

Abstract
The role of the neuropeptide calcitonin gene‐related peptide (CGRP) is well established in nociceptive behaviors. CGRP is highly expressed in the projection pathway from the parabrachial nucleus to the laterocapsular region of the central amygdala (CeC), which plays a critical role in relaying nociceptive information. The CeC is a key structure in pain behavior because it integrates and modulates nociceptive information along with other sensory signals. Previous studies have demonstrated that blockade of the amygdalar CGRP‐signaling cascade attenuates nociceptive behaviors in pain models, while CGRP application facilitates amygdalar synaptic transmission and induces pain behaviors. Despite these lines of evidence, it remains unclear whether endogenous CGRP is involved in the development of nociceptive behaviors accompanied with amygdalar plasticity in a peripheral inflammation model in vivo. To directly address this, we utilized a previously generated CGRP knockout (KO) mouse to longitudinally study formalin‐induced plasticity and nociceptive behavior. We found that synaptic potentiation in the right PB‐CeC pathway that was observed in wild‐type mice was drastically attenuated in the CGRP KO mice 6 h post‐inflammation, when acute nociceptive behavior was no longer observed. Furthermore, the bilateral tactile allodynia 6 h post‐inflammation was significantly decreased in the CGRP KO mice. In contrast, the acute nociceptive behavior immediately after the formalin injection was reduced only at 20–25 min post‐injection in the CGRP KO mice. These results suggest that endogenous CGRP contributes to peripheral inflammation‐induced synaptic plasticity in the amygdala, and this plasticity may underlie the exaggerated nociception–emotion linkage in pain chronification.
Keywords: chronic pain, mouse, parabrachial nucleus, synaptic potentiation
Introduction
The amygdala plays key roles not only in aversive learning and memory (Davis, 1992; Fanselow & LeDoux, 1999; Johansen et al., 2011) but also in nociceptive information processing (Neugebauer et al., 2004, 2009). The laterocapsular subdivision of the central amygdala (CeC) is termed the ‘nociceptive amygdala’ because a large number of CeC neurons respond preferentially or exclusively to noxious stimuli (Bernard et al., 1992; Neugebauer & Li, 2003). The CeC receives excitatory inputs from the external part of the pontine lateral parabrachial (PB) nucleus, which is a major target of nociceptive neurons in the superficial layers of the dorsal horn and trigeminal nucleus (Bernard & Besson, 1990; Bernard et al., 1995; Jasmin et al., 1997; Sarhan et al., 2005; Al‐Khater & Todd, 2009; Han et al., 2015; Sato et al., 2015), through the spino‐parabrachial‐amygdaloid pain pathway (Todd, 2010). CeC neurons also receive excitatory inputs from the lateral and basolateral amygdala. These inputs convey highly processed polymodal sensory information, including pain, from thalamic and/or cortical regions (Sah et al., 2003; Pare et al., 2004). This duplex organization results in pain‐related and sensory information convergence at the CeC, which makes the CeC a modulatory locus of the nociception–emotion linkage. Intriguingly, PB‐CeC synapses exhibit long‐term potentiation following the establishment of various forms of inflammatory and neuropathic pain models as well as threat learning (Ikeda et al., 2007; Fu et al., 2008; Nakao et al., 2012; Watabe et al., 2013; Sugimura et al., 2016). Therefore, synaptic plasticity in the PB‐CeC pathway might be involved in affective aspects of pain. However, the molecular mechanisms of this potentiation and the physiological consequences are not clear.
Calcitonin gene‐related peptide (CGRP) is a 37‐amino acid peptide that activates adenylyl cyclase and protein kinase A (PKA) through G‐protein‐coupled receptors, including the CGRP1 receptor (Van Rossum et al., 1997; Poyner & Marshall, 2001). CGRP consists of two isoforms, αCGRP and βCGRP, which are encoded by separate genes (Russell et al., 2014). αCGRP is widely expressed in the peripheral and central nervous systems, while βCGRP is limited to enteric neurons (Mulderry et al., 1985). While the role of CGRP in peripheral tissues during inflammatory processes is well established in normal animals (Oku et al., 1987; Nakamura‐Craig & Gill, 1991; Saxen et al., 1993; Sun et al., 2004) and in pain models (Kuraishi et al., 1988; Kawamura et al., 1989; Yu et al., 1996; Hirsch et al., 2013), little is known about its role in the central nervous system. CGRP is highly expressed in the PB‐CeC pathway (Dong et al., 2010), and the CeC is one of the brain areas with the highest levels of CGRP receptors (Oliver et al., 1998). Indeed, selective CGRP receptor antagonists in the amygdala attenuate pain‐related behaviors and synaptic potentiation in the PB‐CeC pathway in slices acutely prepared from arthritic rats (Han et al., 2005), suggesting that endogenous CGRP plays a key role in PB‐CeC synaptic potentiation in arthritic rats. Furthermore, the application of exogenous CGRP potentiates PB‐CeC synaptic transmission in brain slices from naïve animals (Han et al., 2010). In addition, αCGRP knockout (KO) mice have been utilized to examine the role of endogenous CGRP in pain models in vivo (Salmon et al., 1999, 2001; Zhang et al., 2001; Ishida et al., 2014).
Despite this evidence, it remains unclear whether endogenous CGRP is involved in the development of nociceptive behaviors accompanied with amygdalar plasticity in peripheral inflammation model in vivo. In this study, we examined this issue using genetically manipulated mice. Here, we utilized previously developed KO mice in which αCGRP but not calcitonin was depleted (Oh‐hashi et al., 2001; Ishida et al., 2014) to analyze synaptic transmission in the PB‐CeC pathway and pain behaviors longitudinally following the intraplantar injection of formalin into the left hind paw. We found that endogenous CGRP was essential in PB‐CeC synaptic potentiation and in the development of hyperalgesic behavioral responses observed 6 h after formalin injection. These results suggest that endogenous CGRP contributes to peripheral inflammation‐induced plasticity in the amygdala, and this plasticity may underlie the exaggerated nociception–emotion linkage in pain chronification.
Materials and methods
The manipulation of the animals was approved by the Institutional Animal Care and Use Committee of Jikei University and conformed to the Guidelines for Proper Conduct of Animal Experiments of the Science Council of Japan (2006) and the guidelines of the International Association for the Study of Pain (Zimmermann, 1983).
Animals
αCGRP KO mice with genetic background of 129SV × C57BL/6 hybrid and backcrossed to C57/BL6 mice and their wild‐type (WT) littermate controls (either sex, 4–10 weeks old, 16 CGRP WT mice [male = 8, female = 8], and 15 CGRP KO mice [male = 8, female = 7]) (Oh‐hashi et al., 2001) were used in this study. The CGRP KO mice were healthy, showed normal behavior, and had no visible phenotypes that differed from the WT mice (Toda et al., 2008). The mice were group‐housed under a 12‐h light/dark cycle and were provided food and water ad libitum. All experiments were performed in a manner blinded to the mouse genotypes during the experiments according to a previously presorted standard protocol (Watabe et al., 2016).
Inflammatory pain model
After the mice were acclimated to the observation chamber for about 20 min, inflammation was induced by injecting 20 μL of 5% formalin (37% formaldehyde solution diluted in saline solution) or saline solution (0.9%) into the intraplantar surface of the left hind paw with a microsyringe with a 30‐gauge needle (Tjølsen et al., 1992; Adedoyin et al., 2010). Each mouse was returned immediately to the observation chamber, and the recording of spontaneous behavior was then started with a web camera (HD Webcam C525; Logicool, Tokyo, Japan). The time spent performing nociceptive behavior, which was defined by licking the injected paw, was measured with a handheld stopwatch for 5‐min intervals up to 1 h after the injection.
von Frey filament test
The paw withdrawal threshold to mechanical stimuli was evaluated according to a previously reported method (Ikeda et al., 2007) by well‐trained experimenters who were blinded to phenotype. Mechanical stimuli were applied with von Frey filaments (North Coast Medical, Gilroy, CA, USA) with different rigidities (0.02–2.0 g). Each mouse was placed on a metal mesh floor (25 cm × 25 cm), and they were allowed to habituate in a 500‐mL glass beaker that was placed upside down for 30 min prior to the experiments. A von Frey filament was applied manually from beneath, and the 50% threshold was estimated with the up‐and‐down method (Chaplan et al., 1994). Care was taken to reduce the number of trials conducted to avoid inflicting unnecessary pain to the animals. Mechanical allodynia was evaluated in the same animals prior to the intraplantar injection and 6 h after the injection. ‘Sensitization index’ was defined as follows: paw withdrawal threshold at pre‐injection divided by that at 6 h post‐injection for each of the hind limbs. Because paw withdrawal threshold decreases as the tactile hypersensitization becomes severe, this sensitization index becomes larger when the tactile hypersensitization progresses compared to the pre‐injection state.
Preparation of the transverse brain slices
Immediately after conducting the final von Frey filament test 6 h after the injection, the mice were first perfused transcardially with ice‐cold cutting solution under isoflurane anesthesia (5% in 100% O2) and then killed. The brain was then removed, and a block of the forebrain containing the amygdala was dissected out and cut at the midline in ice‐cold cutting solution that was composed of the following (in mm): 2.5 KCl, 0.5 CaCl2, 10 MgSO4, 1.25 NaH2PO4, 2 thiourea, 3 sodium pyruvate, 93 N‐methyl‐d‐glucamine, 20 HEPES, 12 N‐acetyl‐l‐cysteine, 25 d‐glucose, 5 l‐ascorbic acid, and 30 NaHCO3. The solution was equilibrated with 95% O2 + 5% CO2 (osmolality, ~ 290 mOsm/kg). The pH of the solution was titrated to 7.1–7.5 with concentrated HCl. The dissected hemisphere was embedded immediately in a 37 °C agarose solution (1.6%; Sigma‐Aldrich, St. Louis, MO, USA) that was solidified by immediately cooling it by covering it with ice‐cold cutting solution. The dissected right hemisphere containing the amygdala was secured on the cutting stage of a vibrating blade slicer (DSK‐1000; Dosaka EM, Kyoto, Japan) with the rostral end upwards, and 400‐μm‐thick coronal slices containing the amygdala were prepared. Previous lines of evidence have demonstrated that pain‐related neuroplasticity occurs only in the right CeC (Carrasquillo & Gereau, 2007; Ji & Neugebauer, 2009) and that activation of the right amygdala is critical for the development of pain sensitization (Kolber et al., 2010; Crock et al., 2012). Therefore, we examined the right hemisphere in this study. The slices were first incubated in a holding chamber with a constant flow of the cutting solution at 34 °C for 10–15 min. The slices were kept at room temperature (20–25 °C) in standard artificial cerebrospinal fluid (ACSF) that was composed of (in mm) 125 NaCl, 3 KCl, 2 CaCl2, 1.3 MgCl2, 1.25 NaH2PO4, 10 d‐glucose, 0.4 l‐ascorbic acid, and 25 NaHCO3 (pH 7.4, bubbled with 95% O2 + 5% CO2; osmolarity, ~ 310 mOsm/kg) until the electrophysiological recordings. Each slice was transferred to a recording chamber (~ 0.4‐mL volume) and fixed with nylon grids to a platinum frame. The slice was submerged in and continuously superfused with standard ACSF at a rate of 1–2 mL/min. To isolate excitatory synaptic inputs, 100 μm of picrotoxin (Sigma‐Aldrich) was dissolved in ACSF.
Patch‐clamp recordings
CeC neurons were visually identified under an upright microscope (BX‐50WI; Olympus, Tokyo, Japan) with oblique illumination optics. The images were captured with a charge‐coupled device camera (IR‐1000; Dage‐MTI, Michigan City, IN, USA) and stored digitally on a computer. The whole‐cell transmembrane current was recorded from neurons in the right CeC (i.e., contralateral to the injection). The patch‐clamp electrodes were made from borosilicate glass pipettes (1B120F‐4; World Precision Instruments, Sarasota, FL, USA). The composition of the internal solution was (in mm) 120 potassium gluconate, 6 NaCl, 1 CaCl2, 2 MgCl2, 2 ATPMg, 0.5 GTPNa, 12 phosphocreatine Na2, 5 EGTA, and 10 HEPES hemisodium (pH 7.2, adjusted with KOH; osmolarity, ~ 310 mOsm/kg). The tip resistance of the electrodes was 3–8 MΩ. The evoked excitatory postsynaptic currents (eEPSCs) were recorded at a holding potential of −60 mV. Input resistance, resting membrane potential, and whole‐cell capacitance were measured immediately after the establishment of the whole‐cell mode by membrane rupture. The membrane currents were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), low‐pass filtered at 2 kHz, and sampled at 10 kHz with a PowerLab interface (ADInstruments, Colorado Springs, CO, USA). To analyze the type of glutamate receptors involved, 50 μm of D(−)‐2‐amino‐5‐phosphonopentanoic acid (APV; a NMDA receptor antagonist) was dissolved in ACSF and bath‐applied for more than 10 min. All recordings were made at room temperature (20–25 °C). All compounds, except those noted above, were purchased from Sigma‐Aldrich or Nacalai Tesque (Kyoto, Japan).
Afferent pathway stimulation
To trigger action potential‐dependent glutamate release from the afferent fibers arising from the PB under microscopic control, we carefully located the custom‐designed bipolar parallel stimulation electrodes (TOG211‐039a, Unique Medical, Tokyo, Japan) on the fiber tract dorsomedial to the CeC (PB tract), as described previously (Watabe et al., 2013). This configuration enabled us to preferentially evoke EPSCs arising from PB tract fibers in CeC. To calculate the paired‐pulse ratio (PPR) of the EPSCs as a measure of the changes in the presynaptic release properties (McKernan & Shinnick‐Gallagher, 1997), two pulses with an interstimulus interval of 100 ms evoked by 1‐mA stimulation were delivered. The PPR was expressed as the second EPSC (EPSC2) amplitude that was normalized by the first EPSC (EPSC1) amplitude. The coefficient of variation of synaptic currents was calculated as the standard deviation of EPSC1 amplitude normalized by the mean EPSC1 amplitude (Manabe et al., 1993) for eight consecutive EPSC responses.
Data and statistical analysis
The recorded membrane current was analyzed offline with Igor Pro 6 (WaveMetrics, Lake Oswego, OR, USA) with programs written by F. K. The peak amplitude was measured based on the averaged waveform of the EPSCs (eight consecutive trials). The values are expressed as the mean ± standard error of the mean. Differences in the values were compared with a one‐way analysis of variance (anova) followed by a post hoc Gabriel's test, Mann–Whitney U‐test with appropriate Bonferroni's correction, Student's t‐test, or paired t‐test. Differences with probability (P) less than 0.05 were considered significant. Correlation analysis was performed with Spearman's rank‐order correlation.
Results
Formalin‐injected WT mice, but not CGRP KO mice, showed increased EPSC amplitude of the PB‐CeC pathway
Previously, we demonstrated that excitatory synaptic transmission at PB‐CeC synapses is potentiated in chronic pain models (Ikeda et al., 2007; Nakao et al., 2012; Sugimura et al., 2016). Previous reports have also shown that CGRP plays a key role in nociceptive transmission from the PB to the amygdala (Han et al., 2005, 2010). Thus, we examined the effects of endogenous CGRP on evoked EPSCs in the PB‐CeC synapses that were recorded at a holding potential of −60 mV in the presence of 100 μm of picrotoxin in formalin‐induced inflammatory pain models using CGRP KO mice. Sixteen CGRP WT mice were divided into formalin‐ and saline‐injected groups (CGRP WT [formalin], n = 11; CGRP WT [saline], n = 5), and 15 CGRP KO mice were divided into formalin‐ and saline‐injected groups (CGRP KO [formalin], n = 10; CGRP KO [saline], n = 5). Six hours after the formalin or saline injection into the left hind paw, brain slices containing the amygdala were prepared for the electrophysiological recordings. We recorded 61 neurons in the CeC under visual guidance (Fig. 1A). The ease with which good‐quality whole‐cell recordings were obtained from neurons did not differ between the groups. No significant differences were observed in the resting membrane potential, input resistance, whole‐cell capacitance, or threshold membrane potential for action potential generation between neurons in the CGRP WT (formalin; n = 19 neurons), CGRP KO (formalin; n = 18 neurons), CGRP WT (saline; n = 11 neurons), and CGRP KO (saline; n = 13 neurons) mice (Table 1). Figure 1B shows representative traces of EPSCs evoked in the PB‐CeC pathway with increasing stimulus intensities in CeC neurons from the formalin‐ (top) and saline‐treated (bottom) mice. When the stimulation intensity was gradually increased in a stepwise manner, PB tract stimulation resulted in EPSCs with larger amplitude in the CeC in the CGRP WT (formalin) mice (thick dashed traces) compared with the CGRP KO (formalin) mice (thick solid traces; Fig. 1B top). The EPSC amplitude in the CGRP WT (formalin) mice was also larger than that in the CeC in the CGRP WT (saline) mice (thin dashed traces) and CGRP KO (saline) mice (thin solid traces; Fig. 1B bottom). Figure 1C indicates the relationship between the intensity of the PB stimulation and the EPSC amplitude. At a 400‐μA stimulus intensity, the PB‐CeC EPSC amplitude was significantly larger in the CGRP WT (formalin) mice (open circles) than in the saline‐treated groups (Fig. 1C; F 3,57 = 5.044, P = 0.004; vs. CGRP WT [saline], P = 0.026; vs. CGRP KO [saline], P = 0.006, anova followed by post hoc Gabriel's test). At stimulus intensities of 600–1000 μA, the EPSC amplitude of the neurons in the CGRP WT (formalin) mice was significantly larger than that in the neurons in the other groups (Fig. 1C; 600 μA: F 3,57 = 9.147, P < 0.001; vs. CGRP KO [formalin], P = 0.004; vs. CGRP WT [saline], P = 0.001; vs. CGRP KO [saline], P < 0.001; 800 μA: F 3,57 = 13.652, P < 0.001; vs. CGRP KO [formalin], P < 0.001; vs. CGRP WT [saline], P < 0.001; vs. CGRP KO [saline], P < 0.001; 1000 μA: F 3,57 = 16.1, P < 0.001; vs. CGRP KO [formalin], P < 0.001; vs. CGRP WT [saline], P < 0.001; vs. CGRP KO [saline], P < 0.001, anova followed by post hoc Gabriel's test). There were no significant differences between the CGRP KO (formalin) mice and the CGRP WT (saline) mice and CGRP KO (saline) mice. We failed to observe significant difference in EPSC amplitude at 50–200 μA stimulation intensity between CGRP WT and KO groups receiving formalin injection. Because the afferent stimulation in this range gave rise to only small EPSC responses, which are likely to be composed of 1–2 released vesicles (amplitude smaller than 20 pA; Fig. 1B and C), it is likely that the stimulation electrode used in this study recruited only a few fibers with small intensity range, which was insufficient to cause significant EPSC amplitude difference especially when the potentiation is likely to result from changes in release properties. These results strongly indicated that endogenous CGRP was required for formalin‐induced synaptic potentiation in PB‐CeC synapses.
Figure 1.
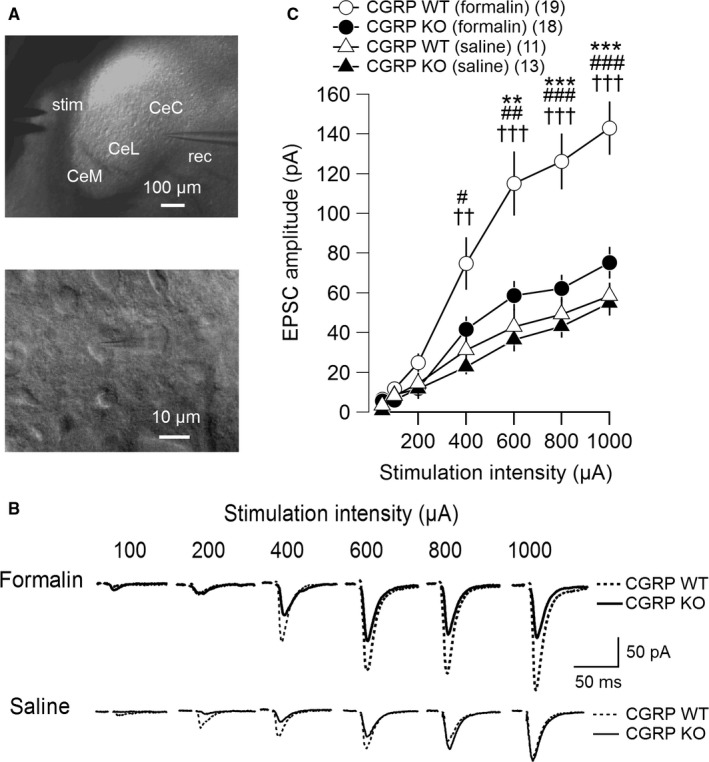
Excitatory postsynaptic currents (EPSCs) evoked at parabrachial (PB)–laterocapsular region of the central amygdala (CeC) synapses in the right amygdala 6 h after intraplantar formalin injection. (A) Micrographs showing the locations of the stimulation and recording electrodes in the central amygdala (CeA) (top) and a representative CeC neuron being recorded (bottom). rec, patch‐clamp recording pipette; stim, parallel bipolar stimulating electrode; CeL, lateral division of the CeA; CeM, medial division of the CeA. (B) Representative traces of averaged EPSC waveforms (n = 8 consecutive responses) evoked by the PB tract stimulation at different intensities. The top and bottom traces, recordings from mice injected with formalin (thick traces) and saline (thin traces), respectively, and the traces in dashed and solid lines are those from the CGRP WT and KO mice, respectively. (C) Relationship between stimulation intensity (abscissa) and evoked EPSC amplitude (ordinate). The open and filled markers indicate the values of neurons in the CeC of the CGRP WT and KO mice, respectively. The circles, after formalin injection (CGRP WT, n = 19 neurons from 11 mice; CGRP KO, n = 18 neurons from 10 mice); the triangles, saline treated (CGRP WT, n = 11 neurons from five mice; CGRP KO, n = 13 neurons from five mice). Mean ± standard error of the mean (SEM). ***P < 0.001 and **P < 0.01 (vs. CGRP KO [formalin]); ### P < 0.001, ## P < 0.01, and # P < 0.05 (vs. CGRP WT [saline]); ††† P < 0.001 and †† P < 0.01 (vs. CGRP KO [saline]), anova followed by post hoc Gabriel's test. Numbers in parentheses indicate the number of neurons. CGRP, calcitonin gene‐related peptide; WT, wild‐type; KO, knockout.
Table 1.
Membrane properties of the central amygdala neurons recorded
| CGRP WT (formalin) | CGRP KO (formalin) | CGRP WT (saline) | CGRP KO (saline) | |
|---|---|---|---|---|
| Resting potential (mV) | −54.3 ± 1.3 | −54.2 ± 1.5 | −56.0 ± 2.2 | −53.5 ± 1.7 |
| Input resistance (MΩ) | 226.4 ± 19.6 | 210.8 ± 16.7 | 192.2 ± 21.1 | 250.6 ± 19.2 |
| Cell capacitance (pF) | 11.1 ± 0.5 | 11.2 ± 0.4 | 12.5 ± 0.7 | 10.6 ± 0.5 |
| Action potential threshold (mV) | −42.2 ± 1.1 | −43.9 ± 0.8 | −41.7 ± 0.8 | −41.7 ± 0.7 |
| n | 19 | 18 | 11 | 13 |
All data were obtained immediately after the establishment of whole‐cell mode. Data are presented as the mean ± SEM. CGRP, calcitonin gene‐related peptide; WT, wild‐type; KO, knockout.
We previously showed that the synaptic potentiation at PB‐CeC synapses was not attributable to the potentiation of the NMDA component in neuropathic pain models (Ikeda et al., 2007). Therefore, we next examined whether the potentiated PB‐CeC EPSCs involved the NMDA receptor component in the formalin‐treated CGRP WT mice. The application of APV (50 μm) did not significantly affect the amplitude of the PB‐CeC EPSCs in the CeC in the CGRP WT (formalin) mice (CGRP WT [formalin] APV‐, 115.3 ± 11.2 pA, n = 5; CGRP WT [formalin] APV+, 100.6 ± 6.7 pA, n = 5, P = 0.203, paired t‐test). The EPSC amplitude of the neurons in the CeC in the CGRP WT (formalin) mice was markedly larger than that in the CGRP WT (saline) mice, not only in the absence of APV (F 2,18 = 14.402, P < 0.001; CGRP WT [formalin] APV‐, 115.3 ± 11.2 pA, n = 5; CGRP WT [saline], 58.4 ± 6.7 pA, n = 11, P < 0.001, anova followed by post hoc Gabriel's test) but also in the presence of APV (F 2,18 = 14.402, P < 0.001; CGRP WT [formalin] APV+, 100.6 ± 6.7 pA, n = 5; CGRP WT [saline], 58.4 ± 6.7 pA, n = 11, P = 0.005, anova followed by post hoc Gabriel's test). These data indicated that the increased EPSC amplitude in PB‐CeC synaptic transmission in the formalin‐injected CGRP WT mice did not involve the NMDA receptor component.
Presynaptic changes in release probabilities accompanied with the PB‐CeC synaptic potentiation
We previously reported that changes in the PPR, which is a parameter affected by changes in the release probabilities in presynaptic terminals (Xu‐Friedman & Regehr, 2000), did not accompany PB‐CeC synaptic potentiation in neuropathic pain models. This result suggested that presynaptic release probabilities did not play a principal role in the neuropathic pain (Ikeda et al., 2007). To investigate whether synaptic potentiation in the inflammatory pain model involved changes in release probabilities, we examined PPR in this study. PB tract stimulation, which was delivered twice with an interval of 100 ms, induced paired‐pulse facilitation in CGRP WT mice (left in Fig. 2A) and CGRP KO mice (right in Fig. 2A). In the CGRP WT (formalin) mice, the PPR, which was expressed as EPSC2/EPSC1, decreased significantly compared with the PPR in the CGRP WT (saline) mice (Fig. 2B left; P = 0.023, Student's t‐test). In contrast, the PPR did not significantly differ between the CGRP KO (formalin) mice and CGRP KO (saline) mice (Fig. 2B right; P = 0.063, Student's t‐test). These results suggest that changes in presynaptic release probabilities accompany the synaptic potentiation observed in the CGRP WT mice. To further identify the origin of increase in the EPSC amplitude, we next analyzed the coefficient of variation (CV) of synaptic currents in CGRP WT mice (left panel in Fig. 2C) and CGRP KO mice (right panel in Fig. 2C). CV has been used as an index of presynaptic processes which reflects the trial‐to‐trial variations in the combination of multiple release sites activated by each stimulation on an assumption that a portion of these sites successfully releases transmitters. CV decreases when the release probability from the terminals of these fibers and/or the number of release sites is increased, both of which result from presynaptic changes (Bekkers & Stevens, 1990; Manabe et al., 1993). CV of EPSC1 amplitude was significantly smaller in the CGRP WT (formalin) mice than that in the CGRP WT (saline) mice (left in Fig. 2D; P = 0.002, Mann–Whitney U‐test). In contrast, CV did not significantly differ between CGRP KO (formalin) mice and CGRP KO (saline) mice (right panel in Fig. 2D; P = 0.161, Mann–Whitney U‐test). These results of the analysis of CV of synaptic currents further suggest that the synaptic potentiation observed in the CGRP WT mice was presynaptically expressed.
Figure 2.
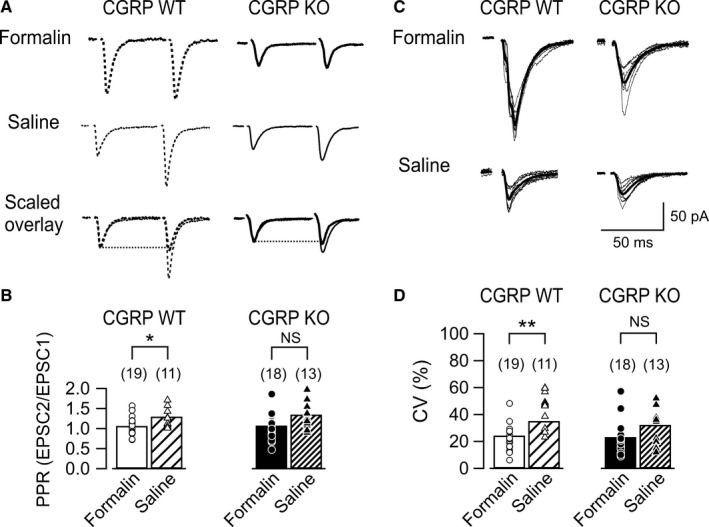
Responses to paired‐pulse stimulation at PB‐CeC synapses. (A) Averaged waveforms (n = 8) of excitatory postsynaptic currents (EPSCs) evoked by paired stimuli (100 ms interstimulus interval) of the PB pathway. The left and right panels show representative responses of CeC neurons to the PB pathway stimulation in the CGRP WT (dashed line) and KO (solid line) mice, respectively. Top, typical recordings in neurons of mice injected with formalin (thick line); middle, those of the mice injected with saline (thin line); bottom, overlaid displays of the responses scaled to the peak of EPSC1. (B) Summary of the paired‐pulse ratio (PPR; EPSC2 amplitude normalized by that of EPSC1) in neurons from CGRP WT (left) and KO (right) mice after formalin (open and filled bars) and saline (hatched bars) injection. Bars, mean; markers, values for each neuron. *P < 0.05 (Student's t‐test). NS, not significantly different. Numbers in parentheses indicate the number of neurons. The PPRs in formalin‐ and saline‐treated mice did not significantly differ in the CGRP KO mice, unlike in the CGRP WT mice. (C) Representative traces of eight consecutive responses of CeC neuron (thin lines) and averaged waveforms of EPSC1s (bold lines). The left and right panels show representative responses of neurons in the CGRP WT and KO mice, respectively. Top, typical recordings in neurons of mice injected with formalin; bottom, those of the mice injected with saline. (D) Summary of the coefficient of variation (CV) of synaptic currents from CGRP WT (left) and KO (right) mice after formalin (open and filled bars) and saline (hatched bars) injection. Bars, mean; markers, values for each neuron. **P < 0.01 (Mann–Whitney U‐test). NS, not significantly different. Numbers in parentheses indicate the number of neurons. The CV in formalin‐ and saline‐treated mice differed significantly in the CGRP WT mice but not in the CGRP KO mice. CGRP, calcitonin gene‐related peptide; WT, wild‐type; KO, knockout.
Acute nociceptive behavior was reduced only during the early part of the second phase of the post‐formalin injection period in the CGRP KO mice
The above results suggest that CGRP is necessary for the potentiation in the EPSC amplitude in the CeC following formalin injection. A possible hypothesis to account for this reduced synaptic potentiation in the CeC in the CGRP KO animals is that the acute effect of formalin in producing peripheral inflammation was smaller in the CGRP KO mice, which simply resulted in reduced CeC potentiation. Therefore, we analyzed the licking behaviors appearing immediately after the formalin injection in the animals used for the above electrophysiological experiments. In agreement with the previous reports, formalin, but not saline, injection produced biphasic nocifensive behaviors which last for 60 min in the CGRP WT mice (Fig. 3A; 0–5 min, F 3,27 = 5.359, P = 0.005; CGRP WT [formalin] vs. CGRP WT [saline], P = 0.049; 20–25 min, F 3,27 = 9.824, P < 0.001; CGRP WT [formalin] vs. CGRP WT [saline], P = 0.001; 25–30 min, F 3,27 = 7.067, P = 0.001; CGRP WT [formalin] vs. CGRP WT [saline], P = 0.005; 30–35 min, F 3,27 = 4.675, P = 0.009; CGRP WT [formalin] vs. CGRP WT [saline], P = 0.034, anova followed by post hoc Gabriel's test). This formalin‐induced biphasic behavioral pattern was significantly altered in the CGRP KO mice in the second phase but not in the first phase (Fig. 3A; 0–5 min, CGRP KO [formalin] vs. CGRP KO [saline], P = 0.039; 20–25 min, CGRP KO [formalin] vs. CGRP KO [saline], P = 0.905; 25–30 min, CGRP KO [formalin] vs. CGRP KO [saline], P = 0.546; 30–35 min, CGRP KO [formalin] vs. CGRP KO [saline], P = 0.143, anova followed by post hoc Gabriel's test). In particular, the CGRP KO (formalin) mice showed attenuated nocifensive behaviors only for a short period of time (20–25 min) compared with the CGRP WT (formalin) mice (Fig. 3A; CGRP WT [formalin] vs. CGRP KO [formalin], P = 0.003, anova followed by post hoc Gabriel's test). These attenuated behaviors were limited to the first half of the second phase. Therefore, we divided the second phase into two phases: the early (early second phase; 20–30 min post‐injection) and later part of the second phase (late second phase; 30–60 min post‐injection). The formalin‐induced acute nociceptive behaviors were significantly attenuated in the CGRP KO mice during the early second phase compared with the CGRP WT mice (Fig. 3B; CGRP WT [formalin] vs. CGRP KO [formalin], P < 0.05, Mann–Whitney U‐test with appropriate Bonferroni's correction), but the levels of these formalin‐induced nocifensive behaviors were indistinguishable between the CGRP WT mice and CGRP KO mice during the first phase (0–5 min post‐injection) (Fig. 3B; CGRP WT [formalin] vs. CGRP KO [formalin], P > 0.05, Mann–Whitney U‐test with appropriate Bonferroni's correction) and the late second phase (Fig. 3B; CGRP WT [formalin] vs. CGRP KO [formalin], P > 0.05, Mann–Whitney U‐test with appropriate Bonferroni's correction). These results suggest that CGRP may contribute to formalin‐induced acute behavioral responses only during this limited period of the phase (early second phase). If the attenuated synaptic potentiation measured in CGRP KO mice in slices prepared at 6 h post‐formalin was a consequence of the reduced acute nocifensive behaviors appearing within this early second phase in these mice, it is expected that the mice showing stronger acute response to the formalin would also show stronger synaptic potentiation. To directly confirm this hypothesis, we analyzed the correlation between the mean EPSC amplitude in slices prepared at 6 h and the total value of the licking behavior in each mouse observed within the early second phase (20–30 min post‐injection) (Fig. 3C). Although the CGRP WT mice showed significantly larger EPSC amplitude and longer licking time than in the CGRP KO mice as shown above, they were not significantly correlated. This result supports a notion that the attenuated synaptic potentiation in CGRP KO mice (Fig. 1) is not a simple result of reduced nociception in the early second phase. It is more likely that CGRP plays a specific role in potentiating the PB‐CeC transmission.
Figure 3.
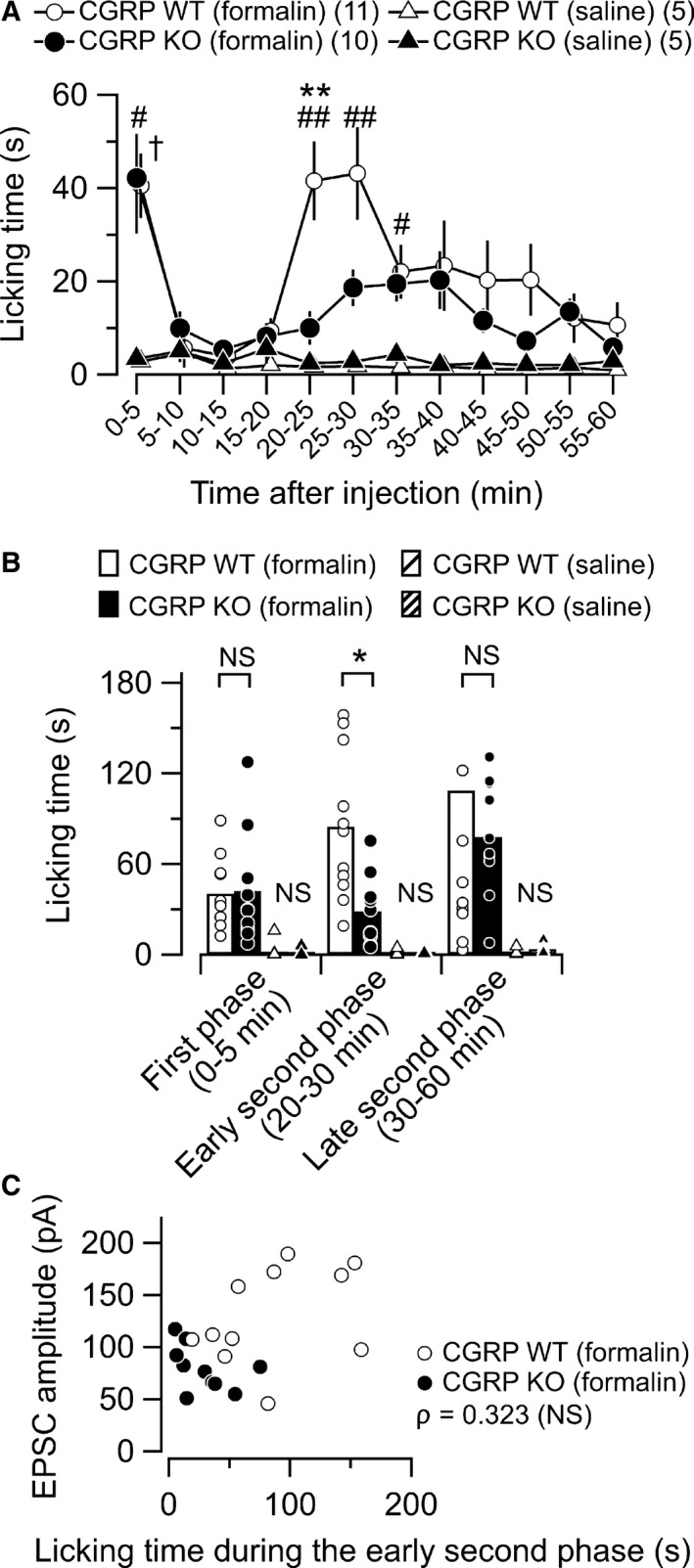
Formalin‐induced acute nociceptive behavior. (A) Summary of the time course of the licking behavior in the mice after intraplantar injection of formalin (5%) or saline into the left hind paw. The open and filled markers indicate the responses of the CGRP WT and KO mice, respectively. The circles, after formalin injection (CGRP WT, n = 11; CGRP KO, n = 10); the triangles, saline treated (CGRP WT, n = 5; CGRP KO, n = 5). Mean ± SEM. The formalin‐injected CGRP KO mice showed significantly reduced nocifensive behaviors only for a specific time window (20–25 min) following the intraplantar formalin injection. ## P < 0.01 and # P < 0.05 (CGRP WT [formalin] vs. CGRP WT [saline]); † P < 0.05 (CGRP KO [formalin] vs. CGRP KO [saline]); **P < 0.01 (CGRP WT [formalin] vs. CGRP KO [formalin]), anova followed by post hoc Gabriel's test. Numbers in parentheses indicate the number of mice. (B) Cumulative licking time in the first (0–5 min), early second (20–30 min), and late second phases (late second phase, 30–60 min) following the intraplantar injection. The open and filled bars show the responses of the formalin‐injected CGRP WT (n = 11) and KO mice (n = 10), respectively. The hatched bars show the responses of the saline‐injected CGRP WT (n = 5) and KO mice (n = 5), respectively. The bars show the mean and the markers show the values from each mouse. *P < 0.05 (Mann–Whitney U‐test with appropriate Bonferroni's correction). NS, not significantly different. (C) Relationship between the nociceptive behaviors 20–30 min after the formalin injection and synaptic potentiation 6 h after the injection in the CGRT WT and KO mice. Abscissa, the time spent performing nociceptive behaviors in the early second phase (see Fig. 3A, B); ordinate, the mean amplitude of excitatory postsynaptic currents (EPSCs) in the each mouse. No significant correlation was found between the time spent performing nociceptive behaviors in the early second phase and synaptic potentiation in formal‐injected group (ρ = 0.323, P = 0.152, n = 21, Spearman's rank‐order correlation). NS, not significantly different; CGRP, calcitonin gene‐related peptide; WT, wild‐type; KO, knockout.
Bilateral tactile allodynia was established 6 h after the formalin injection in the WT mice but not in the CGRP KO mice
Previous studies have demonstrated that CGRP‐mediated synaptic plasticity in the CeC is accompanied by changes in spinal reflex in both naive and pain model animals (Han et al., 2005, 2010). Therefore, we next examined whether endogenous CGRP affected hypersensitivity in the formalin‐induced inflammatory pain models. The paw withdrawal threshold was measured with von Frey filaments prior to the formalin injection (pre‐) and immediately before decapitation (6 h post‐injection; 6 h). Bilateral paw withdrawal thresholds were not influenced by intraplantar saline injection, and there was no significant difference between pre‐ and 6 h post‐injection in either CGRP WT mice (Fig. 4A left panel; right, P = 0.906; left, P = 0.829, Mann–Whitney U‐test) or CGRP KO mice (Fig. 4A right panel; right, P = 0.814; left, P = 0.577, Mann–Whitney U‐test). In contrast, the paw withdrawal threshold in CGRP WT mice differed significantly between pre‐ and 6 h post‐injection in both the left (i.e., ipsilateral to the injection) and right hind paw (i.e., contralateral to the injection) following formalin injection (Fig. 4B left panel; right, P = 0.004; left, P < 0.001, Mann–Whitney U‐test). The degree of decrease in the paw withdrawal threshold at 6 h post‐formalin was significantly smaller in CGRP KO mice (Fig. 4B right panel; right, P = 0.777; left, P = 0.535, Mann–Whitney U‐test).
Figure 4.
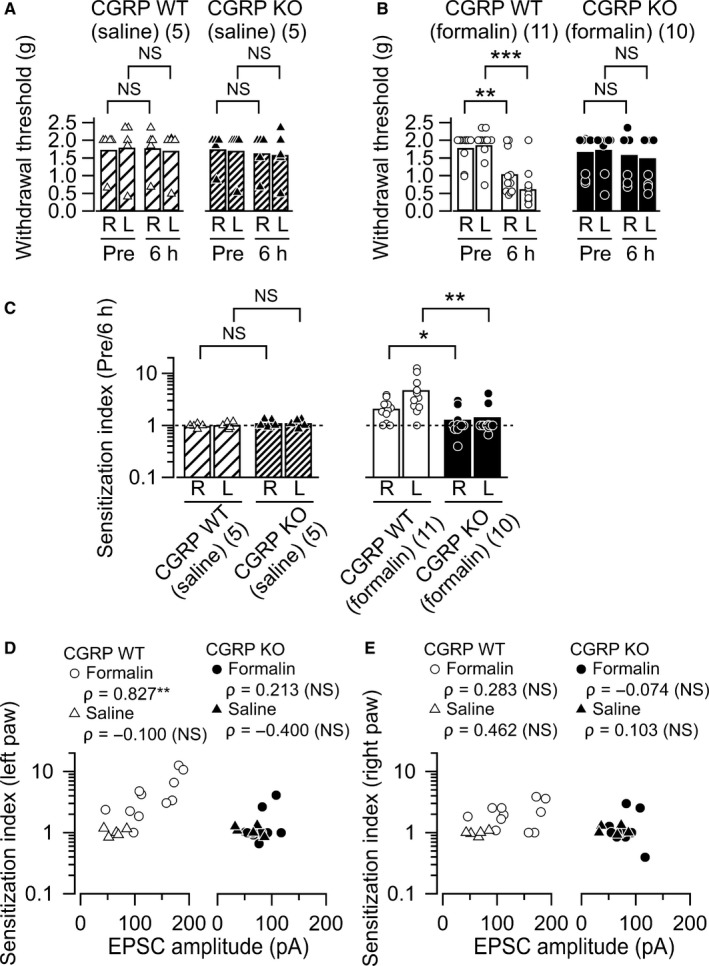
Bilateral mechanical allodynia induced by formalin. (A) and (B) Summary of the paw withdrawal threshold as measured with von Frey filaments. Left in each figure, CGRP WT mice; right in each figure, KO mice. (A) The data before (pre‐) and 6 h after saline injection; (B) the data after formalin injection. R, values from the right hind limb; L, from the left. The bars show the mean and the markers show the values from each mouse. **P < 0.01; ***P < 0.001; NS, not significantly different (Mann–Whitney U‐test). (C) The sensitization index (the paw withdrawal threshold at pre‐injection normalized by that at 6 h post‐injection). Left, effects of CGRP KO on the sensitization index in saline‐injected group; right, those in formalin‐injected. The sensitization index in the bilateral hind paws increased significantly following the formalin injection in the CGRP WT but not significantly in KO mice. *P < 0.05 and **P < 0.01 (Mann–Whitney U‐test). (D) Relationship between synaptic potentiation 6 h after the injection and the sensitization index of the left paw in the CGRT WT (left) and KO (right) mice after formalin (circles) and saline (triangles) injection. Abscissa, the mean amplitude of excitatory postsynaptic currents (EPSCs) in each mouse; ordinate, the sensitization index (see Fig. 4C). There was a positive correlation between these two variables in the CGRP WT (formalin) mice (P = 0.003, n = 11). No significant correlation was found between synaptic potentiation 6 h after the injection and the sensitization index in both the CGRP WT (saline) mice and KO mice (CGRP WT [saline], P = 0.950, n = 5; CGRP KO [formalin], P = 0.554, n = 10, CGRP KO [saline], P = 0.517, n = 5). **P < 0.01 (Spearman's rank‐order correlation). NS, not significantly different. (E) Relationship between synaptic potentiation 6 h after the injection and the sensitization index of the right paw in the CGRT WT (left) and KO (right) mice after formalin (circles) and saline (triangles) injection. No significant correlation was found between synaptic potentiation 6 h after the injection and the sensitization index in both the CGRP WT and KO mice (CGRP WT [formalin], P = 0.399, n = 11; CGRP WT [saline], P = 0.434, n = 5; CGRP KO [formalin], P = 0.839, n = 10, CGRP KO [saline], P = 0.870, n = 5, Spearman's rank‐order correlation). NS, not significantly different; CGRP, calcitonin gene‐related peptide; WT, wild‐type; KO, knockout.
The decrease in the paw withdrawal threshold at 6 h post‐formalin would represent expression of sensitization resulting from peripheral/central (in the limb ipsilateral to the inflammation) or central (in the limb contralateral to the inflammation) mechanisms. To directly evaluate the changes in the paw withdrawal threshold after intraplantar injection of saline or formalin solution, we examined ‘sensitization index’ for bilateral hind limbs and compared the values between WT and KO mice. The sensitization index in both hind paws did not differ significantly between CGRP WT and CGRP KO mice following the saline injection (Fig. 4C left panel; right, P = 0.332; left, P = 0.402, Mann–Whitney U‐test). In contrast, sensitization index values in both hind paws increased significantly following formalin injection in CGRP WT mice compared with the CGRP KO mice (Fig. 4C right panel; right, P = 0.033; left, P = 0.003, Mann–Whitney U‐test). These results strongly suggest that bilateral tactile allodynia was established 6 h after the formalin injection in the WT mice but not in the CGRP KO mice. Next, we asked whether the synaptic potentiation observed in the CeC is correlated with the expression of sensitization in the hind paw because lines of evidence suggest the latent central amygdala (CeA) activation is functionally related to the expression of tactile sensitization (Carrasquillo & Gereau, 2007). We found a significant correlation between the mean EPSC amplitude of the CeC neurons recorded in each mouse and the sensitization index of the left hind paw only in CGRP WT group but not in CGRP KO group (left in Fig. 4D). Such significant correlation was not evident for the sensitization of the right hind paw at 6 h post‐formalin despite the significantly larger sensitization index in the right hind paw (Fig. 4C right and 4E). These results suggest that, while formalin injection induces tactile allodynia bilaterally, the molecular mechanisms underlying the hypersensitivity in non‐injured side might be different from the allodynia observed in the injured side. Exploring precise mechanisms for this ectopic hypersensitivity would be of future interests. In any of the cohorts, correlation between the EPSC amplitude and sensitization index at 6 h post‐saline was not significantly correlated (Fig. 4D and E). These results suggest that endogenous CGRP is necessary for the full expression of the post‐inflammatory synaptic and behavioral plasticity leading to exaggerated pain‐related behaviors.
Discussion
The involvement of CGRP in nociceptive behaviors and plasticity is well established (Wimalawansa, 1997; Russell et al., 2014). While the formalin test is widely used as a tonic nociceptive pain model (Tjølsen et al., 1992), the molecular mechanisms underlying the induction and/or development of pain behavior are not fully understood. In the present study, we used mice with a genetically manipulated αCGRP gene to longitudinally study formalin‐induced amygdala plasticity and nociceptive behaviors. We made the following novel findings in the CGRP KO mice compared with their littermate CGRP WT mice. (i) Synaptic potentiation at the PB‐CeC excitatory pathway 6 h post‐inflammation was drastically attenuated in the right amygdala (Figs 1 and 2). (ii) Formalin‐induced acute nocifensive behavior was reduced only 20–25 min, but reached a level that was indistinguishable from that in WT mice by 60 min after the formalin injection (Fig. 3). (iii) Bilateral tactile allodynia observed 6 h after the formalin injection was significantly suppressed (Fig. 4). These findings revealed a pivotal role of endogenous αCGRP in formalin‐induced plasticity in spino‐parabrachial‐amygdaloid networks, and suggest that αCGRP might be involved in the long‐term changes in central sensitization‐induced pain behaviors, potentially contributing to the exaggerated nociception–emotion linkage in pain chronification.
Amygdalar plasticity correlates with various kinds of chronic pain (Neugebauer et al., 2003; Bird et al., 2005; Ikeda et al., 2007; Fu et al., 2008; Ren et al., 2013). In this study, we found that the amplitude of the eEPSCs recorded from the CeC in slices prepared 6 h after formalin injection, when acute nocifensive behavior such as licking was no longer observed, was significantly larger in the right CeC than that of mice receiving saline injection (Fig. 1). Furthermore, the enhanced PB‐CeC synaptic transmission that was observed in the formalin‐injected CGRP WT mice was drastically attenuated in the formalin‐injected CGRP KO mice (Fig. 1C), demonstrating that endogenous CGRP was involved in the formalin‐induced synaptic plasticity in the PB‐CeC. These results strongly suggest that endogenous CGRP regulates intraplantar formalin injection‐induced synaptic potentiation in right PB‐CeC synapses. A previous study has demonstrated that the blockade of CGRP receptors in the amygdala attenuates nociceptive behaviors and amygdalar plasticity in an arthritis pain model (Han et al., 2005) and that CGRP application in the amygdala facilitates synaptic transmission at PB‐CeC synapses and increases CeC neuron excitability and pain behaviors in normal rats (Han et al., 2010). These studies strongly support the notion that endogenous CGRP is the critical regulator for amygdalar plasticity in a formalin‐induced inflammatory pain model in vivo.
Synaptic potentiation can be induced by postsynaptic and presynaptic mechanisms (Nicoll & Schmitz, 2005; Citri & Malenka, 2008). In the present study, we found that the synaptic potentiation in PB‐CeC pathway was accompanied with changes in PPR and CV, suggesting that the potentiation can be at least partly attributed to presynaptic changes in vesicular release probability (Fig. 2). In a previous study, Han et al. (2010) demonstrated that exogenous application of CGRP potentiates PB‐CeC EPSCs with no significant changes in PPR. Thus, one interesting possibility is that CGRP modulates synaptic transmission via different mechanisms with a different time course, such that CGRP causes postsynaptic modification when applied acutely, while presynaptic modification occurs via long‐term effect. Recently, we found that exogenous application of CGRP also modulates NMDA receptor‐mediated EPSCs in a PKA‐dependent manner (Okutsu et al., 2017), further supporting the notion of acute CGRP effects on postsynaptic mechanisms.
Intraplantar formalin injection induces two‐phase nociceptive responses in rodents (Dubuisson & Dennis, 1977; Tjølsen et al., 1992). The acute phase (first phase) starts immediately after the formalin injection, lasts approximately 5 min, and is then followed by an intermittent period in which little nociceptive behavior is observed (interphase). Following the interphase, a tonic phase (second phase) gradually develops around 20 min to typically 60 min after the injection. The first phase probably results from peripheral inflammation with increased sensitivity of primary afferent nociceptors at the site of injury through the direct activation of transient receptor potential channels (sensory sensitization; McNamara et al., 2007; Tian et al., 2009), whereas the second phase depends on the combination of peripheral inflammation and increased excitability of dorsal horn neurons (spinal sensitization; Tjølsen et al., 1992). Various chemical mediators are thought to be involved in the first and/or second phase, including substance P and bradykinin in the first phase and histamine, serotonin, and prostaglandins in the second phase (Shibata et al., 1989). Similar to previous studies, we observed a biphasic pattern of nociceptive behavior following intraplantar formalin injection (Fig. 3A). Interestingly, only the early part of the second phase was attenuated in CGRP KO mice, but both CGRP KO and CGRP WT mice exhibited indistinguishable levels of nociceptive behavior in the later part of the second phase (Fig. 3B). These results suggest that CGRP plays a critical role in spinal nociceptive processing as well. We did not find any significant correlation between the spinal sensitization 20 min post‐injection and the synaptic transmission 6 h post‐injection (Fig. 3C). Because we used conventional knockout mice with global lack of CGRP in the present study, it is impossible to dissociate the site of action and potential interaction in the molecular mechanisms and pathways involved in the CGPR‐mediated spinal pain processing and the amygdala synaptic potentiation. Studies with region‐specific conditional knockout mice would be an interesting future study to dissect out these problems. CGRP is released in the lumbar spinal cord following noxious stimulation or peripheral inflammation (Morton & Hutchison, 1989; Schaible et al., 1994; Galeazza et al., 1995). Thus, one possible explanation for the attenuated latent nociceptive responses in the CGRP KO mice is that formalin injection triggers the endogenous CGRP release, which contributes to the spinal sensitization during the second phase. Another possibility is that endogenously released CGRP may suppress unknown factors that inhibit the transition from the interphase to the second phase in CGRP WT mice. Therefore, CGRP KO mice exhibited a prolonged interphase due to the absence of such suppression. Our results were partially consistent with those of a previous report on the effects of intraplantar formalin injection‐related pain responses in CGRP KO mice (Salmon et al., 2001). Like our study, they showed a decrease in formalin‐induced nocifensive behavior in CGRP KO mice in the second phase. However, they reported that pain behavior was also reduced in the first phase. This difference might be attributable to the different mouse strains and/or gene knockout strategies used to generate the CGRP KO mice or to differences in the formalin concentrations (2% in their report and 5% in this study). The inflammatory mediators involved in formalin‐induced nocifensive behaviors may vary according to formalin concentration. Thus, a plausible explanation for the present observation is that endogenous CGRP is specifically involved in nocifensive behaviors in the early second phase but not in the first phase following the administration of 5% formalin solution. Further studies are necessary to clarify the molecular events underlying the regulation of the acute and tonic phases of inflammation‐induced pain behaviors.
In addition to spontaneous nocifensive behavioral responses, formalin injection into paw produces latent long‐lasting tactile hypersensitivity (Fu et al., 2000, 2001; Vierck et al., 2008). In the present study, tactile hypersensitivity was significantly reduced 6 h after the formalin injection in the CGRP KO mice, and these results were similar to those of a previous study that reported decreased latencies of paw withdrawal to heat stimulation 24–72 h after subcutaneous injection of complete Freund's adjuvant in CGRP KO mice (Ishida et al., 2014). Intriguingly, this hypersensitivity occurs not only in the injected paw but also at sites distant from the injection site, including the tail (Wiertelak et al., 1994) and undamaged tissue surrounding the injection site (Nozaki‐Taguchi & Yaksh, 1998), and in uninjected paw (Carrasquillo & Gereau, 2007; Vierck et al., 2008; Ambriz‐Tututi et al., 2009, 2013), suggesting the involvement of a central mechanism. In the present study, we also observed latent hypersensitivity in the left‐injected hind paw as well as in the right‐uninjected hind paw 6 h after the formalin injection (Fig. 4B left). Furthermore, we found that this bilateral tactile allodynia was almost completely diminished in the CGRP KO mice (Fig. 4B right). Injection of formalin into the hind paw causes sensitization of dorsal horn neurons and activates c‐Fos expression and extracellular signal‐regulated kinase (ERK) phosphorylation in the spinal cord, which are critical for central sensitization (Presley et al., 1990; Tjølsen et al., 1992; Ji et al., 1999). However, the latent long‐lasting hypersensitivity may not be solely attributable to inflammation‐induced central sensitization of the dorsal horn because the time courses of both c‐Fos expression and pERK increases in the dorsal horn are much faster than the onset of the latent hypersensitivity (Ji et al., 1999; Gao & Ji, 2009; Hunter et al., 2015). Furthermore, formalin‐induced hyperalgesia has been reported to be mediated by the ERK signaling cascade in the CeC (Carrasquillo & Gereau, 2007). Importantly, they showed that ERK inhibition in the CeC decreases formalin‐induced hyperalgesia, while ERK activation in the CeC produces mechanical hyperalgesia in naive animals even without actual peripheral inflammation (Carrasquillo & Gereau, 2007). In addition, CGRP‐induced neuronal plasticity and pain‐related behavior, such as vocalizations and spinal reflexes, depend on the activation of PKA, and not protein kinase C, in the CeC (Han et al., 2005, 2010). Therefore, together with the results of previous reports showing that the PB terminal projections to the CeC contain CGRP and that CeC neurons express the CGRP receptor (Oliver et al., 1998; Dong et al., 2010; Han et al., 2015), the results of the present study suggest that endogenous CGRP plays a critical role in inducing plastic changes in the CeC via ERK and/or PKA activation, which may be involved in the development of persistent tactile hypersensitivity through descending pain pathways (Basbaum & Fields, 1984; Morgan et al., 2008).
It remains undetermined how hypersensitivity is regulated in uninjured sites (Fig. 4C–E). The CeA is well situated to be critically involved in both ascending and descending pain pathways, including the periaqueductal gray matter (PAG) and rostroventromedial medulla (Tracey & Mantyh, 2007; Neugebauer et al., 2009). Although little attention has been paid to the facilitatory effects of the descending pathway compared with the inhibitory effects, electrical stimulation of the rostroventromedial medulla can inhibit and/or facilitate pain behavior depending on the stimulation frequency (Zhuo & Gebhart, 1992, 1997). Furthermore, the neural transmission of the CeA‐PAG pathway is enhanced by stress (Adamec et al., 2001). Therefore, one intriguing possibility is that CGRP modulates the synaptic potentiation in the CeA, which may contribute to an exaggerated nociception–emotion link, such that animals are more susceptible to exhibiting hyperalgesia and enhanced anxiety during persistent pain, through the descending pain pathways. Thus, the CeA does not necessarily have a major role in the acute nociception state at the basal level per se, but rather in the chronic pain state (Veinante et al., 2013). This view is further supported by the results of the present study and other studies that showed that saline‐injected CGRP KO mice did not exhibit any differences in hypersensitivity and synaptic potentiation compared with saline‐injected CGRP WT mice (Figs 1 and 4A).
CGRP has now been established as a key molecule in migraine, such that CGRP antagonists are emerging as a new generation of migraine treatment drugs (Russo, 2015; Schuster & Rapoport, 2016). However, there are still many unanswered questions. The results of the present study further support a role of CGRP in the pain system.
Conflict of interest
The authors declare no conflict of interests.
Author contributions
K.S., A.M.W., and F.K. designed the research project; K.S. performed the experiments; K.S., Y.O., M.N., and Y.T. performed the analyses of electrophysiological and behavioral data; H.K. generated a critical reagent (CGRP KO mice); K.S. and A.M.W. wrote the manuscript. K.S., M.N., Y.T., H.K., A.M.W., and F.K. edited and revised the manuscript. All authors read and approved the final version of the manuscript. We are grateful for the technical assistance of N. Nozaki, T. Tokita and T. Tarumi‐Matsuo.
Data accessibility
All raw data are stored in the academic repository of The Jikei University School of Medicine and are available upon request to the corresponding author.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- CeA
central amygdala
- CeC
laterocapsular region of the central amygdala
- CGRP
calcitonin gene‐related peptide
- EPSC
excitatory postsynaptic current
- ERK
extracellular signal‐regulated kinase
- KO
knockout
- PAG
periaqueductal gray matter
- PB
parabrachial nucleus
- PKA
protein kinase A
- PPR
paired‐pulse ratio
- WT
wild type
Supporting information
Acknowledgements
This work was supported by a Grant‐in‐Aid for Exploratory Research from the Ministry of Education, Culture, Sports, Science and Technology to F.K. (No. 23650208), MEXT‐Supported Program for the Strategic Research Foundation at Private Universities (S1311009) to F.K., Grant‐in‐Aid for Scientific Research (B) to F.K (25293136), Grant‐in‐Aid for Scientific Research (C) to A.M.W (25430015), Strategic Research Program for Brain Sciences to A.M.W. and F.K., Japan Science and Technology Agency, PRESTO to A.M.W., Grants‐in‐Aid for Scientific Research on Innovative Areas “Memory dynamism” (26115523) and “Microendophenotypes of psychiatric disorders” (15H01295) to A.M.W., and Grant‐in‐Aid for Young Scientists (B) to Y.T.
Edited by Masahiko Watanabe
Reviewed by Yuki Hashimotodani, University of Tokyo, Japan; and Volker Neugebauer, Texas Tech University Health Sciences Center, USA
The associated peer review process communications can be found in the online version of this article.
References
- Adamec, R.E. , Blundell, J. & Collins, A. (2001) Neural plasticity and stress induced changes in defense in the rat. Neurosci. Biobehav. R., 25, 721–744. [DOI] [PubMed] [Google Scholar]
- Adedoyin, M.O. , Vicini, S. & Neale, J.H. (2010) Endogenous N‐acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model. Mol. Pain., 6, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Khater, K.M. & Todd, A.J. (2009) Collateral projections of neurons in laminae I, III, and IV of rat spinal cord to thalamus, periaqueductal gray matter, and lateral parabrachial area. J. Comp. Neurol., 515, 629–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambriz‐Tututi, M. , Rocha‐González, H.I. , Castañeda‐Corral, G. , Araiza‐Saldaña, C.I. , Caram‐Salas, N.L. , Cruz, S.L. & Granados‐Soto, V. (2009) Role of opioid receptors in the reduction of formalin‐induced secondary allodynia and hyperalgesia in rats. Eur. J. Pharmacol., 619, 25–32. [DOI] [PubMed] [Google Scholar]
- Ambriz‐Tututi, M. , Palomero‐Rivero, M. , Ramirez‐López, F. , Millán‐Aldaco, D. & Drucker‐Colín, A.R. (2013) Role of glutamate receptors in the dorsal reticular nucleus in formalin‐induced secondary allodynia. Eur. J. Neurosci., 38, 3008–3017. [DOI] [PubMed] [Google Scholar]
- Basbaum, A.I. & Fields, H.L. (1984) Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu. Rev. Neurosci., 7, 309–338. [DOI] [PubMed] [Google Scholar]
- Bekkers, J.M. & Stevens, C.F. (1990) Presynaptic mechanism for long‐term potentiation in the hippocampus. Nature, 346, 724–729. [DOI] [PubMed] [Google Scholar]
- Bernard, J.F. & Besson, J.M. (1990) The spino(trigemino)pontoamygdaloid pathway: electrophysiological evidence for an involvement in pain processes. J. Neurophysiol., 63, 473–490. [DOI] [PubMed] [Google Scholar]
- Bernard, J.F. , Huang, G.F. & Besson, J.M. (1992) Nucleus centralis of the amygdala and the globus pallidus ventralis: electrophysiological evidence for an involvement in pain processes. J. Neurophysiol., 68, 551–569. [DOI] [PubMed] [Google Scholar]
- Bernard, J.F. , Dallel, R. , Raboisson, P. , Villanueva, L. & Le Bars, D. (1995) Organization of the efferent projections from the spinal cervical enlargement to the parabrachial area and periaqueductal gray: a PHA‐L study in the rat. J. Comp. Neurol., 353, 480–505. [DOI] [PubMed] [Google Scholar]
- Bird, G.C. , Lash, L.L. , Han, J.S. , Zou, X. , Willis, W.D. & Neugebauer, V. (2005) Protein kinase A‐dependent enhanced NMDA receptor function in pain‐related synaptic plasticity in rat amygdala neurones. J. Physiol., 564, 907–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo, Y. & Gereau, R.W. (2007) Activation of the extracellular signal‐regulated kinase in the amygdala modulates pain perception. J. Neurosci., 27, 1543–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan, S.R. , Bach, F.W. , Pogrel, J.W. , Chung, J.M. & Yaksh, T.L. (1994) Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Meth., 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Citri, A. & Malenka, R.C. (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology, 33, 18–41. [DOI] [PubMed] [Google Scholar]
- Crock, L.W. , Kolber, B.J. , Morgan, C.D. , Sadler, K.E. , Vogt, S.K. , Bruchas, M.R. & Gereau, R.W. (2012) Central amygdala metabotropic glutamate receptor 5 in the modulation of visceral pain. J. Neurosci., 32, 14217–14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, M. (1992) The role of the amygdala in fear and anxiety. Annu. Rev. Neurosci., 15, 353–375. [DOI] [PubMed] [Google Scholar]
- Dong, Y.L. , Fukazawa, Y. , Wang, W. , Kamasawa, N. & Shigemoto, R. (2010) Differential postsynaptic compartments in the laterocapsular division of the central nucleus of amygdala for afferents from the parabrachial nucleus and the basolateral nucleus in the rat. J. Comp. Neurol., 518, 4771–4791. [DOI] [PubMed] [Google Scholar]
- Dubuisson, D. & Dennis, S.G. (1977) The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain, 4, 161–174. [DOI] [PubMed] [Google Scholar]
- Fanselow, M.S. & LeDoux, J.E. (1999) Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron, 23, 229–232. [DOI] [PubMed] [Google Scholar]
- Fu, K.Y. , Light, A.R. & Maixner, W. (2000) Relationship between nociceptor activity, peripheral edema, spinal microglial activation and long‐term hyperalgesia induced by formalin. Neuroscience, 101, 1127–1135. [DOI] [PubMed] [Google Scholar]
- Fu, K.Y. , Light, A.R. & Maixner, W. (2001) Long‐lasting inflammation and long‐term hyperalgesia after subcutaneous formalin injection into the rat hindpaw. J. Pain, 2, 2–11. [DOI] [PubMed] [Google Scholar]
- Fu, Y. , Han, J. , Ishola, T. , Scerbo, M. , Adwanikar, H. , Ramsey, C. & Neugebauer, V. (2008) PKA and ERK, but not PKC, in the amygdala contribute to pain‐related synaptic plasticity and behavior. Mol. Pain., 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeazza, M.T. , Garry, M.G. , Yost, H.J. , Strait, K.A. , Hargreaves, K.M. & Seybold, V.S. (1995) Plasticity in the synthesis and storage of substance P and calcitonin gene‐related peptide in primary afferent neurons during peripheral inflammation. Neuroscience, 66, 443–458. [DOI] [PubMed] [Google Scholar]
- Gao, Y.J. & Ji, R.R. (2009) c‐Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J., 2, 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J.S. , Li, W. & Neugebauer, V. (2005) Critical role of calcitonin gene‐related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J. Neurosci., 25, 10717–10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J.S. , Adwanikar, H. , Li, Z. , Ji, G. & Neugebauer, V. (2010) Facilitation of synaptic transmission and pain responses by CGRP in the amygdala of normal rats. Mol. Pain., 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, S. , Soleiman, M.T. , Soden, M.E. , Zweifel, L.S. & Palmiter, R.D. (2015) Elucidating an affective pain circuit that creates a threat memory. Cell, 162, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, S. , Corradini, L. , Just, S. , Arndt, K. & Doods, H. (2013) The CGRP receptor antagonist BIBN4096BS peripherally alleviates inflammatory pain in rats. Pain, 154, 700–707. [DOI] [PubMed] [Google Scholar]
- Hunter, D. , Chai, C. & Barr, G.A. (2015) Effects of COX inhibition and LPS on formalin induced pain in the infant rat. Dev. Neurobiol., 75, 1068–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, R. , Takahashi, Y. , Inoue, K. & Kato, F. (2007) NMDA receptor‐independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain, 127, 161–172. [DOI] [PubMed] [Google Scholar]
- Ishida, K. , Kawamata, T. , Tanaka, S. , Shindo, T. & Kawamata, M. (2014) Calcitonin gene‐related peptide is involved in inflammatory pain but not in postoperative pain. Anesthesiology, 121, 1068–1079. [DOI] [PubMed] [Google Scholar]
- Jasmin, L. , Burkey, A.R. , Card, J.P. & Basbaum, A.I. (1997) Transneuronal labeling of a nociceptive pathway, the spino‐(trigemino‐)parabrachio‐amygdaloid, in the rat. J. Neurosci., 17, 3751–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, G. & Neugebauer, V. (2009) Hemispheric lateralization of pain processing by amygdala neurons. J. Neurophysiol., 102, 2253–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, R.R. , Baba, H. , Brenner, G.J. & Woolf, C.J. (1999) Nociceptive‐specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat. Neurosci., 2, 1114–1119. [DOI] [PubMed] [Google Scholar]
- Johansen, J.P. , Cain, C.K. , Ostroff, L.E. & LeDoux, J.E. (2011) Molecular mechanisms of fear learning and memory. Cell, 147, 509–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura, M. , Kuraishi, Y. , Minami, M. & Satoh, M. (1989) Antinociceptive effect of intrathecally administered antiserum against calcitonin gene‐related peptide on thermal and mechanical noxious stimuli in experimental hyperalgesic rats. Brain Res., 497, 199–203. [DOI] [PubMed] [Google Scholar]
- Kolber, B.J. , Montana, M.C. , Carrasquillo, Y. , Xu, J. , Heinemann, S.F. , Muglia, L.J. & Gereau, R.W. (2010) Activation of metabotropic glutamate receptor 5 in the amygdala modulates pain‐like behavior. J. Neurosci., 30, 8203–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraishi, Y. , Nanayama, T. , Ohno, H. , Minami, M. & Satoh, M. (1988) Antinociception induced in rats by intrathecal administration of antiserum against calcitonin gene‐related peptide. Neurosci. Lett., 92, 325–329. [DOI] [PubMed] [Google Scholar]
- Manabe, T. , Wyllie, D.J. , Perkel, D.J. & Nicoll, R.A. (1993) Modulation of synaptic transmission and long‐term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J. Neurophysiol., 70, 1451–1459. [DOI] [PubMed] [Google Scholar]
- McKernan, M.G. & Shinnick‐Gallagher, P. (1997) Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature, 390, 607–611. [DOI] [PubMed] [Google Scholar]
- McNamara, C.R. , Mandel‐Brehm, J. , Bautista, D.M. , Siemens, J. , Deranian, K.L. , Zhao, M. , Hayward, N.J. , Chong, J.A. et al (2007) TRPA1 mediates formalin‐induced pain. Proc. Natl. Acad. Sci. USA, 104, 13525–13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, M.M. , Whittier, K.L. , Hegarty, D.M. & Aicher, S.A. (2008) Periaqueductal gray neurons project to spinally projecting GABAergic neurons in the rostral ventromedial medulla. Pain, 140, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, C.R. & Hutchison, W.D. (1989) Release of sensory neuropeptides in the spinal cord: studies with calcitonin gene‐related peptide and galanin. Neuroscience, 31, 807–815. [DOI] [PubMed] [Google Scholar]
- Mulderry, P.K. , Ghatei, M.A. , Bishop, A.E. , Allen, Y.S. , Polak, J.M. & Bloom, S.R. (1985) Distribution and chromatographic characterisation of CGRP‐like immunoreactivity in the brain and gut of the rat. Regul. Peptides, 12, 133–143. [DOI] [PubMed] [Google Scholar]
- Nakamura‐Craig, M. & Gill, B.K. (1991) Effect of neurokinin A, substance P and calcitonin gene related peptide in peripheral hyperalgesia in the rat paw. Neurosci. Lett., 124, 49–51. [DOI] [PubMed] [Google Scholar]
- Nakao, A. , Takahashi, Y. , Nagase, M. , Ikeda, R. & Kato, F. (2012) Role of capsaicin‐sensitive C‐fiber afferents in neuropathic pain‐induced synaptic potentiation in the nociceptive amygdala. Mol. Pain., 8, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer, V. & Li, W. (2003) Differential sensitization of amygdala neurons to afferent inputs in a model of arthritic pain. J. Neurophysiol., 89, 716–727. [DOI] [PubMed] [Google Scholar]
- Neugebauer, V. , Li, W. , Bird, G.C. , Bhave, G. & Gereau, R.W. (2003) Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J. Neurosci., 23, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer, V. , Li, W. , Bird, G.C. & Han, J.S. (2004) The amygdala and persistent pain. Neuroscientist, 10, 221–234. [DOI] [PubMed] [Google Scholar]
- Neugebauer, V. , Galhardo, V. , Maione, S. & Mackey, S.C. (2009) Forebrain pain mechanisms. Brain Res. Rev., 60, 226–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll, R.A. & Schmitz, D. (2005) Synaptic plasticity at hippocampal mossy fibre synapses. Nat. Rev. Neurosci., 6, 863–876. [DOI] [PubMed] [Google Scholar]
- Nozaki‐Taguchi, N. & Yaksh, T.L. (1998) A novel model of primary and secondary hyperalgesia after mild thermal injury in the rat. Neurosci. Lett., 254, 25–28. [DOI] [PubMed] [Google Scholar]
- Oh‐hashi, Y. , Shindo, T. , Kurihara, Y. , Imai, T. , Wang, Y. , Morita, H. , Imai, Y. , Kayaba, Y. et al (2001) Elevated sympathetic nervous activity in mice deficient in CGRP. Circ. Res., 89, 983–990. [DOI] [PubMed] [Google Scholar]
- Oku, R. , Satoh, M. , Fujii, N. , Otaka, A. , Yajima, H. & Takagi, H. (1987) Calcitonin gene‐related peptide promotes mechanical nociception by potentiating release of substance P from the spinal dorsal horn in rats. Brain Res., 403, 350–354. [DOI] [PubMed] [Google Scholar]
- Okutsu, Y. , Takahashi, Y. , Nagase, M. , Shinohara, K. , Ikeda, R. & Kato, F. (2017) Potentiation of NMDA receptor‐mediated synaptic transmission at the parabrachial‐central amygdala synapses by CGRP in mice. Mol. Pain., 13, 174480691770920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver, K.R. , Wainwright, A. , Heavens, R.P. , Hill, R.G. & Sirinathsinghji, D.J.S. (1998) Distribution of novel CGRP receptor and adrenomedullin receptor mRNAs in the rat central nervous system. Mol. Brain Res., 57, 149–154. [DOI] [PubMed] [Google Scholar]
- Pare, D. , Quirk, G.J. & Ledoux, J.E. (2004) New vistas on amygdala networks in conditioned fear. J. Neurophysiol., 92, 1–9. [DOI] [PubMed] [Google Scholar]
- Poyner, D. & Marshall, I. (2001) CGRP receptors: beyond the CGRP(1)‐CGRP(2) subdivision? Trends Pharmacol. Sci., 22, 223. [DOI] [PubMed] [Google Scholar]
- Presley, R.W. , Menétrey, D. , Levine, J.D. & Basbaum, A.I. (1990) Systemic morphine suppresses noxious stimulus‐evoked Fos protein‐like immunoreactivity in the rat spinal cord. J. Neurosci., 10, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, W. , Kiritoshi, T. , Grégoire, S. , Ji, G. , Guerrini, R. , Calo, G. & Neugebauer, V. (2013) Neuropeptide S: a novel regulator of pain‐related amygdala plasticity and behaviors. J. Neurophysiol., 110, 1765–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, F.A. , King, R. , Smillie, S.‐J. , Kodji, X. & Brain, S.D. (2014) Calcitonin gene‐related peptide: physiology and pathophysiology. Physiol. Rev., 94, 1099–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo, A.F. (2015) CGRP as a neuropeptide in migraine: lessons from mice. Brit. J. Clin. Pharmaco., 80, 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah, P. , Faber, E.S. , Lopez De Armentia, M. & Power, J. (2003) The amygdaloid complex: anatomy and physiology. Physiol. Rev., 83, 803–834. [DOI] [PubMed] [Google Scholar]
- Salmon, A.M. , Damaj, I. , Sekine, S. , Picciotto, M.R. , Marubio, L. & Changeux, J.P. (1999) Modulation of morphine analgesia in alphaCGRP mutant mice. Neuroreport, 10, 849–854. [DOI] [PubMed] [Google Scholar]
- Salmon, A.M. , Damaj, M.I. , Marubio, L.M. , Epping‐Jordan, M.P. , Merlo‐Pich, E. & Changeux, J.P. (2001) Altered neuroadaptation in opiate dependence and neurogenic inflammatory nociception in alpha CGRP‐deficient mice. Nat. Neurosci., 4, 357–358. [DOI] [PubMed] [Google Scholar]
- Sarhan, M. , Freund‐Mercier, M.‐J. & Veinante, P. (2005) Branching patterns of parabrachial neurons projecting to the central extended amgydala: Single axonal reconstructions. J. Comp. Neurol., 491, 418–442. [DOI] [PubMed] [Google Scholar]
- Sato, M. , Ito, M. , Nagase, M. , Sugimura, Y.K. , Takahashi, Y. , Watabe, A.M. & Kato, F. (2015) The lateral parabrachial nucleus is actively involved in the acquisition of fear memory in mice. Mol. Brain, 8, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxen, M.A. , Welch, S.P. & Dewey, W.L. (1993) The mouse paw withdrawal assay: a method for determining the effect of calcitonin gene‐related peptide on cutaneous heat nociceptive latency time. Life Sci., 53, 397–405. [DOI] [PubMed] [Google Scholar]
- Schaible, H.G. , Freudenberger, U. , Neugebauer, V. & Stiller, R.U. (1994) Intraspinal release of immunoreactive calcitonin gene‐related peptide during development of inflammation in the joint in vivo – a study with antibody microprobes in cat and rat. Neuroscience, 62, 1293–1305. [DOI] [PubMed] [Google Scholar]
- Schuster, N.M. & Rapoport, A.M. (2016) New strategies for the treatment and prevention of primary headache disorders. Nat. Rev. Neurol., 12, 635–650. [DOI] [PubMed] [Google Scholar]
- Shibata, M. , Ohkubo, T. , Takahashi, H. & Inoki, R. (1989) Modified formalin test: characteristic biphasic pain response. Pain, 38, 347–352. [DOI] [PubMed] [Google Scholar]
- Sugimura, Y.K. , Takahashi, Y. , Watabe, A.M. & Kato, F. (2016) Synaptic and network consequences of monosynaptic nociceptive inputs of parabrachial nucleus origin in the central amygdala. J. Neurophysiol., 115, 2721–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, R.Q. , Tu, Y.J. , Lawand, N.B. , Yan, J.Y. , Lin, Q. & Willis, W.D. (2004) Calcitonin gene‐related peptide receptor activation produces PKA‐ and PKC‐dependent mechanical hyperalgesia and central sensitization. J. Neurophysiol., 92, 2859–2866. [DOI] [PubMed] [Google Scholar]
- Tian, L.J. , Du, Y.R. , Xiao, Y. , Lv, Z.M. , Yu, Y.Q. , Cui, X.Y. & Chen, J. (2009) Mediating roles of the vanilloid receptor TRPV1 in activation of rat primary afferent nociceptive neurons by formaldehyde. Sheng Li Xue Bao, 61, 404–416. [PubMed] [Google Scholar]
- Tjølsen, A. , Berge, O.‐G.G. , Hunskaar, S. , Rosland, J.H. & Hole, K. (1992) The formalin test: an evaluation of the method. Pain, 51, 5–17. [DOI] [PubMed] [Google Scholar]
- Toda, M. , Suzuki, T. , Hosono, K. , Hayashi, I. , Hashiba, S. , Onuma, Y. , Amano, H. , Kurihara, Y. et al (2008) Neuronal system‐dependent facilitation of tumor angiogenesis and tumor growth by calcitonin gene‐related peptide. Proc. Natl. Acad. Sci. USA, 105, 13550–13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, A.J. (2010) Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci., 11, 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey, I. & Mantyh, P.W. (2007) The cerebral signature for pain perception and its modulation. Neuron, 55, 377–391. [DOI] [PubMed] [Google Scholar]
- Van Rossum, D. , Hanisch, U.K. & Quirion, R. (1997) Neuroanatomical localization, pharmacological characterization and functions of CGRP, related peptides and their receptors. Neurosci. Biobehav. R., 21, 649–678. [DOI] [PubMed] [Google Scholar]
- Veinante, P. , Yalcin, I. & Barrot, M. (2013) The amygdala between sensation and affect: a role in pain. J. Mol. Psychiat., 1, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierck, C.J. , Yezierski, R.P. & Light, A.R. (2008) Long‐lasting hyperalgesia and sympathetic dysregulation after formalin injection into the rat hind paw. Neuroscience, 153, 501–506. [DOI] [PubMed] [Google Scholar]
- Watabe, A.M. , Ochiai, T. , Nagase, M. , Takahashi, Y. , Sato, M. & Kato, F. (2013) Synaptic potentiation in the nociceptive amygdala following fear learning in mice. Mol. Brain, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe, A.M. , Nagase, M. , Hagiwara, A. , Hida, Y. , Tsuji, M. , Ochiai, T. , Kato, F. & Ohtsuka, T. (2016) SAD‐B kinase regulates pre‐synaptic vesicular dynamics at hippocampal Schaffer collateral synapses and affects contextual fear memory. J. Neurochem., 136, 36–47. [DOI] [PubMed] [Google Scholar]
- Wiertelak, E.P. , Furness, L.E. , Horan, R. , Martinez, J. , Maier, S.F. & Watkins, L.R. (1994) Subcutaneous formalin produces centrifugal hyperalgesia at a non‐injected site via the NMDA‐nitric oxide cascade. Brain Res., 649, 19–26. [DOI] [PubMed] [Google Scholar]
- Wimalawansa, S.J. (1997) Amylin, calcitonin gene‐related peptide, calcitonin, and adrenomedullin: a peptide superfamily. Crit. Rev. Neurobiol., 11, 167–239. [DOI] [PubMed] [Google Scholar]
- Xu‐Friedman, M.A. & Regehr, W.G. (2000) Probing fundamental aspects of synaptic transmission with strontium. J. Neurosci., 20, 4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L.C. , Hansson, P. , Brodda‐Jansen, G. , Theodorsson, E. & Lundeberg, T. (1996) Intrathecal CGRP8‐37‐induced bilateral increase in hindpaw withdrawal latency in rats with unilateral inflammation. Brit. J. Pharmacol., 117, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Hoff, A.O. , Wimalawansa, S.J. , Cote, G.J. , Gagel, R.F. & Westlund, K.N. (2001) Arthritic calcitonin/alpha calcitonin gene‐related peptide knockout mice have reduced nociceptive hypersensitivity. Pain, 89, 265–273. [DOI] [PubMed] [Google Scholar]
- Zhuo, M. & Gebhart, G.F. (1992) Characterization of descending facilitation and inhibition of spinal nociceptive transmission from the nuclei reticularis gigantocellularis and gigantocellularis pars alpha in the rat. J. Neurophysiol., 67, 1599–1614. [DOI] [PubMed] [Google Scholar]
- Zhuo, M. & Gebhart, G.F. (1997) Biphasic modulation of spinal nociceptive transmission from the medullary raphe nuclei in the rat. J. Neurophysiol., 78, 746–758. [DOI] [PubMed] [Google Scholar]
- Zimmermann, M. (1983) Ethical guidelines for investigations of experimental pain in conscious animals. Pain, 16, 109–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are stored in the academic repository of The Jikei University School of Medicine and are available upon request to the corresponding author.