming growth factor (TGF)-β by diabetes could also contribute to development of intestinal hyperplasia and hypertrophy since amelioration of diabetes through metabolic surgery in patients is accompanied by increased expression of these genes in non-excluded segments of the intestine [30]. Also increased by metabolic surgery is the secretion of glucagon like peptide-1 (GLP-1), a primary incretin hormone secreted from the intestine to stimulate insulin secretion by pancreatic β cells after ingestion of glucose or nutrients [30]. All our treatments failed to reverse these morphological changes (Tables 1, 2) suggesting that prolonged treatment is necessary for this to occur.
The morphometric data (Figure 1 and Tables 1 and 2) suggest the proximal SI (jejunum and the duodenum) to be the main site of diabetes-induced SI changes. Hyperphagia is a characteristic of diabetes and the disease itself has been suggested to be a trigger of intestinal growth that is further enhanced by dietary fibre and increased GLP-2 secretion [31] hence the changes observed in the proximal SI could be attributable to the absorptive nature of the jejunum. In contrast, the villi in the ileum were not affected by either diabetes or its treatment and this could be due to minimal or no function in nutrient absorption of this intestinal segment. Since LDD caused none to very little villi changes in contrast to HDD (Figure 1) a role of blood glucose level in diabetes induced villi growth is suggested particularly since chronically elevated glucose levels are associated with increased small intestinal enterocyte mass and increased enterocyte loss [29]. The surprisingly profound increase in height and surface area of jejunal villi of normal rats that results from OA treatment is reported here we believe, for the first time. The mechanism involved in OA-induced villi changes in the SI of normal animals appears different from that in diabetic animals in that the weight or length of the whole intestine was only slightly affected by OA treatment, despite gross villi changes, which suggests minimal hypertrophy. Suppression of apoptosis in a caspase 3-related mechanism has been suggested for diabetes induced SI changes [32]. However, some in vitro studies suggest that other structurally similar pentacyclic triterpenes such as maslinic acid do not alter the cell cycle or induce caspase-3 dependent apoptosis in the non tumoural intestine cell lines IEC-6 and IEC-18 [33]. Therefore, changes in small intestinal morphology during diabetes and those caused by OA are most likely due to multiple factors. Since SI hypertrophy and mucosal changes appear to occur via different mechanisms it is important to investigate these mechanisms considering the fact that metabolic surgeries are becoming a successful alternative for treatment of diabetes and obesity [30,34].
Remodeling of the small intestine as discussed above is a process that inherently requires energy. The results of our study (Figure 2) show a non-significant reduction in the activity of intestinal PDG due to diabetes in both HDD and LDD studies. Previous animal studies have shown that in the early stages of fasting and in diabetes there can occur decrease, increase or no effect, as well as decrease followed by a rebound in intestinal PDG activity [16,17,35]. Watford and colleagues [17] showed that an increase in intestinal PDG activity can occur without an increase glutamine utilization due to diminished arterial levels of the amino acid. Their work also negated the suggestion of ketone bodies becoming a major source of energy in the intestine in the diabetic state since ketone body infusions did not alter glutamine utilization, and the activity of the key enzyme involved in ketone body utilization was not increased. The question of how the intestine meets the energy requirements for the profound changes that occur in diabetes is vexing considering that the key enzyme that initiates glutamine oxidation (a major source of energy) remains largely unchanged or when changed, this does not result in increased substrate utilization. PDG may, however, be amenable to short-term regulation by allosteric activators like ADP [36]. The significant decrease in PDG activity in normal animals treated with OA is also puzzling in view of the profound changes in jejunal villi of these animals.
In current study, jejunal activities of alanine and aspartate aminotransferases (ALT and AST) and glutamate dehydrogenase (GDH) were greatly increased in both HDD and LDD (Figures 3, 4) in contrast to PDG. It has been shown previously that isolated intestinal mitochondria from normal rats generate glutamate from glutamine at a rate far exceeding the ability of mitochondrial aminotransferases (ALT and AST) and exogenous alanine aminotransferase (in presence of exogenous pyruvate) to metabolize it further [37]. The latter was added since alanine is the main amino acid product in jejunal glutamine oxidation in vivo [38]. Further, arterio-venous measurements of metabolites across portal vein-drained viscera following infusions of glutamine in diabetic rats showed decreased glutamate release but increased alanine and ammonia release by the viscera when compared to normal animals [17]. Taken together, the results of these studies suggest that, in diabetes, increased glutamine oxidation could occur without change in PDG activity and be due to increased alanine aminotransferase activity and elevated GDH activity, also observed in the current study. The observation that elevated activities of ALT and AST were reduced by treatment of diabetic animals, and in the case of ALT returned to normal by treatment with insulin or insulin + OA in HDD diabetic rats shows that the reversal of diabetes-induced structural changes in the intestine lag behind those of intestinal aminotransferase enzymes. Intestinal glutamate dehydrogenase activity which was highly elevated in diabetic animals was also reduced by treatment with insulin in the HDD study; or by treatment with OA in LDD study, which suggests that flux through GDH is enhanced in diabetes. Under normal conditions flux through intestinal glutamate dehydrogenase is known to be minimal and less important than the aminotransferases pathway for glutamate utilization [39]. However, under conditions such as starvation, generation of ammonia from glutamate in the intestine has been reported to be increased as an adaptive mechanism for modulating redox potential of the whole body [40]. Hence, the GDH pathway could also be increased, and important, in diabetes to generate more ammonia for redox control of diabetes-induced ketoacidosis and to provide α-ketoglutarate for oxidation.
Conclusion
In conclusion, the morphological changes that occur in early diabetes suggest a role of blood glucose level in intestinal hyperplasia. The morphological changes in villi are more pronounced in HDD and these were not reversed by the two-week treatments except in the duodenum in which they were significantly reduced. This was probably due to early encounter of the duodenum with the anti-hyperglycaemic agents. Further, OA treatment leads to profound villi growth in the absence of diabetes via a mechanism that seems different to that involved in diabetes. Since PDG activity was largely unaffected by diabetes it is suggested that the intestine may meet its energy needs in diabetes by increasing flux through the downstream enzymes (ALT, AST, GDH) whose elevated activities were reduced by treatment with OA, insulin or OA + insulin. However the return to normal of intestinal structures appears to lag behind that of tissue enzymes.
Acknowledgements
We thank the University of KwaZulu-Natal for financial support through Productivity Awards to Dr B Masola and the Education Task Fund (ETF) desk office, Umaru Musa Yar’adua University, Katsina, Nigeria for a study fellowship award to Murtala Bindawa Isah.
Disclosure of conflict of interest
None.
References
- 1.Guariguata L, Whiting D, Hambleton I, Beagley J, Linnenkamp U, Shaw J. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pr. 2014;103:137–149. doi: 10.1016/j.diabres.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Bailey CJ, Mynett KJ, Page T. Importance of the intestine as a site of metformin-stimulated glucose utilization. Brit J Pharmacol. 1994;112:671–675. doi: 10.1111/j.1476-5381.1994.tb13128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lebovitz HE. Alpha-glucosidase inhibitors. Endocrin Metab Clin. 1997;26:539–551. doi: 10.1016/s0889-8529(05)70266-8. [DOI] [PubMed] [Google Scholar]
- 4.Cheng AY, Fantus IG. Oral antihyperglycemic therapy for type 2 diabetes mellitus. Can Med Assoc J. 2005;172:213–226. doi: 10.1503/cmaj.1031414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollier J, Goossens A. Oleanolic acid. Phytochemistry. 2012;77:10–15. doi: 10.1016/j.phytochem.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 6.Castellano JM, Guinda A, Delgado T, Rada M, Cayuela JA. Biochemical basis of the antidiabetic activity of oleanolic acid and related pentacyclic triterpenes. Diabetes. 2013;62:1791–1799. doi: 10.2337/db12-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukundwa A, Mukaratirwa S, Masola B. Effects of oleanolic acid on the insulin signaling pathway in skeletal muscle of streptozotocininduced diabetic male Sprague-Dawley rats. J Diabetes. 2016;8:98–108. doi: 10.1111/1753-0407.12260. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Liu R, Zhang W, Zhang X, Liao N, Wang Z, Li W, Qin X, Hai C. Oleanolic acid improves hepatic insulin resistance via antioxidant, hypolipidemic and anti-inflammatory effects. Mol Cell Endocrinol. 2013;376:70–80. doi: 10.1016/j.mce.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 9.Khathi A, Masola B, Musabayane CT. Effects of Syzygium aromaticum-derived oleanolic acid on glucose transport and glycogen synthesis in the rat small intestine. J Diabetes. 2013;5:80–87. doi: 10.1111/j.1753-0407.2012.00230.x. [DOI] [PubMed] [Google Scholar]
- 10.Mayhew TM. Adaptive remodelling of intestinal epithelium assessed using stereology: correlation of single cell and whole organ data with nutrient transport. Histol Histopathol. 1996;11:729–741. [PubMed] [Google Scholar]
- 11.Zhao M, Liao D, Zhao J. Diabetes-induced mechanophysiological changes in the small intestine and colon. World J Diabetes. 2017;8:249–269. doi: 10.4239/wjd.v8.i6.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mingrone G, Castagneto-Gissey L. Type 2 diabetes mellitus in 2013: a central role of the gut in glucose homeostasis. Nat Rev Endocrinol. 2014;10:73–74. doi: 10.1038/nrendo.2013.241. [DOI] [PubMed] [Google Scholar]
- 13.Adachi T, Mori C, Sakurai K, Shihara N, Tsuda K, Yasuda K. Morphological changes and increased sucrase and isomaltase activity in small intestines of insulin-deficient and type 2 diabetic rats. Endocr J. 2003;50:271–279. doi: 10.1507/endocrj.50.271. [DOI] [PubMed] [Google Scholar]
- 14.Windmueller HG, Spaeth AE. Identification of ketone bodies and glutamine as the major respiratory fuels in vivo for postabsorptive rat small intestine. J Biol Chem. 1978;253:69–76. [PubMed] [Google Scholar]
- 15.Pinkus LM, Windmueller HG. Phosphatedependent glutaminase of small intestine: localization and role in intestinal glutamine metabolism. Arch Biochem Biophys. 1977;182:506–517. doi: 10.1016/0003-9861(77)90531-8. [DOI] [PubMed] [Google Scholar]
- 16.Nagy LE, Kretchmer N. Effect of diabetic ketosis on jejunal glutaminase. Arch Biochem Biophys. 1986;248:80–88. doi: 10.1016/0003-9861(86)90403-0. [DOI] [PubMed] [Google Scholar]
- 17.Watford M, Erbelding EJ, Smith E. The regulation of glutamine and ketone-body metabolism in the small intestine of the long-term (40-day) streptozotocin-diabetic rat. Biochem J. 1987;242:61–68. doi: 10.1042/bj2420061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishigaki A, Noma H, Kakizawa T. The relations between doses of streptozotocin and pathosis in induced diabetes mellitus. Shika Gakuho. 1989;89:639–662. [PubMed] [Google Scholar]
- 19.Wilson RD, Islam MS. Fructose-fed streptozotocin-injected rat: an alternative model for type 2 diabetes. Pharmacol Rep. 2012;64:129–139. doi: 10.1016/s1734-1140(12)70739-9. [DOI] [PubMed] [Google Scholar]
- 20.Musabayane CT, Tufts MA, Mapanga RF. Synergistic antihyperglycemic effects between plant-derived oleanolic acid and insulin in streptozotocin-induced diabetic rats. 2010;32:832–839. doi: 10.3109/0886022X.2010.494802. [DOI] [PubMed] [Google Scholar]
- 21.DeSesso JM, Jacobson CF. Anatomical and physiological parameters affecting gastrointestinal absorption in humans and rats. Food Chem Toxicol. 2001;39:209–228. doi: 10.1016/s0278-6915(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 22.Curthoys NP, Weiss RF. Regulation of renal ammoniagenesis subcellular localization of rat kidney glutaminase isoenzymes. J Biol Chem. 1974;249:3261–3266. [PubMed] [Google Scholar]
- 23.Masola B, Zvinavashe E. Phosphate-dependent glutaminase in enterocyte mitochon-dria and its regulation by ammonium and other ions. Amino acids. 2003;24:427–434. doi: 10.1007/s00726-002-0312-x. [DOI] [PubMed] [Google Scholar]
- 24.Volman-Mitchell H, Parsons DS. Distribution and activities of dicarboxylic amino acid transaminases in gastrointestinal mucosa of rat, mouse, hamster, guinea pig, chicken and pigeon. BBA-Enzymol. 1974;334:316–327. [Google Scholar]
- 25.Schmidt E. Glutamate dehydrogenase UV assay. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. vol. II. New York and London: Academic Press; 1974. pp. 650–656. [Google Scholar]
- 26.Schedl HP, Wilson HD. Effects of diabetes on intestinal growth and hexose transport in the rat. Am J Physiol. 1971;220:1739–1745. doi: 10.1152/ajplegacy.1971.220.6.1739. [DOI] [PubMed] [Google Scholar]
- 27.Nakabou Y, Okita C, Takano Y, Hagihira H. Hyperplastic and hypertrophic changes of the small intestine in alloxan diabetic rats. J Nutr Sci Vitaminol. 1974;20:227–234. doi: 10.3177/jnsv.20.227. [DOI] [PubMed] [Google Scholar]
- 28.Fischer KD, Dhanvantari S, Drucker DJ, Brubaker PL. Intestinal growth is associated with elevated levels of glucagon-like peptide 2 in diabetic rats. Am J Physiol. 1997;273:E815–E820. doi: 10.1152/ajpendo.1997.273.4.E815. [DOI] [PubMed] [Google Scholar]
- 29.Verdam FJ, Greve JW, Roosta S, van Eijk H, Bouvy N, Buurman WA, Rensen SS. Small intestinal alterations in severely obese hyperglycemic subjects. J Clin Endocrinol Metab. 2011;96:E379–E383. doi: 10.1210/jc.2010-1333. [DOI] [PubMed] [Google Scholar]
- 30.da Silva Rodrigues MR, Santo MA, Favero GM, Vieira EC, Artoni RF, Nogaroto V, de Moura EG, Lisboa P, Milleo FQ. Metabolic surgery and intestinal gene expression: digestive tract and diabetes evolution considerations. World J Gastroenterol. 2015;21:6990–6998. doi: 10.3748/wjg.v21.i22.6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thulesen J, Hartmann B, Nielsen C, Holst JJ, Poulsen SS. Diabetic intestinal growth adaptation and glucagon-like peptide 2 in the rat: effects of dietary fibre. Gut. 1999;45:672–678. doi: 10.1136/gut.45.5.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noda T, Iwakiri R, Fujimoto K, Yoshida T, Utsumi H, Sakata H, Hisatomi A, Aw TY. Suppression of apoptosis is responsible for increased thickness of intestinal mucosa in streptozotocin-induced diabetic rats. Metabolism. 2001;50:259–264. doi: 10.1053/meta.2001.21030. [DOI] [PubMed] [Google Scholar]
- 33.Reyes FJ, Centelles JJ, Lupiáñez JA, Cascante M. (2α, 3β)-2, 3-Dihydroxyolean-12-en-28-oic acid, a new natural triterpene from Olea europea, induces caspase dependent apoptosis selectively in colon adenocarcinoma cells. FEBS Lett. 2006;580:6302–6310. doi: 10.1016/j.febslet.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 34.Rubino F, Schauer PR, Kaplan LM, Cummings DE. Metabolic surgery to treat type 2 diabetes: clinical outcomes and mechanisms of action. Annu Rev Med. 2010;61:393–411. doi: 10.1146/annurev.med.051308.105148. [DOI] [PubMed] [Google Scholar]
- 35.Mithieux G, Bady I, Gautier A, Croset M, Rajas F, Zitoun C. Induction of control genes in intestinal gluconeogenesis is sequential during fasting and maximal in diabetes. Am J Physiol-Endoc M. 2004;286:E370–E375. doi: 10.1152/ajpendo.00299.2003. [DOI] [PubMed] [Google Scholar]
- 36.Masola B, Ngubane N. The activity of phosphate-dependent glutaminase from the rat small intestine is modulated by ADP and is dependent on integrity of mitochondria. Arch Biochem Biophys. 2010;504:197–203. doi: 10.1016/j.abb.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 37.Masola B, Peters TJ, Evered DF. Transamination pathways influencing L-glutamine and L-glutamate oxidation by rat enterocyte mitochondria and the subcellular localization of L-alanine aminotransferase and L-aspartate aminotransferase. BBA-Gen Subjects. 1985;843:137–143. doi: 10.1016/0304-4165(85)90060-1. [DOI] [PubMed] [Google Scholar]
- 38.Windmueller HG, Spaeth AE. Respiratory fuels and nitrogen metabolism in vivo in small intestine of fed rats. Quantitative importance of glutamine, glutamate, and aspartate. J Biol Chem. 1980;255:107–112. [PubMed] [Google Scholar]
- 39.Newsholme EA, Carrie AL. Quantitative aspects of glucose and glutamine metabolism by intestinal cells. Gut. 1994;35(Suppl):S13–7. doi: 10.1136/gut.35.1_suppl.s13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Souba WW. Intestinal glutamine metabolism and nutrition. J Nutr Biochem. 1993;4:2–9. [Google Scholar]