

Abstract
Selective inhibition of exclusively transcription‐regulating PTEFb/CDK9 is a promising new approach in cancer therapy. Starting from lead compound BAY‐958, lead optimization efforts strictly focusing on kinase selectivity, physicochemical and DMPK properties finally led to the identification of the orally available clinical candidate atuveciclib (BAY 1143572). Structurally characterized by an unusual benzyl sulfoximine group, BAY 1143572 exhibited the best overall profile in vitro and in vivo, including high efficacy and good tolerability in xenograft models in mice and rats. BAY 1143572 is the first potent and highly selective PTEFb/CDK9 inhibitor to enter clinical trials for the treatment of cancer.
Keywords: antitumor agents, CDK, drug design, PTEFb, sulfoximines
Introduction
The cyclin‐dependent kinase (CDK) family consists of members that are key regulators of the cell‐division cycle (cell‐cycle CDKs), involved in regulation of gene transcription (transcriptional CDKs), and of members with other functions (e.g., CDK5). Whereas cell‐cycle CDKs 1, 2, 3, 4 and 6 are required for the correct timing and order of events of the cell‐division cycle, transcriptional CDK7 and CDK9 regulate the activity of RNA polymerase II via phosphorylation of the carboxy‐terminal domain (CTD). Since their discovery, CDKs have been considered strong prospective targets for a new generation of anticancer drugs.1 Although numerous pharmaceutical companies have initiated drug discoverd xy efforts to identify low‐molecular‐weight CDK inhibitors for cancer therapy, most pan‐CDK inhibitors have failed rigorous clinical testing. Nevertheless, the recent approvals of the selective CDK4/6 inhibitors palbociclib and ribociclib demonstrate that CDK inhibitors with narrow selectivity profiles can have therapeutic clinical utility.1c, 2
Positive transcription elongation factor b (PTEFb) is a heterodimer of CDK9 and one of four cyclin partners, cyclin T1, cyclin K, cyclin T2a or cyclin T2b. Whereas CDK9 is exclusively involved in transcriptional regulation, CDK7 in addition participates in cell‐cycle regulation as a CDK‐activating kinase. Transcription of genes by RNA polymerase II is initiated by assembly of the preinitiation complex at the promoter region and phosphorylation of Ser5 and Ser7 of the CTD by CDK7/cyclin H. For most genes, RNA polymerase II stops mRNA transcription after it has moved 20–40 nucleotides along the DNA template. This promoter‐proximal pausing of RNA polymerase II is mediated by negative elongation factors and is recognized as a major control mechanism to regulate expression of rapidly induced genes in response to a variety of stimuli.3 PTEFb is crucially involved in overcoming promoter‐proximal pausing of RNA polymerase II and transition into a productive elongation state by phosphorylation of Ser2 of the CTD, as well as by phosphorylation and inactivation of negative elongation factors. Deregulated kinase activity of CDK9 of the PTEFb heterodimer is associated with a variety of human pathological conditions such as hyperproliferative diseases (e.g., cancer), virally induced infectious diseases and cardiovascular diseases.4 PTEFb‐mediated transcription of short‐lived, antiapoptotic survival proteins, such as Mcl‐1, and oncogenes, such as c‐MYC, plays a critical role in cancer cell growth and survival. In addition, these proteins exhibit important functions in the development of resistance to chemotherapy. Inhibition of PTEFb/CDK9 results in the rapid depletion of short‐lived mRNA transcripts of these important survival proteins and oncogenes. Thus, selective, transient inhibition of exclusively transcription‐regulating PTEFb/CDK9 is a promising new approach in cancer therapy.5
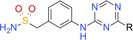
We now report the identification of atuveciclib (BAY 1143572, Figure 1), the first potent and highly selective PTEFb/CDK9 inhibitor that entered clinical trials. Starting from lead compound BAY‐958, which displayed potent PTEFb‐inhibitory activity and high kinase selectivity in vitro, lead optimization by a collaborative effort involving medicinal chemistry, pharmacology, DMPK, structural biology and computational chemistry led to the identification of the orally available clinical candidate BAY 1143572. Structurally characterized by an unusual benzyl sulfoximine group, BAY 1143572 exhibited the most promising overall profile with respect to potency, kinase selectivity, physicochemical and DMPK properties, and antitumour efficacy in animal models.
Figure 1.
Structures of lead compound BAY‐958 and clinical candidate atuveciclib (BAY 1143572).
Results and Discussion
Search for low‐molecular‐weight PTEFb/CDK9 inhibitors
Attracted by the encouraging biological rationale, we set course to identify a potent, highly selective and orally applicable PTEFb/CDK9 inhibitor exclusively targeting oncogenic transcription for the treatment of cancer. Special attention was paid to the aspect of high selectivity with respect to kinases in general as well as within the CDK family, in particular high CDK9 selectivity. Such CDK9 selectivity was anticipated as being crucial for the differentiation of a potential candidate from cell‐cycle‐addressing selective CDK inhibitors (like the CDK4/6 inhibitor palbociclib6) and from pan‐CDK inhibitors (like dinaciclib7 or roniciclib8), because a differentiated activity and tolerability profile can be expected. Furthermore, favorable physicochemical and DMPK properties, which have been key hurdles in many kinase inhibitor projects,9 were of high importance.
In the search for a suitable lead structure, triazine BAY‐958 (LDC 526, Figure 1) attracted our attention, as this compound proved to be a potent PTEFb/CDK9 inhibitor that also displayed very high kinase selectivity, even within the CDK family (in‐house kinase panel: IC50 CDK9/CycT1: 11 nm, selectivity vs. CDK2: 98, see Table 3; Millipore panel: IC50 CDK9/CycT1: 5 nm, selectivity vs. other CDKs: >90, see Table 4). BAY‐958 also exhibited good antiproliferative activity in vitro, for example against HeLa cells (IC50: 1000 nm) and MOLM‐13 cells (IC50: 280 nm). In vitro pharmacokinetic studies with BAY‐958 demonstrated high metabolic stability in rat hepatocytes (ratHep) and liver microsomes (ratLM), resulting in a low predicted blood clearance (CLb) of 0.33 L h−1 kg−1 and 0.48 L h−1 kg−1, respectively (see Table 3).
Table 3.
Properties of PTEFb inhibitors BAY‐958, benzyl sulfone 4, and BAY 1143572.
| BAY‐958 | 4 | BAY 1143572 | |
|---|---|---|---|
| CDK9/CycT1 IC50 [nm] | 11 | 24 | 13 |
| Selectivity vs. CDK2, ratio of IC50 values | 98 | 67 | 100 |
| HeLa IC50 [nm][a] | 1000 | 1500 | 920 |
| MOLM‐13 IC50 [nm][a] | 280 | 130 | 310 |
| TPSA[b] | 120.0 | 94.1 | 100.9 |
| Sw, pH 6.5 [mg L−1][c] | 11 | 4 | 479 |
| P app A→B [nm s−1] | 22 | 143 | 35 |
| Efflux ratio | 15 | 1.2 | 6 |
| CLb, ratHep [L h−1 kg−1] | 0.33 | 0.79 | 0.17 |
| CLb, ratLM [L h−1 kg−1] | 0.48 | 0.58 | 0.15 |
| CLb, rat in vivo, i.v. [L h−1 kg−1] | 0.50 | 2.7 | 1.1 |
| V ss, rat in vivo, i.v. [L kg−1] | 1.4 | 1.5 | 1.0 |
| t 1/2, rat in vivo, i.v. [h] | 0.7 | 0.3 | 0.6 |
| AUC,[d] rat in vivo, p.o. [mg h−1 L−1] | 0.11 | 0.16 | 0.28 |
| C max,[d] rat in vivo, p.o. [mg L−1] | 0.029 | 0.059 | 0.058 |
| F, rat in vivo, p.o. [%] | 10 | 53 | 54 |
| Blood/plasma ratio (rat) | 3.0 | 1.2 | 1.1 |
| CYP inhibition [μm] | >20 | >20 | >20 |
| CYP1A2 induction | NOEL[e] ≤5 μg L−1 | no (up to 370 μg L−1) | no (up to 370 μg L−1) |
[a] Cells were treated with test compounds for 96 h. [b] Topological polar surface area.20 [c] The solid state of the test compounds was not characterized. [d] Normalized to 1 mg kg−1. [e] No‐observed‐effect‐level.
Table 4.
CDK‐inhibitory activity of lead compound BAY‐958 and clinical candidate BAY 1143572 in the Merck Millipore KinaseProfilerTM panel.
| IC50 [nm] | BAY‐958 | BAY 1143572 |
|---|---|---|
| CDK9/CycT1(h) | 5 | 6 |
| CDK1/CycB(h) | 690 | 1100 |
| CDK2/CycE(h) | 470 | 1000 |
| CDK3/CycE(h) | 570 | 890 |
| CDK5/p35(h) | 800 | 1600 |
| CDK6/CycD3(h) | 4400 | >10 000 |
| CDK7/CycH/MAT1(h) | >10 000 | >10 000 |
On the other hand, BAY‐958 has a rather low aqueous solubility of 11 mg L−1 at pH 6.5. Moreover, we recorded a very moderate permeability coefficient (P app A→B) of 22 nm s−1 and a high efflux ratio of 15 in Caco‐2 cells, as well as a blood/plasma partitioning in rats of about 3:1 (see Table 3). Pharmacokinetic studies in rats in vivo revealed a low blood clearance (CLb: 0.5 L h−1 kg−1), a high volume of distribution (V ss: 1.4 L kg−1) and a short half‐life (t 1/2: 0.7 h) of BAY‐958. After oral administration, a low bioavailability of only 10 % was observed.
Due to the slow absorption and low bioavailability of BAY‐958 observed in the rat pharmacokinetic studies, the hydrochloride of BAY‐958 was used in a subsequent mouse xenograft study to analyze the in vivo efficacy. Daily oral administration of BAY‐958 hydrochloride at 30 or 40 mg kg−1 resulted in a marked inhibition of tumor growth with treatment‐to‐control (T/C) ratios of 0.16 and 0.12, respectively, at the end of the experiment (Figure 2). Additionally, there was excellent tolerability upon treatment with BAY‐958 hydrochloride, with a body weight change of less than 10 % and no fatal toxicities. Nevertheless, due to the unfavorable physicochemical and DMPK properties of BAY‐958, we elected to search for a compound with an improved overall profile.
Figure 2.
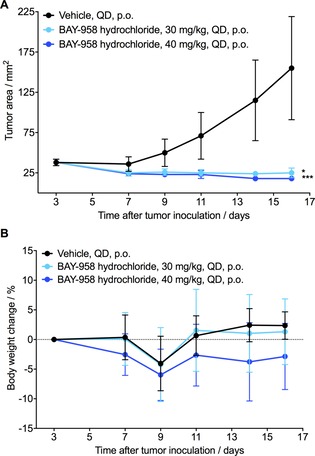
Antitumor efficacy of BAY‐958 hydrochloride in an MOLM‐13 human AML model in mice. Treatments were started three days after tumor cell inoculation. A) Tumor growth. Asterisks indicate statistical significance relative to the vehicle control, calculated using the mean tumor areas at the time point when the vehicle group was sacrificed (*p<0.047, ***p<0.001). B) Body weight change expressed as a percentage of the starting weight.
Structural variation of lead compound BAY‐958
Lead compound BAY‐958 and structurally related derivatives were easily accessible by straightforward chemistry (Scheme 1). In the first step, commercial 2,4‐dichloro‐1,3,5‐triazine (1) was reacted with a suitable aniline under basic conditions to give the corresponding N‐aryl‐4‐chloro‐1,3,5‐triazin‐2‐amine 2. In the second step, the crude coupling product 2 was reacted with a boronic acid derivative to give the desired product 3.
Scheme 1.
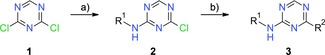
General synthesis of triazine‐based PTEFb inhibitors. Reagents and conditions: a) R1NH2, DIPEA, THF/iPrOH (1:1), −40 °C to 0 °C; b) R2B(OR)2, Pd(dppf)Cl2⋅CH2Cl2, K3PO4, dioxane/water (10:1), 140–145 °C, microwave oven or R2B(OR)2, Pd(PPh3)4, K2CO3 (aq), 1,2‐dimethoxyethane, 100 °C.
The binding mode of lead compound BAY‐958 with CDK9 was investigated by docking experiments (Figure 3). According to these modeling studies, binding to the hinge region of CDK9 is mediated by the triazine core and the aniline NH. The benzyl sulfonamide moiety is directed toward the exit of the ATP binding pocket, whereas the methoxyphenyl substituent points toward the ribose pocket. Favorable interactions include two hydrogen bonds to the hinge region and two hydrogen bonds formed by the amino part of the sulfonamide group, as well as a π‐stacking interaction of the methoxyphenyl moiety with the Phe103 gatekeeper residue. In addition, a weak hydrogen bond between the para‐fluoro substituent and the catalytic Lys48 may be postulated.10 However, the high CDK9 selectivity of BAY‐958, versus other CDKs, could not be rationalized.
Figure 3.
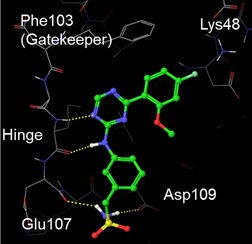
Docking mode of BAY‐958 in complex with CDK9. The compound was docked into a published X‐ray complex of CDK9/CycT1 (PDB ID: 3MY1) using the Glide docking program. The sulfonamide moiety forms two hydrogen bonds, one to Glu107 (main chain) and the other to Asp109 (side chain). For the substituted phenyl ring attached to the triazine, a π‐stacking interaction with Phe103 and a weak hydrogen bond to Lys48 may be postulated.
Analyzing the chemical structure of lead compound BAY‐958, the chemistry team was well‐disposed toward inclusion of the central triazine core. The benzyl sulfonamide group, however, raised concerns: due to its polarity, high number of heteroatoms and multiple hydrogen‐bonding properties, it was suspected of contributing significantly to the low aqueous solubility of BAY‐958, the rather moderate Caco‐2 permeability and the high efflux ratio. Furthermore, the acidity properties and potential instability of the benzyl sulfonamide group were considered and ultimately triggered the evaluation of potential structural alternatives at this position (Table 1).
Table 1.
Selected examples of lead optimization efforts on the western side and corresponding key in vitro properties.
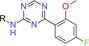
| ||||
|---|---|---|---|---|
| Compound | R | CDK9/CycT1 IC50 [nm] | Selectivity vs. CDK2, ratio of IC50 values | HeLa IC50 [nm][a] |
| BAY‐958 |
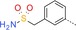
|
11 | 98 | 1000 |
| 4 |
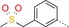
|
24 | 67 | 1500 |
| 5 |
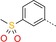
|
32 | 19 | 1340 |
| 6 |
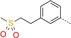
|
49 | 12 | 2200 |
| 7 |
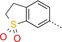
|
7 | 22 | 550 |
| 8 |
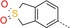
|
51 | <1 | 11 000 |
| 9 |
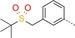
|
47 | 43 | 2900 |
| 10 |
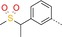
|
85 | 21 | 3200 |
| 11 |
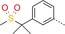
|
1000 | >20 | ND |
| 12 |
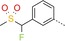
|
20 | 13 | 890 |
| 13 |
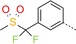
|
67 | 19 | 4200 |
[a] Cells were treated with test compounds for 96 h; ND: not determined.
Sulfones aroused our attention, because analogue 4 exhibited a similar CDK9‐inhibitory activity as BAY‐958 in our biochemical assay in vitro [IC50 CDK9/CycT1 (in‐house), 4: 24 nm vs. BAY‐958: 11 nm, see Table 1], even though the sulfone group cannot form similar hydrogen bonds as the amino part of the sulfonamide group (Figure 3). Sulfone 4 also exhibited similar selectivity against cell‐cycle kinase CDK2 (ratio of IC50 values CDK2/CDK9, 4: 67 vs. BAY‐958: 98) and antiproliferative activity against HeLa cells (IC50 4: 1.5 μm vs. BAY‐958: 1.0 μm). To address the initial concerns regarding the stability and acidity of a polar functional group at the benzylic position, analogues 5 and 6 were synthesized; however, in this sulfone series, the removal of the methylene group from 4 (compound 5) or the introduction of an additional methylene group to 4 (compound 6) resulted in both decreased PTEFb/CDK9 activity and significantly reduced selectivity against cell‐cycle kinase CDK2 (Table 1). This finding was quite surprising, as the sulfone group is directed toward the exit of the ATP binding pocket. Furthermore, the introduction of methyl or even fluoro substituent(s) at the benzylic position was not well‐tolerated with regard to activity against CDK9 (compounds 10, 11, 13), as well as selectivity against CDK2 (compounds 10–13). Inclusion of an annulated ring system (compounds 7, 8) also resulted in poor CDK9 selectivity.
Extensive structural variation on the eastern side of lead compound BAY‐958 also revealed a rather steep SAR with respect to selectivity against CDK2 and potency (Table 2).
Table 2.
Selected examples of lead optimization efforts on the eastern side and corresponding key in vitro properties.
| ||||
|---|---|---|---|---|
| Compound | R | CDK9/CycT1 IC50 [nm] | Selectivity vs. CDK2, ratio of IC50 values | HeLa IC50 [nm][a] |
| BAY‐958 |
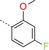
|
11 | 98 | 1000 |
| 14 |
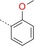
|
23 | 62 | 1600 |
| 15 |
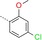
|
27 | 108 | 3800 |
| 16 |
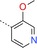
|
1800 | 14 | ND |
| 17 |
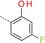
|
130 | 6 | 2900 |
| 18 |
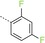
|
270 | 16 | >3000 |
| 19 |
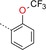
|
620 | 20 | >3000 |
| 20 |
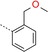
|
1700 | >11 | ND |
| 21 |
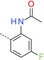
|
3600 | <1 | 3000 |
| 22 |
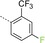
|
15 000 | 1 | ND |
| 23 |
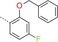
|
4 | 34 | 100 |
[a] Cells were treated with test compounds for 96 h; ND: not determined.
The para‐fluoro substituent on the phenyl ring of BAY‐958 proved to be beneficial for antiproliferative activity in vitro: analogues 14 (p‐H) and 15 (p‐Cl) both displayed decreased IC50 values against HeLa cells (IC50 14: 1.6 μm, 15: 3.8 μm vs. BAY‐958: 1.0 μm). Pyridyl analogue 16 exhibited significantly reduced activity against CDK9 in the biochemical assay (IC50 16: 1800 nm). Moreover, an ortho‐alkoxy substituent was crucial for the desired CDK9 selectivity, as well as in vitro potency; various functional groups were evaluated as potential structural alternatives, but all such analogues displayed significantly reduced selectivity against CDK2 and inhibitory activity against CDK9 (see Table 2 for selected examples; e.g., 17–22). Introduction of a sterically larger alkoxy substituent, especially a benzyloxy group, resulted in significantly improved potency; however, most of these compounds suffered from very low metabolic stability in vitro. For instance, whilst benzyl analogue 23 proved to be a highly potent CDK9 inhibitor, it exhibited low metabolic stability in liver microsome preparations of human, mouse and rat origin, with recovery of the parent compound after incubation for 60 minutes of 0 %, 0.2 % and 0 %, respectively.
Identification of BAY 1143572
Since the late discovery of the sulfoximine group in 1949,11 sulfoximine chemistry12 has been rather a niche discipline. Applications have mainly centered around the use of sulfoximines as either chiral auxiliaries13 or ligands in asymmetric catalysis.14 Until very recently, the sulfoximine group has rarely been used in life science approaches, even though it offers a unique combination of interesting properties, namely high stability, favorable physicochemical properties, hydrogen‐bond acceptor/donor functionalities and structural diversity.15 Lately, however, there has been a rapidly increasing interest in sulfoximines as pharmacophores in the life sciences.15, 16 Our prior, long‐standing interest in sulfoximines in medicinal chemistry had been exclusively focused on aryl sulfoximines;17 nevertheless, we then decided to evaluate a benzyl sulfoximine analogue of BAY‐958.
In the corresponding synthesis (Scheme 2), thioether 25, prepared from the commercial benzyl chloride 24, was oxidized and the resulting sulfoxide 26 was converted into sulfoximine 27 using the rhodium‐catalyzed method of Okamura and Bolm.18 Next, a protecting group was introduced at the sulfoximine nitrogen to provide 28. The nitro group of 28 was reduced and the resulting aniline 29 reacted with 2,4‐dichloro‐1,3,5‐triazine (1) under basic conditions. The crude coupling product 30 was used in a subsequent Suzuki reaction to yield compound 31. Finally, deprotection and chiral HPLC separation provided the (R)‐sulfoximine BAY 1143572 and its enantiomer 32.19 The stereochemistry at the sulfur of the sulfoximine group was determined by X‐ray crystallography of the corresponding N‐acetyl derivative 33 (Figure 4).
Scheme 2.
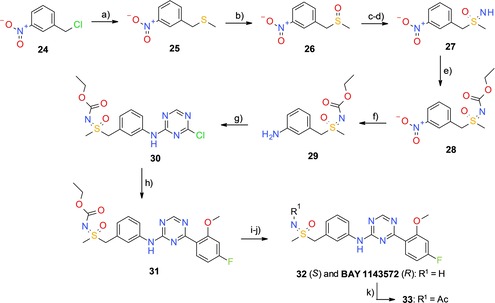
Synthesis of BAY 1143572. Reagents and conditions: a) NaSMe, EtOH, −15 °C, then RT, 3 h; b) H5IO6, FeCl3, MeCN, RT, 90 min, 70 % (2 steps); c) CF3C(O)NH2, PhI(OAc)2, MgO, Rh2(OAc)4, CH2Cl2, RT, 16 h; d) K2CO3, MeOH, RT, 1 h, 79 % (2 steps); e) ClC(O)OEt, pyridine, 0 °C to RT, 24 h; f) TiCl3, THF, RT, 18 h, 94 % (2 steps); g) 2,4‐dichloro‐1,3,5‐triazine (1), DIPEA, THF/iPrOH (1:1), −40 °C to 0 °C, 3 h; h) 4‐fluoro‐2‐methoxyphenylboronic acid, Pd(PPh3)4, K2CO3 (aq), 1,2‐dimethoxyethane, 100 °C, 80 min, 36 %; i) NaOEt, EtOH, 60 °C, 7 h, 90 %; j) preparative chiral HPLC; k) AcCl, TEA, CH2Cl2, 0 °C to RT, 3 h, 57 %.
Figure 4.
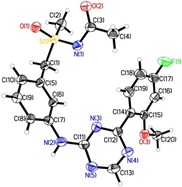
X‐ray structure of N‐acetyl derivative 33.
In comparison with BAY‐958 and benzyl sulfone 4, benzyl sulfoximine BAY 1143572 clearly exhibited the best overall profile in vitro and in vivo (Table 3). BAY 1143572 is a potent and highly selective CDK9 inhibitor (IC50 CDK9/CycT1: 13 nm, ratio of IC50 values CDK2/CDK9: 100). The selectivity of BAY 1143572 within the CDK family is even higher than that of lead compound BAY‐958 (Table 4). Outside the CDK family, submicromolar inhibitory activity was only recorded against GSK3 kinase (IC50 GSK3α: 45 nm, GSK3β: 87 nm) (see the Supporting Information). BAY 1143572 also demonstrated similar antiproliferative activity as lead compound BAY‐958, for example against HeLa cells (IC50 BAY 1143572: 920 nm, BAY‐958: 1000 nm) and MOLM‐13 cells (IC50 BAY 1143572: 310 nm, BAY‐958: 280 nm). In comparison with BAY 1143572, the (S)‐enantiomer 32 revealed very similar in vitro properties, well within the limits of measurement accuracy; however, with multiple batches of enantiomer 32 there was a trend toward a slightly lower activity against CDK9 in the biochemical assay (IC50 CDK9/CycT1: 16 nm) and antiproliferative activity against HeLa cells (IC50: 1100 nm). Surprisingly, BAY 1143572 has a high aqueous solubility of 479 mg L−1, whereas both BAY‐958 and benzyl sulfone 4 have a low aqueous solubility of 11 mg L−1 and 4 mg L−1, respectively. Nevertheless, BAY 1143572 demonstrated improved Caco‐2 permeability and a decreased efflux ratio (P app A→B: 35 nm s−1, ER: 6) relative to lead compound BAY‐958 (P app A→B: 22 nm s−1, ER: 15).
In an in vivo pharmacokinetic study in rats, BAY 1143572 showed low blood clearance (CLb 1.1 L h−1 kg−1), whereas benzyl sulfone 4 exhibited a significantly higher blood clearance (2.7 L h−1 kg−1) (Table 3). The volumes of distribution (V ss) of BAY‐958 (1.4 L kg−1), benzyl sulfone 4 (1.5 L kg−1) and BAY 1143572 (1.0 L kg−1) proved to be rather similar, but, in contrast to lead compound BAY‐958, benzyl sulfone 4 and BAY 1143572 showed significantly improved oral bioavailability (4: 53 %, BAY 1143572: 54 %). Furthermore, benzyl sulfone 4 and BAY 1143572 exhibited blood/plasma ratios of about 1. Relative to BAY 1143572, its enantiomer 32 revealed very similar rat PK properties in vivo (CLb: 1.2 L h−1 kg−1, V ss: 1.2 L kg−1, t 1/2: 0.6 h, F: 53 %). All three key CDK9 inhibitors, BAY‐958, benzyl sulfone 4 and BAY 1143572, did not show significant inhibition of cytochrome P450 activity, with IC50 values >20 μm; however, the switch from benzyl sulfonamide BAY‐958 to benzyl sulfone 4 and benzyl sulfoximine BAY 1143572 removed a potential CYP1A2 in vitro induction liability. Induction of CYP3A4 in vitro was not observed.
For the subsequent in vivo studies, two xenograft models of human acute myeloid leukemia (AML) were selected, as it is well known from the preclinical literature21 that this indication is dependent on oncogenic transcription and also the only indication where nonselective CDK inhibitors with CDK9 inhibitory activity have reached randomized phase 2 clinical trials (NCT01349972, NCT00634244, NCT00795002). Of note, BAY 1143572 did not show relevant activity against Flt3 in the Merck Millipore KinaseProfilerTM panel (see the Supporting Information); therefore, the possibility that in vivo activity of the compound might be partially mediated by Flt3 inhibition in Flt3‐alterated MOLM‐13 and MV4‐11 cells can be excluded.
In in vivo efficacy studies in the MOLM‐13 xenograft model in mice, BAY 1143572 demonstrated greater potency and higher antitumor efficacy than benzyl sulfone 4 (Figure 5 A and Table 5). Daily treatment with benzyl sulfone 4 at the maximum tolerated dose of 20 mg kg−1 revealed no efficacy (T/C ratio of 0.73, p=0.073). In contrast, daily administration of BAY 1143572 at 6.25 or 12.5 mg kg−1 resulted in a dose‐dependent antitumor efficacy with a T/C ratio of 0.64 and 0.49, respectively (p<0.001). In a separate experiment with a higher daily dose of 20 or 25 mg kg−1 BAY 1143572, antitumor efficacy with a T/C ratio of 0.41 and 0.31, respectively, was observed (p<0.001). The 25 mg kg−1 once daily dose is the maximum tolerated dose in nude mice. Furthermore, BAY 1143572 administered at 25 or 35 mg kg−1, three days on / two days off, resulted in a T/C ratio of 0.33 and 0.20, respectively (p<0.001), suggesting a possible flexibility for the selection of a clinical dosing regimen (Figure 5 B and Table 5). Treatment with BAY 1143572 was well‐tolerated, as demonstrated by less than 10 % mean body weight reduction throughout the study.
Figure 5.
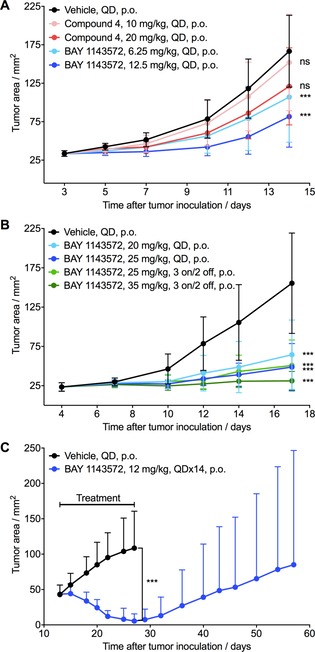
Antitumor efficacy in two AML models in mice and rats. A, B: Antitumor efficacy of BAY 1143572 in an MOLM‐13 human AML model in mice. A) Tumor growth in mice treated with compound 4 or BAY 1143572. Treatments were started three days after tumor cell inoculation. B) Tumor growth in mice treated with BAY 1143572 once daily (QD) or with an intermittent 3 days on/2 days off dosing schedule. Treatments were started 4 days after tumor cell inoculation. C) Antitumor efficacy of BAY 1143572 in an MV4‐11 human AML model in rats. Tumor growth in rats treated with vehicle or BAY 1143572. Treatments were started 13 days after tumor cell inoculation and were continued for 14 days, until the vehicle group was sacrificed. Asterisks indicate statistical significance relative to the vehicle control, calculated using the mean tumor areas at the time point when the vehicle group was sacrificed (***p<0.001, ns=not significant).
Table 5.
Antitumor efficacy of compound 4 and BAY 1143572 in human AML models in female NMRI nu/nu mice (MOLM‐13) and athymic rats (MV4‐11).
| Animal model | Treatment | Dose [mg kg−1] | Schedule | T/Carea [a] | Fatal toxicity | Max BWC [%][b] |
|---|---|---|---|---|---|---|
| MOLM‐13 model in mice | Vehicle | 0 | QD | 1.00 | 0/10 | +13 |
| Compound 4 | 10 | QD | 0.91 | 0/10 | +11 | |
| 20 | QD | 0.73 | 0/10 | +11 | ||
| BAY 1143572 | 6.25 | QD | 0.64* | 0/10 | +10 | |
| 12.5 | QD | 0.49* | 1/10 | +10 | ||
| MOLM‐13 model in mice | Vehicle | 0 | QD | 1.00 | 1/13 | 0 |
| BAY 1143572 | 20 | QD | 0.41* | 0/13 | −1 | |
| 25 | QD | 0.31* | 0/13 | −3 | ||
| 25 | 3 on/2 off | 0.33* | 0/13 | −2 | ||
| 35 | 3 on/2 off | 0.20* | 0/13 | −4 | ||
| MV4‐11 model in rats | Vehicle | 0 | QD | 1.00 | 1/12 | −1 |
| BAY 1143572 | 12 | QD×14 | 0.05* | 0/12 | −1 |
[a] Treatment‐to‐control (T/C) ratios and statistical significances were calculated using the mean tumor areas at the time point when the vehicle group was sacrificed. Asterisks indicate statistical significance (p<0.001) relative to the vehicle control. [b] The maximum body weight change (BWC) expressed as a percentage of the starting weight, for duration of the treatment.
To further substantiate the antitumor efficacy, BAY 1143572 was evaluated in an MV4‐11 human acute myeloid leukemia model in nude rats. Of note, the antiproliferative activity of BAY 1143572 against MV4‐11 cells (IC50: 890 nm) and HeLa cells (IC50: 920 nm) in vitro is similar, and is slightly improved against MOLM‐13 cells (IC50: 310 nm). Daily oral administration of BAY 1143572 at 12 mg kg−1 for 14 days (maximum tolerated dose) resulted in almost complete tumor remission, followed by delayed regrowth after cessation of the treatment (Figure 5 C). At the conclusion of the experiment (57 days after initial inoculation), there was no tumor regrowth in nine of the 12 test animals.
Due to the promising overall profile in vitro and in vivo, benzyl sulfoximine BAY 1143572 was selected as the development candidate and entered phase 1 clinical trials in patients with advanced cancer and leukemia (NCT01938638, NCT02345382).22
Conclusions
The benzyl sulfoximine atuveciclib (BAY 1143572) is a potent and highly selective, oral PTEFb/CDK9 inhibitor. During lead optimization, a surprisingly steep SAR with regard to the key optimization parameters kinase selectivity and potency was recorded. BAY 1143572 clearly exhibited the most promising overall profile with respect to potency, selectivity, physicochemical properties, and in vivo PK as well as in vivo potency. Notably, in contrast to lead compound BAY‐958 and benzyl sulfone 4, BAY 1143572 combines high aqueous solubility with moderate Caco‐2 permeability and moderate efflux, as well as low blood clearance and moderate bioavailability in rats in vivo. Surprisingly, the switch from the benzyl sulfonamide BAY‐958 to the benzyl sulfoximine BAY 1143572 removed an in vitro CYP1A2 induction liability. BAY 1143572 is efficacious in human xenograft tumor models of acute myeloid leukemia in both mice and rats. The compound is active upon once‐daily dosing, as well as upon intermittent dosing schedules, providing valuable options to optimize human dosing schedules with respect to efficacy and tolerability. BAY 1143572 is the first selective PTEFb/CDK9 inhibitor that entered clinical evaluation. The introduction of the uncommon benzyl sulfoximine group was crucial for overcoming hurdles in this project, which underlines the constant need for novel chemical functionalities and methodologies as a means to solve biological problems.
Experimental Section
Kinase assays
CDK9/CycT1: Recombinant full‐length His‐tagged human CDK9 and CycT1, expressed in insect cells and purified by Ni‐NTA affinity chromatography, were purchased from Invitrogen (Cat. no. PV4131). As substrate for the kinase reaction, the biotinylated peptide biotin‐Ttds‐YISPLKSPYKISEG (C‐terminus in amide form) was used, which is commercially available (e.g., from JERINI Peptide Technologies, Berlin, Germany). For assays, 50 nL of a 100‐fold concentrated solution of the test compound in DMSO was pipetted into a black, low‐volume, 384‐well microtiter plate (Greiner Bio‐One, Frickenhausen, Germany); 2 μL of a solution of CDK9/CycT1 in aqueous assay buffer [50 mm Tris⋅HCl pH 8.0, 10 mm MgCl2, 1.0 mm dithiothreitol, 0.1 mm sodium orthovanadate, 0.01 % (v/v) Nonidet P‐40 (Sigma)] was added, and the mixture was incubated for 15 min at 22 °C to allow pre‐binding of the test compound to the enzyme before the start of the kinase reaction. Then, the kinase reaction was started by the addition of 3 μL of a solution of adenosine triphosphate (ATP, 16.7 μm; final concn in the 5 μL assay volume: 10 μm) and substrate (1.67 μm; final concn in the 5 μL assay volume: 1 μm) in assay buffer, and the resulting mixture was incubated for 25 min at 22 °C. The concentration of CDK9/CycT1 was adjusted depending on the activity of the enzyme lot and was chosen appropriate to have the assay in the linear range: typical concentrations were in the order of 1 μg mL−1. The reaction was stopped by the addition of 5 μL of a solution of TR‐FRET detection reagents [0.2 μm streptavidin‐XL665 (Cisbio Bioassays, Codolet, France), 1 nm anti‐RB(pSer807/pSer811) antibody (BD Pharmingen, Cat. no. 558389) and 1.2 nm LANCE EU‐W1024 labeled anti‐mouse IgG antibody (PerkinElmer, product no. AD0077)] in an aqueous EDTA solution [100 mm EDTA, 0.2 % (w/v) bovine serum albumin (BSA) in 100 mm HEPES/NaOH pH 7.0]. The resulting mixture was incubated for 1 h at 22 °C to allow the formation of a complex between the phosphorylated biotinylated peptide and the detection reagents. Subsequently, the amount of phosphorylated substrate was evaluated by measurement of the resonance energy transfer from the europium chelate to the streptavidin‐XL. Therefore, the fluorescence emissions at 620 nm and 665 nm, after excitation at 350 nm, were measured with a TR‐FRET reader [e.g., PHERAstar (BMG Labtechnologies, Offenburg, Germany) or ViewLux (PerkinElmer)]. The ratio of the emissions at 665 nm and 620 nm was taken as the measure of the amount of phosphorylated substrate. The data were normalized (enzyme reaction without inhibitor=0 % inhibition, all other assay components but no enzyme=100 % inhibition). Usually the test compound was tested on the same microtiter plate at 11 different concentrations in the range of 20 μm to 0.1 nm (20 μm, 5.9 μm, 1.7 μm, 0.51 μm, 0.15 μm, 44 nm, 13 nm, 3.8 nm, 1.1 nm, 0.33 nm and 0.1 nm; the dilution series was prepared separately before the assay on the 100‐fold concentrated solution in DMSO by serial 1:3.4 dilutions) in duplicate for each concentration. IC50 values were calculated using a four‐parameter fit.
CDK2/CycE: Recombinant fusion proteins of GST and human CDK2 and of GST and human CycE, expressed in insect cells (Sf9) and purified by Glutathione‐Sepharose affinity chromatography, were purchased from ProQinase GmbH (Freiburg, Germany). As substrate for the kinase reaction, the biotinylated peptide biotin‐Ttds‐YISPLKSPYKISEG (C‐terminus in amide form) was used, which is commercially available (e.g., from JERINI Peptide Technologies, Berlin, Germany). For assays, 50 nL of a 100‐fold concentrated solution of the test compound in DMSO was pipetted into a black, low volume, 384‐well microtiter plate (Greiner Bio‐One, Frickenhausen, Germany); 2 μL of a solution of CDK2/CycE in aqueous assay buffer [50 mm Tris⋅HCl pH 8.0, 10 mm MgCl2, 1.0 mm dithiothreitol, 0.1 mm sodium orthovanadate, 0.01 % (v/v) Nonidet P‐40 (Sigma)] was added, and the mixture was incubated for 15 min at 22 °C to allow pre‐binding of the test compound to the enzyme before the start of the kinase reaction. Then, the kinase reaction was started by the addition of 3 μL of a solution of ATP (16.7 μm; final concn in the 5 μL assay volume: 10 μm) and substrate (1.25 μm; final concn in the 5 μL assay volume: 0.75 μm) in assay buffer, and the resulting mixture was incubated for 25 min at 22 °C. The concentration of CDK2/CycE was adjusted depending on the activity of the enzyme lot and was chosen appropriate to have the assay in the linear range; typical concentrations were in the order of 130 ng mL−1. The reaction was stopped by the addition of 5 μL of a solution of TR‐FRET detection reagents [0.2 μm streptavidin‐XL665 (Cisbio Bioassays, Codolet, France), 1 nm anti‐RB(pSer807/pSer811) antibody (BD Pharmingen, Cat. no. 558389) and 1.2 nm LANCE EU‐W1024 labeled anti‐mouse IgG antibody (PerkinElmer, product no. AD0077)] in an aqueous EDTA solution [100 mm EDTA, 0.2 % (w/v) BSA in 100 mm HEPES/NaOH pH 7.0]. The resulting mixture was incubated for 1 h at 22 °C to allow the formation of a complex between the phosphorylated biotinylated peptide and the detection reagents. Subsequently, the amount of phosphorylated substrate was evaluated by measurement of the resonance energy transfer from the europium chelate to the streptavidin‐XL. Therefore, the fluorescence emissions at 620 nm and 665 nm, after excitation at 350 nm, were measured with a TR‐FRET reader [e.g., PHERAstar (BMG Labtechnologies, Offenburg, Germany) or ViewLux (PerkinElmer)]. The ratio of the emissions at 665 nm and 620 nm was taken as the measure of the amount of phosphorylated substrate. The data were normalized (enzyme reaction without inhibitor=0 % inhibition, all other assay components but no enzyme=100 % inhibition). Usually the test compound was tested on the same microtiter plate at 11 different concentrations in the range of 20 μm to 0.1 nm (20 μm, 5.9 μm, 1.7 μm, 0.51 μm, 0.15 μm, 44 nm, 13 nm, 3.8 nm, 1.1 nm, 0.33 nm and 0.1 nm; the dilution series was prepared separately before the assay on the 100‐fold concentrated solution in DMSO by serial 1:3.4 dilutions) in duplicate for each concentration. IC50 values were calculated using a four‐parameter fit.
Merck Millipore CDK assays: Assays were performed according to the Merck Millipore KinaseProfilerTM standard protocols, with an ATP concentration of 10 μm.
Proliferation assay
HeLa human cervical tumor cells (CCL‐2) were obtained from the American Type Culture Collection (Manassas, USA) and MOLM‐13 human acute myeloid leukemia cells (ACC 554) were obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Authentication of cell lines was conducted at the German Collection of Microorganisms and Cell Cultures via PCR‐based DNA profiling of polymorphic short tandem repeats. Cells were propagated under the suggested growth conditions in a humidified 37 °C incubator. Proliferation assays were conducted in 96‐well plates at densities of 3000 (HeLa) and 5000 (MOLM‐13) cells per well in the growth medium containing 10 % fetal calf serum (FCS). Cells were treated in quadruplicate with serial dilutions of test compounds for 96 h. Relative cell numbers were quantified by crystal violet staining (HeLa)23 or CellTitre‐Glo Luminescent Cell Viability Assay (Promega) (MOLM‐13). IC50 values (inhibitory concentration at 50 % of maximal effect) were determined by means of a four‐parameter fit on measurement data which were normalized to vehicle (DMSO) treated cells (=100 %) and measurement readings taken immediately before compound exposure (=0 %).
Equilibrium shake flask solubility assay
The thermodynamic solubility of compounds in water was determined by an equilibrium shake flask method.24 A saturated solution of the test compound was prepared and the solution was mixed for 24 h to ensure that equilibrium was reached. The solution was centrifuged to remove the insoluble fraction and the concentration of the compound in solution was determined using a standard calibration curve. To prepare the test sample, solid compound (2 mg) was weighed in a 4‐mL glass vial. Phosphate buffer pH 6.5 (1 mL) was added and the suspension was stirred for 24 h at RT. Then, the solution was centrifuged. To prepare the sample for the standard calibration, solid sample (2 mg) was dissolved in MeCN (30 mL). After sonication, the solution was diluted with water to 50 mL. Sample and standard were quantified by HPLC with UV detection. For each test sample, two injection volumes (5 and 50 μL) in triplicate were made. Three injection volumes (5 μL, 10 μL and 20 μL) were made for the standard. HPLC conditions: column: Xterra MS C18 2.5 μm, 4.6×30 mm; injection volume: sample: 3×5 μL and 3×50 μL, standard: 5 μL, 10 μL and 20 μL; flow: 1.5 mL min−1; mobile phase: acidic gradient: eluent A: water/0.01 % TFA, eluent B: MeCN/0.01 % TFA, 0 min 95 % A 5 % B; 0–3 min 35 % A 65 % B linear gradient, 3–5 min 35 % A 65 % B isocratic, 5–6 min 95 % A 5 % B isocratic; UV detection: wavelength near the absorption maximum (between 200 and 400 nm). The areas of sample and standard injections, as well as the calculation of the solubility values (in mg L−1), were determined using Waters Empower 2 FR software.
Pharmacokinetic studies
For the metabolic stability assay in rat hepatocytes, liver cells were distributed in Williams’ Medium E containing 5 % FCS to glass vials at a density of 1.0×106 vital cells mL−1. The test compound was added at a final concentration of 1 μm. During incubation, the hepatocyte suspensions were continuously shaken at 580 rpm and aliquots were removed at 2, 8, 16, 30, 45 and 90 min, to which an equal volume of cold MeCN was immediately added. Samples were frozen at −20 °C overnight, then centrifuged for 15 min at 3000 rpm. The supernatants were analyzed by liquid chromatography‐tandem mass spectrometry (LC–MS/MS) using an Agilent 1200 HPLC system with an Ascentis Express column and water/MeCN (isocratic) as eluent. The half‐life of a test compound was determined from the concentration‐time plot and the intrinsic clearances were calculated. Together with the additional parameters liver blood flow and amount of liver cells in vivo and in vitro, and application of the “well‐stirred” liver model,25 the hepatic in vivo blood clearance (CLb) and the maximal oral bioavailability (F max) were calculated.
The inhibitory potency of the test compounds toward cytochrome P450 dependent metabolic pathways was determined in human liver microsomes by applying individual CYP isoform‐selective standard probes (CYP1A2, phenacetin; CYP2C8, amodiaquine; CYP2C9, diclofenac; CYP2D6, dextromethorphan; CYP3A4, midazolam). Reference inhibitors were included as positive controls. Incubation conditions (protein and substrate concentration, incubation time) were optimized with regard to linearity of metabolite formation. Assays were processed in 96‐well plates at 37 °C by using a Genesis Workstation (Tecan, Crailsheim, Germany). After protein precipitation, the metabolite formation was quantified by LC–MS/MS analysis followed by inhibition evaluation and IC50 calculation.
To evaluate the CYP induction potential in vitro, cultured human hepatocytes from three separate livers were treated once daily for three consecutive days with vehicle control, one of eight concentrations of test compound and known human CYP inducers (e.g., omeprazole, phenobarbital, rifampin). After treatment, the cells were incubated in situ with the appropriate marker substrates for the analysis of CYP3A4 and CYP1A2 activity by LC–MS/MS.
Caco‐2 cells (purchased from the German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) were seeded at a density of 4.5×104 cells per well on 24‐well insert plates, 0.4 μm pore size, and grown for 15 days in DMEM supplemented with 10 % FCS, 1 % GlutaMAX (100x, GIBCO), 100 U mL−1 penicillin, 100 μg mL−1 streptomycin (GIBCO) and 1 % nonessential amino acids (100×). Cells were maintained at 37 °C in a humidified 5 % CO2 atmosphere. The medium was changed every 2–3 d. Before running the permeability assay, the culture medium was replaced with an FCS‐free HEPES/carbonate transport buffer pH 7.2. For assessment of monolayer integrity, the transepithelial electrical resistance was measured. Test compounds were predissolved in DMSO and added either to the apical or basolateral compartment at a final concentration of 2 μm. Before and after incubation for 2 h at 37 °C, samples were taken from both compartments. LC–MS/MS analysis of compound content was undertaken after precipitation with MeOH. Permeability (P app) was calculated in the apical to basolateral (A→B) and basolateral to apical (B→A) directions. The efflux ratio basolateral (B) to apical (A) was calculated by dividing P app B→A by P app A→B.
For rat PK studies, test compounds were administered to male Wistar rats intravenously at low doses of 0.3 to 0.5 mg kg−1 and intragastrically at doses of 0.6 to 1 mg kg−1 formulated as solutions using solubilizers such as PEG 400 in well‐tolerated amounts. Animals were catheterized at the jugular vein and samples were taken via the catheter. Plasma samples were collected after 2 min (only intravenously), 8 min, 15 min, 30 min, 45 min, 1 h, 2 h, 4 h, 6 h, 8 h and 24 h post‐application, and precipitated with ice‐cold MeCN (1:5). Supernatants were analyzed by LC–MS/MS. Pharmacokinetic parameters were based on the plasma concentration‐time data and calculated (e.g., using the linear‐log trapezoidal rule for AUC estimation) with KinEx, an Excel‐based program.
MOLM‐13 and MV4–11 human AML tumor models in mice and rats
All animal experiments were conducted in accordance with the German Animal Welfare Law and approved by local authorities. For the acute myeloid leukemia (AML) mouse model, 2×106 MOLM‐13 human AML cells were suspended in 100 % MatrigelTM Basement Membrane Matrix (BD Biosciences) and inoculated subcutaneously to the left flank of female NMRI nu/nu mice (18–21 g, 5–6 weeks, Taconic M&B). For the AML model in rats, 2×106 MV4‐11 human AML cells were suspended in 100 % MatrigelTM and inoculated subcutaneously to the left flank of female athymic nude rats (160–200 g, 5–6 weeks, Harlan). Animals were stratified into treatment and control groups (n=8–13/group for mice, n=12/group for rats) based on primary tumor size. Treatments were started 3–13 days after tumor cell inoculation when the average tumor sizes were 23–38 mm2 and 43 mm2 for mice and rats, respectively.
BAY 1143572 was administered either using a once daily (QD) or an intermittent 3 days on/2 days off treatment schedule. BAY‐958 hydrochloride and compound 4 were administered QD. PEG 400/water 80:10 was used as the vehicle control. Unless otherwise indicated, all treatments were administered orally (p.o.) and were continued until the end of the experiment. Body weight and tumor areas (longest diameter multiplied by its perpendicular) measured by caliper were determined at least twice weekly. Treatment‐to‐control (T/C) ratios were calculated by dividing the mean tumor area of the treatment group by the mean tumor area of the vehicle group at the time point when the vehicle group was sacrificed.
All statistical analyses were performed using statistical software R (version 3.3.2) by comparing the treatment groups to the vehicle group at the time point when the vehicle group was sacrificed. Statistical analysis of the log‐transformed tumor area data was performed using one‐way ANOVA followed by Dunnett's contrasts or Kruskal‐Wallis test followed by Dunn's test with Holm‐Bonferroni correction. In the MV4‐11 model with only two groups, the analysis was performed using Student's t test. In all cases, p<0.05 was considered as being statistically significant.
Docking
Docking calculations were performed using Glide26 in standard precision mode and default settings. The X‐ray complex of CDK9/CycT1 with a benzimidazole inhibitor (PDB ID: 3MY1) was used.27 Prior to the docking procedure, all water molecules and ligands in the protein‐ligand complex were removed. Ligands were pre‐processed using LigPrep.28 Docking poses were selected based on Glide scoring.
Synthetic procedures
General methods and materials: Commercially available reagents and anhydrous solvents were used as supplied, without further purification. All air‐ and moisture‐sensitive reactions were carried out in oven‐dried (at 120 °C) glassware under an inert atmosphere of argon. A Biotage® Initiator Classic microwave reactor was used for reactions conducted in a microwave oven. Reactions were monitored by TLC and UPLC analysis with a Waters Acquity UPLC MS Single Quad system; column: Acquity UPLC BEH C18 1.7 μm, 50×2.1 mm; basic conditions: eluent A: H2O+0.2 vol% aq. NH3 (32 %), eluent B: MeCN; gradient: 0–1.6 min 1–99 % B, 1.6–2.0 min 99 % B; flow: 0.8 mL min−1; acidic conditions: eluent A: H2O+0.1 vol % formic acid (99 %), eluent B: MeCN; gradient: 0–1.6 min 1–99 % B, 1.6–2.0 min 99 % B; flow: 0.8 mL min−1; temperature: 60 °C; DAD scan: 210–400 nm. Analytical TLC was carried out on aluminum‐backed plates coated with Merck Kieselgel 60 F254, with visualization under UV light at 254 nm. Flash chromatography was carried out using a Biotage® IsoleraTM One system with 200–400 nm variable detector. Preparative HPLC was carried out with a Waters AutoPurification MS Single Quad system; column: Waters XBridge C18 5 μm, 100×30 mm; basic conditions: eluent A: H2O+0.2 vol % aq. NH3 (32 %), eluent B: MeCN; gradient: 0–0.5 min 5 % B, flow: 25 mL min−1; 0.51–5.50 min 10–100 % B, flow: 70 mL min−1; 5.51–6.5 min 100 % B, flow: 70 mL min−1; acidic conditions: eluent A: H2O+0.1 vol % formic acid (99 %), eluent B: MeCN; gradient: 0–0.5 min 5 % B, flow: 25 mL min−1; 0.51–5.50 min 10–100 % B, flow: 70 mL min−1; 5.51–6.5 min 100 % B, flow: 70 mL min−1; temperature: 25 °C; DAD scan: 210–400 nm. NMR spectra were recorded at ambient temperature (22±1 °C), unless otherwise noted, on Bruker Avance III HD spectrometers. 1H NMR spectra were obtained at 300, 400, 500 or 600 MHz, and referenced to the residual solvent signal (7.26 ppm for CDCl3, 2.50 ppm for [D6]DMSO). 13C NMR spectra were obtained at 125 MHz and also referenced to the residual solvent signal (39.52 ppm for [D6]DMSO). 1H NMR data are reported as follows: chemical shift (δ) in ppm, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, br=broad, m=multiplet) and integration. High‐resolution mass spectra were recorded on a Xevo® G2‐XS Tof (Waters) instrument. Low‐resolution mass spectra (electrospray ionization) were obtained via HPLC–MS (ESI) using a Waters Acquity UPLC system equipped with an SQ 3100 Mass Detector; column: Acquity UPLC BEH C18 1.7 μm, 50×2.1 mm; eluent A: H2O+0.05 % formic acid (99 %), eluent B: MeCN+0.05 % formic acid (99 %); gradient: 0–0.5 min 5 % B, 0.5–2.5 min 5–100 % B, 2.5–4.5 min 100 % B; total run time: 5 min; flow: 0.5 mL min−1. Melting points were determined with a Büchi B‐540 melting point apparatus. Optical rotations were recorded on a JASCO P‐2000 polarimeter. The purity of all target compounds was >97 %, as determined by 1H NMR spectroscopy.
3‐[(4‐Chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide: To a solution of 2,4‐dichloro‐1,3,5‐triazine (1; 900 mg, 6.0 mmol) in anhyd. THF/iPrOH (1:1, 8 mL) at −20 °C under N2 atmosphere was added a cooled solution (−20 °C) of (3‐aminophenyl)methanesulfonamide (1117 mg, 6.0 mmol) and DIPEA (2.07 mL, 12 mmol) in anhyd. THF/iPrOH (1:1, 8 mL). The reaction mixture was stirred for 2 h at −10 °C. The mixture was concentrated under reduced pressure and the crude product was dried in vacuo for 15 h. The white solid was used in the next step without further purification: 1H NMR (400 MHz, [D6]DMSO): δ=4.26 (s, 2 H), 6.89 (s, 2 H), 7.15 (d, 1 H), 7.36 (d, 1 H), 7.66 (d, 1 H), 8.64 (s, 1 H), 9.75 (br s, 1 H), 10.83 ppm (s, 1 H); MS (ESI) m/z: 300 [M+H]+.
1‐(3‐{[4‐(4‐Fluoro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐yl]amino}phenyl)methanesulfonamide (BAY‐958): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (1767 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (1504 mg, 8.85 mmol) and K3PO4 (2505 mg, 11.82 mmol) in dioxane/water (10:1, 66 mL) was degassed with a stream of nitrogen for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (489 mg, 0.6 mmol) was added and the reaction mixture was heated for 90 min at 145 °C in a microwave oven. The mixture was diluted with EtOAc and washed with sat. aq. NaHCO3. The organic phase was dried (Na2SO4), filtered and concentrated. The crude was purified by flash chromatography (EtOAc/MeOH, 100:0 to 5:1). Finally, precipitation from EtOAc yielded BAY‐958 as a white solid (129 mg, 0.33 mmol): mp: 234 °C; 1H NMR (400 MHz, [D6]DMSO): δ=3.87 (s, 3 H), 4.22 (s, 2 H), 6.82–6.91 (m, 3 H), 7.07 (m, 2 H), 7.34 (t, 1 H), 7.72 (s, 1 H), 7.89 (br s, 2 H), 8.78 (s, 1 H), 10.32 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C17H17FN5O3S: 390.1036, found: 390.1039.
1‐(3‐{[4‐(4‐Fluoro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐yl]amino}phenyl)methanesulfonamide hydrochloride (BAY‐958⋅HCl): BAY‐958 (914 mg, 2.35 mmol) was suspended in aq. 1 n HCl (2.35 mL) and the mixture was stirred for 4 h at RT, then concentrated under reduced pressure to give the desired hydrochloride as a white solid (980 mg, 2.30 mmol, 98 %): mp: 180 °C; 1H NMR (400 MHz, [D6]DMSO): δ=3.90 (s, 3 H), 4.23 (s, 2 H), 6.91 (m, 3 H), 7.13 (m, 2 H), 7.37 (m, 1 H), 7.81 (m, 3 H), 8.82 (s, 1 H), 10.83 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C17H17FN5O3S: 390.1036, found: 390.1038.
1‐[(Methylsulfanyl)methyl]‐3‐nitrobenzene: Sodium methanethiolate (13.5 g, 192 mmol) was added in two portions to a stirred solution of 1‐(chloromethyl)‐3‐nitrobenzene (30.0 g, 175 mmol) in EtOH (360 mL) at −15 °C. The cold bath was removed and the mixture was stirred at RT for 3 h. Then, it was diluted with brine and extracted with EtOAc (2×). The combined organic phases were washed with water, dried (Na2SO4), filtered and concentrated to give the desired product (32.2 g) that was used without further purification: 1H NMR (400 MHz, CDCl3): δ=2.01 (s, 3 H), 3.75 (s, 2 H), 7.50 (m, 1 H), 7.66 (m, 1 H), 8.11 (m, 1 H), 8.18 ppm (m, 1 H).
1‐[(Methylsulfonyl)methyl]‐3‐nitrobenzene: 3‐Chloroperoxybenzoic acid (77 %; 26.9 g, 120 mmol) was added to a stirred solution of 1‐[(methylsulfanyl)methyl]‐3‐nitrobenzene (10.0 g) in CH2Cl2 (1305 mL) at 0 °C. The mixture was stirred at 0 °C for 30 min and then at RT for 2.5 h. Then, it was diluted with water (300 mL), and NaHCO3 (11.0 g) was added. The mixture was extracted with CH2Cl2 (2×). The combined organic phases were filtered using a Whatman filter and concentrated. The residue was purified by flash chromatography (CH2Cl2/EtOH, 95:5) and finally recrystallized from EtOAc to give the desired product (6.2 g, 28.9 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=2.93 (s, 3 H), 4.68 (s, 2 H), 7.69 (m, 1 H), 7.83 (m, 1 H), 8.22 (m, 1 H), 8.28 ppm (m, 1 H).
3‐[(Methylsulfonyl)methyl]aniline: TiCl3 solution (ca. 15 % in ∼10 % HCl, 162 mL) was added to a stirred solution of 1‐[(methylsulfonyl)methyl]‐3‐nitrobenzene (5.1 g, 23.8 mmol) in THF (250 mL) at RT, and the mixture was stirred for 16 h. The pH was increased to 10 by the addition of 1 n NaOH, then the reaction mixture was extracted with EtOAc (2×). The combined organic phases were washed with brine, filtered using a Whatman filter and concentrated to give the desired product (4.5 g) that was used without further purification: 1H NMR (400 MHz, [D6]DMSO): δ=2.83 (s, 3 H), 4.23 (s, 2 H), 5.13 (br, 2 H), 6.51 (m, 3 H), 6.97 ppm (m, 1 H).
4‐Chloro‐N‐{3‐[(methylsulfonyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine: DIPEA (3.7 mL, 21.3 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 1.60 g, 10.7 mmol) in THF/iPrOH (1:1, 20 mL) at −40 °C. Then, a suspension of 3‐[(methylsulfonyl)methyl]aniline (1.97 g, 10.7 mmol) in THF/iPrOH (1:1, 10 mL) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 3 h to 0 °C. The mixture was concentrated in vacuo to give the crude product (5.2 g) that was used without further purification.
4‐(4‐Fluoro‐2‐methoxyphenyl)‐N‐{3‐[(methylsulfonyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine (4): A mixture of crude 4‐chloro‐N‐{3‐[(methylsulfonyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine (1000 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (569 mg, 3.35 mmol) and tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4; 580 mg, 0.50 mmol] in 1,2‐dimethoxyethane (10.3 mL) and aq. 2 m K2CO3 (3.4 mL) was degassed using argon. The mixture was stirred under argon for 90 min at 100 °C. After cooling, the mixture was diluted with EtOAc and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by flash chromatography (CH2Cl2 to CH2Cl2/EtOH, 95:5) to give 4 as a white solid (402 mg, 1.03 mmol): 1H NMR (400 MHz, CDCl3): δ=2.79 (s, 3 H), 3.94 (s, 3 H), 4.26 (s, 2 H), 6.75–6.81 (m, 2 H), 7.16 (d, 1 H), 7.38–7.46 (m, 2 H), 7.69–7.87 (m, 2 H), 7.96 (br s, 1 H), 8.82 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C18H18FN4O3S: 389.1084, found: 389.1080.
4‐Chloro‐N‐[3‐(methylsulfonyl)phenyl]‐1,3,5‐triazin‐2‐amine: DIPEA (0.31 mL, 1.75 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 138 mg, 0.88 mmol) in THF/iPrOH (1:1, 1.8 mL) at −40 °C. Then, a suspension of 3‐(methylsulfonyl)aniline (150 mg, 0.88 mmol) in THF/iPrOH (1:1, 0.9 mL) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 90 min to 0 °C. The mixture was concentrated in vacuo to give the crude product (450 mg) that was used without further purification.
4‐(4‐Fluoro‐2‐methoxyphenyl)‐N‐[3‐(methylsulfonyl)phenyl]‐1,3,5‐triazin‐2‐amine (5): A mixture of crude 4‐chloro‐N‐[3‐(methylsulfonyl)phenyl]‐1,3,5‐triazin‐2‐amine (450 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (403 mg, 2.37 mmol) and Pd(PPh3)4 (274 mg, 0.24 mmol) in 1,2‐dimethoxyethane (7.3 mL) and aq. 2 m K2CO3 (1.6 mL) was degassed using argon. The mixture was stirred under argon for 90 min at 100 °C. After cooling, the mixture was diluted with EtOAc and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 5 as a white solid (152 mg, 0.41 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=3.20 (s, 3 H), 3.88 (br s, 3 H), 6.91 (td, 1 H), 7.12 (dd, 1 H), 7.58–7.68 (m, 2 H), 7.91 (br s, 1 H), 8.24 (br s, 1 H), 8.42 (s, 1 H), 8.88 (s, 1 H), 10.68 ppm (br s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C17H16FN4O3S: 375.0927, found: 375.0924.
4‐Chloro‐N‐{3‐[2‐(methylsulfonyl)ethyl]phenyl}‐1,3,5‐triazin‐2‐amine: DIPEA (0.35 mL, 2.01 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 150 mg, 1.00 mmol) in THF/iPrOH (1:1, 2 mL) at −40 °C. Then, a solution of 3‐[2‐(methylsulfonyl)ethyl]aniline (200 mg, 1.00 mmol) in THF/iPrOH (1:1, 1 mL) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 2 h to 0 °C. The mixture was concentrated in vacuo to give the crude product (458 mg) that was used without further purification.
4‐(4‐Fluoro‐2‐methoxyphenyl)‐N‐{3‐[2‐(methylsulfonyl)ethyl]phenyl}‐1,3,5‐triazin‐2‐amine (6): A mixture of crude 4‐chloro‐N‐{3‐[2‐(methylsulfonyl)ethyl]phenyl}‐1,3,5‐triazin‐2‐amine (150 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (122 mg, 0.72 mmol) and Pd(PPh3)4 (83 mg, 0.07 mmol) in 1,2‐dimethoxyethane (1.5 mL) and aq. 2 m K2CO3 (0.5 mL) was degassed using argon. The mixture was stirred under argon for 90 min at 100 °C. After cooling, the mixture was diluted with EtOAc and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 6 as a light beige solid (53 mg, 0.13 mmol): 1H NMR (400 MHz, CDCl3): δ=2.85 (s, 3 H), 3.14–3.26 (m, 2 H), 3.26–3.39 (m, 2 H), 3.94 (s, 3 H), 6.74–6.83 (m, 2 H), 7.00 (br d, 1 H), 7.29–7.39 (m, 1 H), 7.44–7.60 (m, 2 H), 7.65 (s, 1 H), 7.95 (br s, 1 H), 8.82 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C19H20FN4O3S: 403.1240, found: 403.1244.
4‐Chloro‐N‐(1,1‐dioxido‐2,3‐dihydro‐1‐benzothien‐6‐yl)‐1,3,5‐triazin‐2‐amine: DIPEA (0.88 mL, 5.07 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 400 mg, 2.53 mmol) in THF/iPrOH (1:1, 5 mL) at −40 °C. Then, 2,3‐dihydro‐1‐benzothiophen‐6‐amine 1,1‐dioxide (464 mg, 2.53 mmol) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 2 h to 0 °C. The mixture was concentrated in vacuo to give the crude product (1432 mg) that was used without further purification.
N‐( 1,1‐Dioxido‐2,3‐dihydro‐1‐benzothien‐6‐yl)‐4‐(4‐fluoro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐amine (7): A mixture of crude 4‐chloro‐N‐(1,1‐dioxido‐2,3‐dihydro‐1‐benzothien‐6‐yl)‐1,3,5‐triazin‐2‐amine (200 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (115 mg, 0.67 mmol) and Pd(PPh3)4 (117 mg, 0.10 mmol) in 1,2‐dimethoxyethane (2.1 mL) and aq. 2 m K2CO3 (0.7 mL) was degassed using argon. The mixture was stirred under argon for 2 h at 100 °C. After cooling, the mixture was diluted with EtOAc and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 7 as a white solid (40 mg, 0.10 mmol): 1H NMR (400 MHz, CDCl3): δ=3.32–3.42 (m, 2 H), 3.46–3.60 (m, 2 H), 4.00 (s, 3 H), 6.74–6.83 (m, 2 H), 7.28–7.38 (m, 1 H), 7.45–7.69 (m, 2 H), 8.01 (br s, 1 H), 8.57 (br s, 1 H), 8.84 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C18H16FN4O3S: 387.0927, found: 387.0929.
4‐Chloro‐N‐(2,2‐dioxido‐1,3‐dihydro‐2‐benzothien‐5‐yl)‐1,3,5‐triazin‐2‐amine: DIPEA (0.33 mL, 1.90 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 150 mg, 0.95 mmol) in THF/iPrOH (1:1, 2 mL) at −40 °C. Then, 1,3‐dihydro‐2‐benzothiophen‐5‐amine 2,2‐dioxide (174 mg, 0.95 mmol) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 2 h to 0 °C. The mixture was concentrated in vacuo to give the crude product (462 mg) that was used without further purification.
N‐( 2,2‐Dioxido‐1,3‐dihydro‐2‐benzothien‐5‐yl)‐4‐(4‐fluoro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐amine (8): A mixture of crude 4‐chloro‐N‐(2,2‐dioxido‐1,3‐dihydro‐2‐benzothien‐5‐yl)‐1,3,5‐triazin‐2‐amine (75 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (64 mg, 0.38 mmol) and K3PO4 (107 mg, 0.51 mmol) in dioxane/water (20:1, 2.5 mL) was degassed with a stream of argon for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (21 mg, 0.025 mmol) was added and the reaction mixture was heated for 60 min at 140 °C in a microwave oven. The mixture was diluted with dioxane, filtered and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 8 as a white solid (9 mg, 0.02 mmol): 1H NMR (400 MHz, CDCl3): δ=3.93 (s, 3 H), 4.38 (d, 4 H), 6.74–6.83 (m, 2 H), 7.28–7.33 (m, 1 H), 7.46 (s, 1 H), 7.59 (br d, 1 H), 7.86 (s, 1 H), 7.90–8.01 (m, 1 H), 8.83 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C18H16FN4O3S: 387.0927, found: 387.0926.
N ‐{3‐[(tertButylsulfonyl)methyl]phenyl}‐4‐chloro‐1,3,5‐triazin‐2‐amine: DIPEA (0.46 mL, 2.64 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 198 mg, 1.32 mmol) in THF/iPrOH (1:1, 2.6 mL) at −40 °C. Then, 3‐[(tert‐butylsulfonyl)methyl]aniline (300 mg, 1.32 mmol) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 2 h to 0 °C. The mixture was concentrated in vacuo to give the crude product (654 mg) that was used without further purification.
N ‐{3‐[(tertButylsulfonyl)methyl]phenyl}‐4‐(4‐fluoro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐amine (9): A mixture of crude N‐{3‐[(tert‐butylsulfonyl)methyl]phenyl}‐4‐chloro‐1,3,5‐triazin‐2‐amine (150 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (75 mg, 0.44 mmol) and Pd(PPh3)4 (76 mg, 0.07 mmol) in 1,2‐dimethoxyethane (1.4 mL) and aq. 2 m K2CO3 (0.4 mL) was degassed using argon. The mixture was stirred under argon for 90 min at 100 °C. After cooling, the mixture was diluted with EtOAc and THF, and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 9 as a light beige solid (30 mg, 0.07 mmol): 1H NMR (400 MHz, CDCl3): δ=1.46 (s, 9 H), 3.93 (s, 3 H), 4.21 (s, 2 H), 6.74–6.82 (m, 2 H), 7.18–7.25 (m, 1 H), 7.36–7.59 (m, 2 H), 7.72 (s, 1 H), 7.82 (br s, 1 H), 7.99 (br s, 1 H), 8.81 ppm (br s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C21H24FN4O3S: 431.1553, found: 431.1552.
1‐[2‐(Methylsulfonyl)propan‐2‐yl]‐3‐nitrobenzene and (rac)‐1‐[1‐(methylsulfonyl)ethyl]‐3‐nitrobenzene: Under argon, 1 m sodium bis(trimethylsilyl)amide in THF (4.65 mL, 4.65 mmol) was added dropwise to a stirred solution of 1‐[(methylsulfonyl)methyl]‐3‐nitrobenzene (500 mg, 2.32 mmol) in THF (25 mL) at −78 °C. The reaction mixture was stirred for 30 min at this temperature before iodomethane (725 mg, 5.11 mmol) was added. The reaction mixture was stirred for 1 h at −78 °C, then the cold bath was removed and the mixture was slowly warmed to RT. aq. NH4Cl solution was added and the mixture was extracted with EtOAc (3×). The combined organic phases were dried (Na2SO4), filtered and concentrated. The residue was purified by flash chromatography (hexane to hexane/EtOAc, 1:1) to give 1‐[2‐(methylsulfonyl)propan‐2‐yl]‐3‐nitrobenzene (139 mg, 0.57 mmol, 25 %): 1H NMR (400 MHz, CDCl3): δ=1.93 (s, 6 H), 2.62 (s, 3 H), 7.63 (t, 1 H), 8.06 (dt, 1 H), 8.26 (d, 1 H), 8.47 ppm (s, 1 H), and (rac)‐1‐[1‐(methylsulfonyl)ethyl]‐3‐nitrobenzene (84 mg, 0.37 mmol, 16 %): 1H NMR (400 MHz, CDCl3): δ=1.87 (d, 3 H), 2.76 (s, 3 H), 4.31 (q, 1 H), 7.63 (t, 1 H), 7.85 (d, 1 H), 8.25–8.32 ppm (m, 2 H).
(rac)‐3‐[1‐(Methylsulfonyl)ethyl]aniline: TiCl3 solution (ca. 15 % in ∼10 % HCl, 2.4 mL) was added to a stirred solution of (rac)‐1‐[1‐(methylsulfonyl)ethyl]‐3‐nitrobenzene (80 mg, 0.35 mmol) in THF (3.7 mL) at RT, and the mixture was stirred for 16 h. The pH was increased to 10 by the addition of 2 n NaOH, then the reaction mixture was extracted with EtOAc (2×). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated to give the crude product (88 mg) that was used without further purification.
(rac)‐4‐Chloro‐N‐{3‐[1‐(methylsulfonyl)ethyl]phenyl}‐1,3,5‐triazin‐2‐amine: DIPEA (0.15 mL, 0.86 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 65 mg, 0.43 mmol) in THF/iPrOH (1:1, 0.8 mL) at −40 °C. Then, a solution of crude (rac)‐3‐[1‐(methylsulfonyl)ethyl]aniline (86 mg) in THF/iPrOH (1:1, 0.4 mL) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 2 h to 0 °C. The mixture was concentrated in vacuo to give the crude product (233 mg) that was used without further purification.
(rac)‐4‐(4‐Fluoro‐2‐methoxyphenyl)‐N‐{3‐[1‐(methylsulfonyl)ethyl]phenyl}‐1,3,5‐triazin‐2‐amine (10): A mixture of crude (rac)‐4‐chloro‐N‐{3‐[1‐(methylsulfonyl)ethyl]phenyl}‐1,3,5‐triazin‐2‐amine (233 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (127 mg, 0.75 mmol) and Pd(PPh3)4 (129 mg, 0.11 mmol) in 1,2‐dimethoxyethane (2.3 mL) and aq. 2 m K2CO3 (0.7 mL) was degassed using argon. The mixture was stirred under argon for 90 min at 100 °C. After cooling, the mixture was diluted with EtOAc and THF, and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 10 as a light beige solid (34 mg, 0.08 mmol): 1H NMR (400 MHz, CDCl3): δ=1.82 (d, 3 H), 2.69 (s, 3 H), 3.94 (s, 3 H), 4.20 (q, 1 H), 6.74–6.82 (m, 2 H), 7.20 (br d, 1 H), 7.39–7.52 (m, 2 H), 7.80 (s, 2 H), 7.97 (br s, 1 H), 8.82 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C19H20FN4O3S: 403.1240, found: 403.1245.
3‐[2‐(Methylsulfonyl)propan‐2‐yl]aniline: TiCl3 solution (ca. 15 % in ∼10 % HCl, 3.8 mL) was added to a stirred solution of 1‐[2‐(methylsulfonyl)propan‐2‐yl]‐3‐nitrobenzene (135 mg, 0.56 mmol) in THF (5.8 mL) at RT, and the mixture was stirred for 16 h. The pH was increased to 10 by the addition of 2 n NaOH, then the reaction mixture was extracted with EtOAc (2×). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated to give the crude product (148 mg) that was used without further purification.
4‐Chloro‐N‐{3‐[2‐(methylsulfonyl)propan‐2‐yl]phenyl}‐1,3,5‐triazin‐2‐amine: DIPEA (0.24 mL, 1.37 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 103 mg, 0.68 mmol) in THF/iPrOH (1:1, 1.3 mL) at −40 °C. Then, a solution of crude 3‐[2‐(methylsulfonyl)propan‐2‐yl]aniline (146 mg) in THF/iPrOH (1:1, 0.6 mL) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 2 h to 0 °C. The mixture was concentrated in vacuo to give the crude product (354 mg) that was used without further purification.
4‐(4‐Fluoro‐2‐methoxyphenyl)‐N‐{3‐[2‐(methylsulfonyl)propan‐2‐yl]phenyl}‐1,3,5‐triazin‐2‐amine (11): A mixture of crude 4‐chloro‐N‐{3‐[2‐(methylsulfonyl)propan‐2‐yl]phenyl}‐1,3,5‐triazin‐2‐amine (125 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (65 mg, 0.38 mmol) and Pd(PPh3)4 (66 mg, 0.06 mmol) in 1,2‐dimethoxyethane (1.2 mL) and aq. 2 m K2CO3 (0.4 mL) was degassed using argon. The mixture was stirred under argon for 90 min at 100 °C. After cooling, the mixture was diluted with EtOAc and THF, and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 11 as a light beige solid (29 mg, 0.07 mmol): 1H NMR (400 MHz, CDCl3): δ=1.87 (s, 6 H), 2.56 (s, 3 H), 3.93 (s, 3 H), 6.74–6.81 (m, 2 H), 7.28–7.45 (m, 3 H), 7.80 (br s, 1 H), 7.95 (br s, 2 H), 8.82 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C20H22FN4O3S: 417.1397, found: 417.1396.
1‐[Difluoro(methylsulfonyl)methyl]‐3‐nitrobenzene and (rac)‐1‐[fluoro(methylsulfonyl)methyl]‐3‐nitrobenzene: Under argon, 1 m sodium bis(trimethylsilyl)amide in THF (10.22 mL, 10.22 mmol) was added dropwise to a stirred solution of 1‐[(methylsulfonyl)methyl]‐3‐nitrobenzene (1000 mg, 4.65 mmol) in THF (50 mL) at −78 °C. The reaction mixture was stirred for 30 min at this temperature before N‐fluorobenzenesulfonimide (3662 mg, 11.61 mmol) was added. The reaction mixture was stirred for 5 h at −78 °C. aq. NH4Cl solution was added and the cold bath was removed. At RT, the mixture was extracted with EtOAc (3×). The combined organic phases were washed with aq. NaHCO3 solution, dried (Na2SO4), filtered and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 1‐[difluoro(methylsulfonyl)methyl]‐3‐nitrobenzene (173 mg, 0.69 mmol, 15 %): 1H NMR (600 MHz, CDCl3): δ=3.20 (s, 3 H), 7.76 (t, 1 H), 8.01–8.05 (m, 1 H), 8.49 (dd, 1 H), 8.56 ppm (s, 1 H), and (rac)‐1‐[fluoro(methylsulfonyl)methyl]‐3‐nitrobenzene (235 mg, 1.01 mmol, 22 %): 1H NMR (600 MHz, CDCl3): δ=3.07 (d, 3 H), 6.16 (d, 1 H), 7.70 (t, 1 H), 7.91 (dd, 1 H), 8.37–8.41 (m, 1 H), 8.43–8.45 ppm (m, 1 H).
(rac)‐3‐[Fluoro(methylsulfonyl)methyl]aniline: TiCl3 solution (ca. 15 % in ∼10 % HCl, 0.4 mL) was added to a stirred solution of (rac)‐1‐[fluoro(methylsulfonyl)methyl]‐3‐nitrobenzene (14 mg, 0.06 mmol) in THF (0.6 mL) at RT, and the mixture was stirred for 16 h. The pH was increased to 9–10 by the addition of an aq. NaHCO3 solution, then the reaction mixture was extracted with EtOAc/THF (1:1, 3×). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated to give the crude product (20 mg) that was used without further purification.
(rac)‐4‐Chloro‐N‐{3‐[fluoro(methylsulfonyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine: DIPEA (0.03 mL, 0.16 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 12 mg, 0.08 mmol) in THF/iPrOH (1:1, 0.2 mL) at −40 °C. Then, a solution of crude (rac)‐3‐[fluoro(methylsulfonyl)methyl]aniline (16 mg) in THF/iPrOH (1:1, 0.2 mL) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 90 min to 0 °C. The mixture was concentrated in vacuo to give the crude product (38 mg) that was used without further purification.
(rac)‐4‐(4‐Fluoro‐2‐methoxyphenyl)‐N‐{3‐[fluoro(methylsulfonyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine (12): A mixture of crude (rac)‐4‐chloro‐N‐{3‐[fluoro(methylsulfonyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine (38 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (20 mg, 0.12 mmol) and Pd(PPh3)4 (21 mg, 0.02 mmol) in 1,2‐dimethoxyethane (0.4 mL) and aq. 2 m K2CO3 (0.1 mL) was degassed using argon. The mixture was stirred under argon for 90 min at 100 °C. After cooling, the mixture was diluted with EtOAc and THF, and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 12 as a light beige solid (5 mg, 0.01 mmol): 1H NMR (400 MHz, CDCl3): δ=2.96 (s, 3 H), 3.93 (s, 3 H), 6.05 (d, 1 H), 6.73–6.83 (m, 2 H), 7.28–7.37 (m, 1 H), 7.42–7.58 (m, 2 H), 7.85 (br s, 1 H), 7.94 (s, 2 H), 8.83 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C18H17F2N4O3S: 407.0989, found: 407.0990.
3‐[Difluoro(methylsulfonyl)methyl]aniline: TiCl3 solution (ca. 15 % in ∼10 % HCl, 0.4 mL) was added to a stirred solution of 1‐[difluoro(methylsulfonyl)methyl]‐3‐nitrobenzene (16 mg, 0.06 mmol) in THF (0.7 mL) at RT, and the mixture was stirred for 16 h. The pH was increased to 9–10 by the addition of an aq. NaHCO3 solution, then the reaction mixture was extracted with EtOAc/THF (1:1, 3×). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated to give the crude product (18 mg) that was used without further purification.
4‐Chloro‐N‐{3‐[difluoro(methylsulfonyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine: DIPEA (0.03 mL, 0.16 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 12 mg, 0.08 mmol) in THF/iPrOH (1:1, 0.2 mL) at −40 °C. Then, a solution of crude 3‐[difluoro(methylsulfonyl)methyl]aniline (17 mg) in THF/iPrOH (1:1, 0.2 mL) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 90 min to 0 °C. The mixture was concentrated in vacuo to give the crude product (40 mg) that was used without further purification.
N ‐{3‐[Difluoro(methylsulfonyl)methyl]phenyl}‐4‐(4‐fluoro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐amine (13): A mixture of crude 4‐chloro‐N‐{3‐[difluoro(methylsulfonyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine (40 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (20 mg, 0.12 mmol) and Pd(PPh3)4 (21 mg, 0.02 mmol) in 1,2‐dimethoxyethane (0.4 mL) and aq. 2 m K2CO3 (0.1 mL) was degassed using argon. The mixture was stirred under argon for 90 min at 100 °C. After cooling, the mixture was diluted with EtOAc and THF, and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 13 as a white solid (6 mg, 0.01 mmol): 1H NMR (400 MHz, CDCl3): δ=3.14 (s, 3 H), 3.93 (s, 3 H), 6.74–6.81 (m, 2 H), 7.43–7.56 (m, 3 H), 7.95–8.09 (m, 3 H), 8.84 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C18H16F3N4O3S: 425.0895, found: 425.0899.
1‐(3‐{[4‐(2‐Methoxyphenyl)‐1,3,5‐triazin‐2‐yl]amino}phenyl)methanesulfonamide (14): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (103 mg), 2‐methoxyphenylboronic acid (68 mg, 0.45 mmol) and K3PO4 (218 mg, 1.03 mmol) in dioxane/water (10:1, 8 mL) was degassed with a stream of nitrogen for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (25 mg, 0.03 mmol) was added and the reaction mixture was heated for 90 min at 145 °C in a microwave oven. The mixture was diluted with EtOAc and washed with sat. aq. NaHCO3. The organic phase was dried (Na2SO4), filtered and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 14 as a light beige solid (36 mg, 0.10 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=3.87 (s, 3 H), 4.25 (s, 2 H), 6.87 (br s, 2 H), 7.06–7.13 (m, 2 H), 7.21 (d, 1 H), 7.37 (t, 1 H), 7.55 (dt, 1 H), 7.73 (t, 1 H), 7.84 (m, 2 H), 8.82 (s, 1 H), 10.52 ppm (s, 1 H); MS (ESI) m/z: 372 [M+H]+.
1‐(3‐{[4‐(4‐Chloro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐yl]amino}phenyl)methanesulfonamide (15): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (174 mg), 4‐chloro‐2‐methoxyphenylboronic acid (116 mg, 0.62 mmol) and K3PO4 (217 mg, 1.02 mmol) in dioxane/water (10:1, 10 mL) was degassed with a stream of nitrogen for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (28 mg, 0.03 mmol) was added and the reaction mixture was heated for 90 min at 145 °C in a microwave oven. The mixture was diluted with EtOAc and washed with sat. aq. NaHCO3. The organic phase was dried (Na2SO4), filtered and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 15 as a light beige solid (13 mg, 0.03 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=3.90 (s, 3 H), 4.24 (s, 2 H), 6.86 (br s, 2 H), 7.08 (d, 1 H), 7.15 (d, 1 H), 7.28 (d, 1 H), 7.36 (t, 1 H), 7.74 (s, 1 H), 7.88 (m, 2 H), 8.81 (s, 1 H), 10.44 ppm (s, 1 H); MS (ESI) m/z: 406 [M+H]+.
1‐(3‐{[4‐(3‐Methoxypyridin‐4‐yl)‐1,3,5‐triazin‐2‐yl]amino}phenyl)methanesulfonamide (16): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (141 mg), 3‐methoxypyridine‐4‐boronic acid (104 mg, 0.68 mmol) and K3PO4 (213 mg, 1.00 mmol) in dioxane/water (10:1, 10 mL) was degassed with a stream of nitrogen for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (38 mg, 0.05 mmol) was added and the reaction mixture was heated for 90 min at 145 °C in a microwave oven. The mixture was diluted with EtOAc and washed with sat. aq. NaHCO3. The organic phase was dried (Na2SO4), filtered and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 16 as a white solid (51 mg, 0.14 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=3.99 (s, 3 H), 4.25 (s, 2 H), 6.87 (s, 2 H), 7.11 (d, 1 H), 7.36 (dt, 1 H), 7.74 (s, 1 H), 7.83 (m, 2 H), 8.39 (m, 1 H), 8.62 (m, 1 H), 8.87 (m, 1 H), 10.52 ppm (s, 1 H); MS (ESI) m/z: 373 [M+H]+.
1‐(3‐{[4‐(4‐Fluoro‐2‐hydroxyphenyl)‐1,3,5‐triazin‐2‐yl]amino}phenyl)methanesulfonamide (17): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (300 mg), 4‐fluoro‐2‐hydroxyphenylboronic acid (234 mg, 1.50 mmol) and K3PO4 (425 mg, 2.00 mmol) in dioxane/water (20:1, 10 mL) was degassed with a stream of argon for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (82 mg, 0.10 mmol) was added and the reaction mixture was heated for 1 h at 140 °C in a microwave oven. The mixture was diluted with an aq. NaCl solution and extracted with EtOAc. The combined organic phases were dried (Na2SO4), filtered and concentrated. The residue was purified by preparative HPLC (basic conditions) to give 17 as a white solid (91 mg, 0.24 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=4.29 (s, 2 H), 6.64 (s, 2 H), 6.73 (dd, 1 H), 6.75–6.82 (m, 1 H), 7.18 (d, 1 H), 7.38 (t, 1 H), 7.66–7.71 (m, 1 H), 7.75 (s, 1 H), 8.41 (dd, 1 H), 8.78 (s, 1 H), 10.41 (br s, 1 H), 12.95–13.35 ppm (m, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C16H15FN5O3S: 376.0880, found: 376.0883.
1‐(3‐{[4‐(2,4‐Difluorophenyl)‐1,3,5‐triazin‐2‐yl]amino}phenyl)methanesulfonamide (18): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (300 mg), 2,4‐difluorophenylboronic acid (237 mg, 1.50 mmol) and K3PO4 (425 mg, 2.00 mmol) in dioxane/water (20:1, 10 mL) was degassed with a stream of argon for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (82 mg, 0.10 mmol) was added and the reaction mixture was heated for 1 h at 140 °C in a microwave oven. The mixture was diluted with an aq. NaCl solution and extracted with EtOAc. The combined organic phases were dried (Na2SO4), filtered and concentrated. The residue was purified by preparative HPLC (basic conditions) to give 18 as a light pink solid (141 mg, 0.37 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=4.25 (s, 2 H), 6.89 (s, 2 H), 7.10 (d, 1 H), 7.24–7.47 (m, 3 H), 7.80 (br s, 2 H), 8.30 (br s, 1 H), 8.86 (s, 1 H), 10.47 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C16H14F2N5O2S: 378.0836, found: 378.0834.
1‐[3‐({4‐[2‐(Trifluoromethoxy)phenyl]‐1,3,5‐triazin‐2‐yl}amino)phenyl]methanesulfonamide (19): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (300 mg), 2‐(trifluoromethoxy)phenylboronic acid (309 mg, 1.50 mmol) and K3PO4 (425 mg, 2.00 mmol) in dioxane/water (20:1, 10 mL) was degassed with a stream of argon for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (82 mg, 0.10 mmol) was added and the reaction mixture was heated for 1 h at 140 °C in a microwave oven. The mixture was diluted with an aq. NaCl solution and extracted with EtOAc. The combined organic phases were dried (Na2SO4), filtered and concentrated. The residue was purified by preparative HPLC (basic conditions) to give 19 (10 mg, 0.02 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=4.25 (s, 2 H), 6.88 (s, 2 H), 7.10 (d, 1 H), 7.36 (t, 1 H), 7.54 (d, 1 H), 7.59 (t, 1 H), 7.71 (td, 2 H), 7.81 (br s, 1 H), 8.06 (br s, 1 H), 8.88 (s, 1 H), 10.51 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C17H15F3N5O3S: 426.0848, found: 426.0844.
1‐[3‐({4‐[2‐(Methoxymethyl)phenyl]‐1,3,5‐triazin‐2‐yl}amino)phenyl]methanesulfonamide (20): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (400 mg), 2‐(methoxymethyl)phenylboronic acid (221 mg, 1.33 mmol) and K3PO4 (377 mg, 1.80 mmol) in dioxane/water (50:1, 9 mL) was degassed with a stream of nitrogen for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (70 mg, 0.09 mmol) was added and the reaction mixture was heated for 15 h at 140 °C in a sealed tube. The mixture was poured onto ice and extracted with EtOAc. The organic phase was dried (Na2SO4), filtered and concentrated. The residue was purified by preparative TLC (CHCl3/MeOH, 29:1) yielding 20 as a white solid (6 mg, 0.02 mmol): 1H NMR (400 MHz, CDCl3): δ=3.32 (s, 3 H), 4.32 (s, 2 H), 4.82 (s, 2 H), 5.27 (br s, 2 H), 7.11 (d, 1 H), 7.26 (m, 1 H), 7.41 (t, 1 H), 7.52 (t, 1 H), 7.59–7.76 (m, 4 H), 7.93 (br s, 1 H), 8.67 ppm (s, 1 H); MS (ESI) m/z: 386 [M+H]+.
N ‐[5‐Fluoro‐2‐(4‐{[3‐(sulfamoylmethyl)phenyl]amino}‐1,3,5‐triazin‐2‐yl)phenyl]acetamide (21): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (75 mg), 2‐(acetylamino)‐4‐fluorophenylboronic acid (74 mg, 0.38 mmol) and K3PO4 (107 mg, 0.50 mmol) in dioxane/water (20:1, 2.5 mL) was degassed with a stream of argon for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (21 mg, 0.025 mmol) was added and the reaction mixture was heated for 60 min at 140 °C in a microwave oven. The mixture was diluted with dioxane, filtered and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 21 as a white solid (15 mg, 0.04 mmol): 1H NMR (500 MHz, [D6]DMSO, 80 °C): δ=2.08 (br s, 3 H), 4.30 (s, 2 H), 6.68 (s, 2 H), 7.03 (ddd, 1 H), 7.18 (d, 1 H), 7.40 (t, 1 H), 7.67 (br d, 1 H), 7.77 (s, 1 H), 8.38 (dd, 1 H), 8.56 (dd, 1 H), 8.87 (s, 1 H), 10.30 (s, 1 H), 12.33 ppm (br s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C18H18FN6O3S: 417.1145, found: 417.1146.
1‐[3‐({4‐[4‐Fluoro‐2‐(trifluoromethyl)phenyl]‐1,3,5‐triazin‐2‐yl}amino)phenyl]methanesulfonamide (22): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (100 mg), 4‐fluoro‐2‐(trifluoromethyl)phenylboronic acid (104 mg, 0.50 mmol) and K3PO4 (142 mg, 0.67 mmol) in dioxane/water (20:1, 3.3 mL) was degassed with a stream of argon for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (27 mg, 0.033 mmol) was added and the reaction mixture was heated for 60 min at 140 °C in a microwave oven. The mixture was diluted with dioxane, filtered and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 22 as a white solid (18 mg, 0.04 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=4.23 (br s, 2 H), 6.88 (s, 2 H), 7.10 (d, 1 H), 7.28–7.38 (m, 1 H), 7.66–7.77 (m, 3 H), 7.81 (dd, 1 H), 7.91 (br s, 1 H), 8.85 (s, 1 H), 10.52 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C17H14F4N5O2S: 428.0804, found: 428.0813.
1‐[3‐({4‐[2‐(Benzyloxy)‐4‐fluorophenyl]‐1,3,5‐triazin‐2‐yl}amino)phenyl]methanesulfonamide (23): A mixture of crude 3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzenemethanesulfonamide (300 mg), 2‐(benzyloxy)‐4‐fluorophenylboronic acid (369 mg, 1.50 mmol) and K3PO4 (425 mg, 2.00 mmol) in dioxane/water (20:1, 10 mL) was degassed with a stream of argon for 15 min. Pd(dppf)Cl2⋅CH2Cl2 (82 mg, 0.10 mmol) was added and the reaction mixture was heated for 1 h at 140 °C in a microwave oven. The mixture was diluted with an aq. NaCl solution and extracted with EtOAc. The combined organic phases were dried (Na2SO4), filtered and concentrated. The residue was purified by flash chromatography (hexane/EtOAc, 2:1 to EtOAc) to give 23 (32 mg, 0.07 mmol): 1H NMR (400 MHz, [D6]DMSO): δ=4.21 (br s, 2 H), 5.27 (s, 2 H), 6.82–6.95 (m, 3 H), 7.06 (br s, 1 H), 7.14 (dd, 1 H), 7.22–7.49 (m, 6 H), 7.69 (br s, 1 H), 7.84 (br s, 2 H), 8.84 (s, 1 H), 10.36 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C23H21FN5O3S: 466.1349, found: 466.1351.
(rac)‐1‐[(Methylsulfinyl)methyl]‐3‐nitrobenzene (26): FeCl3 (0.55 g, 3.4 mmol) was added to a solution of 1‐[(methylsulfanyl)methyl]‐3‐nitrobenzene (25; 21.6 g, 117.9 mmol) in MeCN (280 mL), and the mixture was stirred at RT for 10 min. Periodic acid (28.8 g, 126.1 mmol) was added under stirring in one portion and the temperature was kept below 30 °C by cooling. The mixture was stirred at RT for 90 min, then added to a stirred solution of sodium thiosulfate pentahydrate (163 g, 660 mmol) in ice‐water (1500 mL). This mixture was saturated with solid NaCl and extracted with THF (2×). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated. The residue was purified by flash chromatography (CH2Cl2/EtOH, 95:5) to give 26 (16.6 g, 83.1 mmol, 70 %): 1H NMR (400 MHz, CDCl3): δ=2.53 (s, 3 H), 3.97 (d, 1 H), 4.10 (d, 1 H), 7.58 (m, 1 H), 7.67 (m, 1 H), 8.17 (m, 1 H), 8.21 ppm (m, 1 H).
(rac)‐2,2,2‐Trifluoro‐N‐[methyl(3‐nitrobenzyl)oxido‐λ6‐sulfanylidene]acetamide: To a suspension of (rac)‐26 (16.6 g, 83.1 mmol), trifluoroacetamide (18.8 g, 166.1 mmol), MgO (13.4 g, 332.3 mmol) and rhodium(II) acetate dimer (1.7 g, 3.9 mmol) in CH2Cl2 (2290 mL) was added iodobenzene diacetate (40.1 g, 124.6 mmol) at RT. The mixture was stirred for 16 h at RT, then filtered and concentrated. The residue was purified by flash chromatography (CH2Cl2/EtOH, 97:3) to give the desired product (25.6 g), still containing minor impurities: 1H NMR (400 MHz, CDCl3): δ=3.28 (s, 3 H), 4.79 (d, 1 H), 4.91 (d, 1 H), 7.69 (m, 1 H), 7.80 (m, 1 H), 8.31 (m, 1 H), 8.36 ppm (m, 1 H).
(rac)‐1‐[(S‐Methylsulfonimidoyl)methyl]‐3‐nitrobenzene (27): K2CO3 (56.9 g, 411.8 mmol) was added to a solution of (rac)‐2,2,2‐trifluoro‐N‐[methyl(3‐nitrobenzyl)oxido‐λ6‐sulfanylidene]acetamide (25.6 g, ∼82.4 mmol) in MeOH (1768 mL) at RT. The mixture was stirred for 1 h at RT, then diluted with EtOAc and brine, and extracted with EtOAc (2×). The combined organic phases were dried (Na2SO4), filtered and concentrated to give (rac)‐27 (13.9 g, 65.1 mmol, 79 %): 1H NMR (400 MHz, CDCl3): δ=2.66 (br, 1 H), 2.99 (s, 3 H), 4.34 (d, 1 H), 4.47 (d, 1 H), 7.63 (m, 1 H), 7.79 (m, 1 H), 8.29 ppm (m, 2 H).
(rac)‐Ethyl [methyl(3‐nitrobenzyl)oxido‐λ6‐sulfanylidene]carbamate (28): Ethyl chlorocarbonate (8.1 mL, 84.6 mmol) was added dropwise to a stirred solution of (rac)‐27 (13.9 g, 65.1 mmol) in pyridine (615 mL) at 0 °C. The mixture was slowly warmed to RT. After 24 h, the mixture was concentrated and the residue was dissolved in EtOAc and washed with brine. The organic phase was filtered using a Whatman filter and concentrated to give (rac)‐28 (19.7 g) that was used without further purification: 1H NMR (400 MHz, CDCl3): δ=1.31 (t, 3 H), 3.07 (s, 3 H), 4.18 (q, 2 H), 4.79 (d, 1 H), 4.88 (d, 1 H), 7.64 (m, 1 H), 7.81 (m, 1 H), 8.30 ppm (m, 2 H).
(rac)‐Ethyl [(3‐aminobenzyl)(methyl)oxido‐λ6‐sulfanylidene]carbamate (29): TiCl3 solution (ca. 15 % in ∼10 % HCl, 118 mL) was added to a stirred solution of (rac)‐28 (5.0 g, ∼17.5 mmol) in THF (220 mL) at RT, and the mixture was stirred for 18 h. The solution was brought to pH 8 by the addition of 2 n NaOH to the reaction mixture, which was cooled with an ice bath. The mixture was saturated with solid NaCl and extracted with EtOAc (3×). The combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated to give (rac)‐29 (4.2 g, 16.5 mmol, 94 %) that was used without further purification: 1H NMR (400 MHz, CDCl3): δ=1.13 (t, 3 H), 3.08 (s, 3 H), 3.95 (m, 2 H), 4.62 (s, 2 H), 5.18 (br, 2 H), 6.53 (m, 3 H), 7.00 ppm (m, 1 H).
(rac)‐Ethyl [{3‐[(4‐chloro‐1,3,5‐triazin‐2‐yl)amino]benzyl}(methyl)oxido‐λ6‐sulfanylidene]carbamate (30): DIPEA (3.1 mL, 17.8 mmol) was added to a stirred solution of 2,4‐dichloro‐1,3,5‐triazine (1; 1.34 g, 8.9 mmol) in THF/iPrOH (1:1, 18 mL) at −40 °C. Then, a solution of (rac)‐29 (2.29 g, ∼8.9 mmol) in THF/iPrOH (1:1, 9 mL) was added at this temperature. Under stirring, the temperature of the reaction mixture was slowly raised over 3 h to 0 °C. The mixture was concentrated to give (rac)‐30 (4.9 g) that was used without further purification.
(rac)‐Ethyl [(3‐{[4‐(4‐fluoro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐yl]amino}benzyl)(methyl)oxido‐λ6‐sulfanylidene]carbamate (31): A mixture of crude (rac)‐30 (400 mg), 4‐fluoro‐2‐methoxyphenylboronic acid (276 mg, 1.62 mmol) and Pd(PPh3)4 (187 mg, 0.16 mmol) in 1,2‐dimethoxyethane (5.0 mL) and aq. 2 m K2CO3 (1.1 mL) was degassed using argon. The mixture was stirred under argon for 80 min at 100 °C. After cooling, the mixture was diluted with EtOAc and washed with brine. The organic phase was filtered using a Whatman filter and concentrated. The residue was purified by flash chromatography (CH2Cl2/EtOH, 95:5) to give (rac)‐31 (178 mg, 0.39 mmol, 36 %): 1H NMR (400 MHz, CDCl3): δ=1.30 (t, 3 H), 3.00 (s, 3 H), 3.93 (s, 3 H), 4.17 (q, 2 H), 4.74 (m, 2 H), 6.77 (m, 2 H), 7.16 (m, 1 H), 7.42 (m, 1 H), 7.53 (s, 1 H), 7.74 (br, 1 H), 7.84 (s, 1 H), 7.94 (m, 1 H), 8.82 ppm (s, 1 H).
(rac)‐4‐(4‐Fluoro‐2‐methoxyphenyl)‐N‐{3‐[(S‐methylsulfonimidoyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine (32 and BAY 1143572): Freshly prepared 1.5 m sodium ethanolate in EtOH (2.9 mL, 4.35 mmol) was added under argon to a solution of (rac)‐31 (500 mg, 1.09 mmol) in EtOH (18.5 mL), and the mixture was stirred at 60 °C for 2 h. Further 1.5 m sodium ethanolate in EtOH (2.9 mL, 4.35 mmol) was added, and the mixture was stirred for an additional 5 h at 60 °C. After cooling, the mixture was diluted with brine and extracted with EtOAc (3×). The combined organic phases were filtered using a Whatman filter and concentrated. The residue was purified by flash chromatography (CH2Cl2/EtOH, 9:1) to give the desired racemic product as a white solid (378 mg, 0.98 mmol, 90 %): 1H NMR (400 MHz, CDCl3): δ=2.71 (s, 1 H), 2.96 (s, 3 H), 3.92 (s, 3 H), 4.26 (d, 1 H), 4.39 (d, 1 H), 6.75 (m, 2 H), 7.15 (m, 1 H), 7.40 (m, 1 H), 7.55 (s, 1 H), 7.77 (m, 2 H), 7.95 (m, 1 H), 8.80 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C18H19FN5O2S: 388.1243, found: 388.1239. The racemate was separated into the single enantiomers by preparative HPLC [system: Dionex P580 pump, Gilson 215 Liquid Handler, Knauer K‐2501 UV detector; column: Chiralpak IC 5 μm, 250×20 mm; eluent: hexane/EtOH (6:4)+0.1 % diethylamine; flow: 40 mL min−1; solution: 2600 mg/44 mL EtOH/DMSO (2:1); injection: 55×0.8 mL; temperature: RT; detection: UV 254 nm]: t R (min): 13.4–15.6 (enantiomer 1: compound 32; ESI‐HRMS: m/z [M+H]+ calcd for C18H19FN5O2S: 388.1243, found: 388.1241), 15.6–18.5 (enantiomer 2: BAY 1143572).
(R)‐4‐(4‐Fluoro‐2‐methoxyphenyl)‐N‐{3‐[(S‐methylsulfonimidoyl)methyl]phenyl}‐1,3,5‐triazin‐2‐amine (BAY 1143572): Mp: 165 °C; =−14.0±0.40 (c=1 in DMSO); 1H NMR (600 MHz, [D6]DMSO): δ=2.81 (br s, 3 H), 3.57 (s, 1 H), 3.88 (br s, 3 H), 4.29–4.40 (m, 2 H), 6.90 (td, 1 H), 7.09 (dd, 1 H), 7.13 (d, 1 H), 7.36 (t, 1 H), 7.62–8.09 (m, 3 H), 8.75–8.83 (m, 1 H), 10.34 ppm (s, 1 H); 13C NMR (125 MHz, [D6]DMSO): δ=40.9, 56.2, 62.3, 100.4, 106.7, 120.2, 122.8, 122.9, 125.8, 128.4, 130.9, 133.2, 138.5, 160.0, 163.1, 164.5, 166.2, 171.1 ppm; IR (KBr): =3325‐3246, 3013, 2960‐2918, 1616‐1403, 1284–1103 cm−1; ESI‐HRMS: m/z [M+H]+ calcd for C18H19FN5O2S: 388.1243, found: 388.1246.
(R)‐N‐[(3‐{[4‐(4‐Fluoro‐2‐methoxyphenyl)‐1,3,5‐triazin‐2‐yl]amino}benzyl)(methyl)oxido‐λ6‐sulfanylidene]acetamide (33): Acetyl chloride (22 mg, 0.28 mmol) was added dropwise to a solution of BAY 1143572 (100 mg, 0.26 mmol) in TEA (31 mg, 0.31 mmol) and CH2Cl2 (3.0 mL) at 0 °C. The ice bath was removed and the mixture was stirred at RT for 3 h. Then, the mixture was diluted with water and extracted with CH2Cl2 (3×). The combined organic phases were filtered using a Whatman filter and concentrated. The residue was purified by preparative HPLC (acidic conditions) to give 33 as a white solid (63 mg, 0.15 mmol, 57 %): 1H NMR (600 MHz, [D6]DMSO): δ=2.14 (s, 3 H), 3.05 (s, 3 H), 3.94 (s, 3 H), 4.67 (d, 1 H), 4.80 (d, 1 H), 6.74–6.83 (m, 2 H), 7.17 (d, 1 H), 7.42 (t, 1 H), 7.82 (br s, 2 H), 7.97 (br s, 1 H), 8.03 (br s, 1 H), 8.83 ppm (s, 1 H); ESI‐HRMS: m/z [M+H]+ calcd for C20H21FN5O3S: 430.1349, found: 430.1346.
X‐ray structure determination of 33
Single crystals of 33 were obtained by slow evaporation of an ethyl acetate solution of 33 at RT. A single crystal was mounted on a CryoLoop using inert oil. Data collection was carried out using a Bruker diffractometer equipped with an APEX II CCD area detector, an IμS microsource with Cu Kα radiation, mirrors as monochromator and a Kryoflex low temperature device (T=110 K). The program APEX II v.2011.8‐0 was used for data collection and reduction.29 Absorption correction and scaling was performed using SADABS.30 The crystal structure solution was achieved using direct methods as implemented in SHELXTL version 6.1431 and visualized using the XP program.31 Missing atoms were subsequently located from difference Fourier synthesis and added to the atom list. Least‐squares refinement on F 2 using all measured intensities was carried out using the program SHELXTL version 6.14.31 All non‐hydrogen atoms were refined including anisotropic displacement parameters. Final data collection and structure refinement parameters: λ=1.54178 Å, T=110 K, space group=P2(1), a=10.3041(8) Å, b=7.9030(9) Å, c=12.8716(11) Å, β=108.990(5)°, Z=2, reflections collected=6794, independent reflections=1261 (Rint=0.0848), completeness=98.2 %, data‐to‐parameter ratio=11.5, R 1 (I>2σ)=0.061, wR 2 (all data)=0.1420, GOF=0.988, Flack parameter=0.05(4), largest difference peak and hole=0.297/‐0.262 e Å−3. CCDC 1565910 (33) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Acknowledgements
We thank K. Sauvageot‐Witzku, R. Golde, A. Glowczewski, C. Pakebusch, N. Gallus, and R. Droschinski for technical support, O. Schenk for HPLC separations, S. Gründemann and G. Depke for analytical support, and U. Ganzer for the measurement of physicochemical properties. M. Bergmann and K. Greenfield are acknowledged for valuable technical support with the manuscript. Activities at the LDC were co‐funded by the Max Planck Foundation on behalf of the Max Planck Society, as well as by a grant from the Ministry for Research and Technology (BMBF, grant number 0315326).
U. Lücking, A. Scholz, P. Lienau, G. Siemeister, D. Kosemund, R. Bohlmann, H. Briem, I. Terebesi, K. Meyer, K. Prelle, K. Denner, U. Bömer, M. Schäfer, K. Eis, R. Valencia, S. Ince, F. von Nussbaum, D. Mumberg, K. Ziegelbauer, B. Klebl, A. Choidas, P. Nussbaumer, M. Baumann, C. Schultz-Fademrecht, G. Rühter, J. Eickhoff, M. Brands, ChemMedChem 2017, 12, 1776.
References
- 1.
- 1a. Wesierska-Gadek J., Chamrad I., Krystof V., Future Med. Chem. 2009, 1, 1561; [DOI] [PubMed] [Google Scholar]
- 1b. Mariaule G., Belmont P., Molecules 2014, 19, 14366; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Cicenas J., Kalyan K., Sorokinas A., Jatulyte A., Valiunas D., Kaupinis A., Valius M., Cancers 2014, 6, 2224; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Santo L., Siu K. T., Raje N., Semin. Oncol. 2015, 42, 788; [DOI] [PubMed] [Google Scholar]
- 1e. Sánchez-Martínez C., Gelbert L. M., Jose Lallena M., de Dios A., Bioorg. Med. Chem. Lett. 2015, 25, 3420; [DOI] [PubMed] [Google Scholar]
- 1f. Asghar U., Witkiewicz A. K., Turner N. C., Knudsen E. S., Nat. Rev. Drug Discovery 2015, 14, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Law M. E., Corsino P. E., Narayan S., Law B. K., Mol. Pharmacol. 2015, 88, 846; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. O′Leary B., Finn R. S., Turner N. C., Nat. Rev. Clin. Oncol. 2016, 13, 417. [DOI] [PubMed] [Google Scholar]
- 3. Cho S., Schroeder S., Ott M., Cell Cycle 2010, 9, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Krystof V., Baumli S., Fürst R., Curr. Pharm. Des. 2012, 18, 2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.See, for example:
- 5a. Beckwith R. E. J., Curtis D. T., Harrington E., Hinrichs J. H.-H., Tallarico J. A. (Novartis AG), Int. PCT Pub. No. WO 2008079933, 2008;
- 5b. Augustin M., Zeitlmann L., Stumm G., Pleiss M. A., Wabnitz P., Allgeier H. (Ingenium Pharmaceuticals AG), Int. PCT Pub. No. WO 2008049856, 2008;
- 5c. Allgeier H., Augustin M., Mueller A., Zeitlmann L., Marquardt A., Pleiss M. A., Heiser U., Niestroj A. J. (Ingenium Pharmaceuticals AG), Int. PCT Pub. No. WO 2009047359, 2009;
- 5d. Barsanti P., Hu C., Jin J., Keyes R., Kucejko R., Lin X., Pan Y., Pfister K. B., Sendzik M., Sutton J., Wan L. (Novartis AG), Int. PCT Pub. No. WO 2011012661, 2011;
- 5e. Nowicki M. W., Walkinshaw M. D., Chem. Biol. 2010, 17, 1047; [DOI] [PubMed] [Google Scholar]
- 5f. Liu X., Shi S., Lam F., Pepper C., Fischer P. M., Wang S., Int. J. Cancer 2012, 130, 1216; [DOI] [PubMed] [Google Scholar]
- 5g. Shao H., Shi S., Huang S., Hole A. J., Abbas A. Y., Baumli S., Liu X., Lam F., Foley D. W., Fischer P. M., Noble M., Endicott J. A., Pepper C., Wang S., J. Med. Chem. 2013, 56, 640; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5h.“AstraZeneca boosts oncology arsenal with CDK9 inhibitor platform”, D. Stanton, in-PharmaTechnologist.com, 07.Jan.2014: http://www.in-pharmatechnologist.com/Drug-Delivery/AstraZeneca-boosts-oncology-arsenal-with-CDK9-inhibitor-platform (accessed July, 2017);
- 5i. Gong J., Tao Z.-F., Tong Y., Zhu G., Penning T., Souers A. J. (Abbvie Inc.), Int. PCT Pub. No. WO 2014151444, 2014;
- 5j. Yin T., Lallena M. J., Kreklau E. L., Fales K. R., Carballares S., Torrres R., Wishart G. N., Ajamie R. T., Cronier D. M., Iversen P. W., Meier T. I., Foreman R. T., Zeckner D., Sissons S. E., Halstead B. W., Lin A. B., Donoho G. P., Qian Y., Li S., Wu S., Aggarwal A., Ye X. S., Starling J. J., Gaynor R. B., de Dios A., Diu J., Mol. Cancer Ther. 2014, 13, 1442; [DOI] [PubMed] [Google Scholar]
- 5k. Morales F., Giordano A., Cell Cycle 2016, 15, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Toogood P. L., Harvey P. J., Repine J. T., Sheehan D. J., VanderWel S. N., Zhou H., Keller P. R., McNamara D. J., Sherry D., Zhu T., Brodfuehrer J., Choi C., Barvian M. R., Fry D. W., J. Med. Chem. 2005, 48, 2388. [DOI] [PubMed] [Google Scholar]
- 7. Parry D., Guzi T., Shanahan F., Davis N., Prabhavalkar D., Wiswell D., Seghezzi W., Paruch K., Dwyer M. P., Doll R., Nomeir A., Windsor W., Fischmann T., Wang Y., Oft M., Chen T., Kirschmeier P., Lees E. M., Mol. Cancer Ther. 2010, 9, 2344. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Siemeister G., Lücking U., Wengner A. M., Lienau P., Steinke W., Schatz C., Mumberg D., Ziegelbauer K., Mol. Cancer Ther. 2012, 11, 2265; [DOI] [PubMed] [Google Scholar]
- 8b. Lücking U., Jautelat R., Krüger M., Brumby T., Lienau P., Schäfer M., Briem H., Schulze J., Hillisch A., Reichel A., Wengner A. M., Siemeister G., ChemMedChem 2013, 8, 1067. [DOI] [PubMed] [Google Scholar]
- 9. Herbrink M., Schellens J. H. M., Beijnen J. H., Nuijen B., J. Controlled Release 2016, 239, 118. [DOI] [PubMed] [Google Scholar]
- 10. Bissantz C., Kuhn B., Stahl M., J. Med. Chem. 2010, 53, 5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bentley H. R., McDermott E. E., Pace J., Whitehead J. K., Moran T., Nature 1949, 163, 675.18120748 [Google Scholar]
- 12.
- 12a. Johnson C. R., Aldrichimica Acta 1985, 18, 3; [Google Scholar]
- 12b. Reggelin M., Zur C., Synthesis 2000, 1. [Google Scholar]
- 13.
- 13a. Johnson C. R., Acc. Chem. Res. 1973, 6, 341; [Google Scholar]
- 13b. Craig D., Grellepois F., White A. J. P., J. Org. Chem. 2005, 70, 6827; [DOI] [PubMed] [Google Scholar]
- 13c. Gais H.-J., Babu G. S., Günter M., Das P., Eur. J. Org. Chem. 2004, 1464; [Google Scholar]
- 13d. Harmata M., Hong X., J. Am. Chem. Soc. 2003, 125, 5754; [DOI] [PubMed] [Google Scholar]
- 13e. Harmata M., Hong X., Barnes C. L., Tetrahedron Lett. 2003, 44, 7261; [Google Scholar]
- 13f. Harmata M., Pavri N., Angew. Chem. Int. Ed. 1999, 38, 2419; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 2577; [Google Scholar]
- 13g. Koep S., Gais H.-J., Raabe G., J. Am. Chem. Soc. 2003, 125, 13243; [DOI] [PubMed] [Google Scholar]
- 13h. Shen X., Miao W., Ni C., Hu J., Angew. Chem. Int. Ed. 2014, 53, 775; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 794; [Google Scholar]
- 13i. Shen X., Liu Q., Zhang W., Hu J., Eur. J. Org. Chem. 2016, 906. [Google Scholar]
- 14.
- 14a. Bolm C., Simic O., J. Am. Chem. Soc. 2001, 123, 3830; [DOI] [PubMed] [Google Scholar]
- 14b. Harmata M., Ghosh S. K., Org. Lett. 2001, 3, 3321; [DOI] [PubMed] [Google Scholar]
- 14c. Bolm C., Martin M., Simic O., Verrucci M., Org. Lett. 2003, 5, 427; [DOI] [PubMed] [Google Scholar]
- 14d. Bolm C., Felder M., Müller J., Synlett 1992, 439; [Google Scholar]
- 14e. Bolm C., Verrucci M., Simic O., Cozzi P. G., Raabe G., Okamura H., Chem. Commun. 2003, 2826; [DOI] [PubMed] [Google Scholar]
- 14f. Langner M., Bolm C., Angew. Chem. Int. Ed. 2004, 43, 5984; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 6110; [Google Scholar]
- 14g. Langner M., Remy P., Bolm C., Chem. Eur. J. 2005, 11, 6254; [DOI] [PubMed] [Google Scholar]
- 14h. Reetz M. T., Bondarev O. G., Gais H.-J., Bolm C., Tetrahedron Lett. 2005, 46, 5643. [Google Scholar]
- 15. Lücking U., Angew. Chem. Int. Ed. 2013, 52, 9399; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9570. [Google Scholar]
- 16.
- 16a. Frings M., Bolm C., Blum A., Gnamm C., Eur. J. Med. Chem. 2017, 126, 225; [DOI] [PubMed] [Google Scholar]
- 16b. Sirvent J. A., Lücking U., ChemMedChem 2017, 12, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.See, for example:
- 17a. Lücking U., Krueger M., Jautelat R., Siemeister G. (Schering AG), Int. PCT Pub. No. WO 2005037800, 2005;
- 17b. Lücking U., Siemeister G., Jautelat R. (Schering Aktiengesellschaft), Int. PCT Pub. No. WO 2006099974, 2006;
- 17c. Lücking U. (Schering Aktiengesellschaft), EP 1710246, 2006;
- 17d. Lücking U., Kettschau G., Briem H., Schwede W., Schäfer M., Thierauch K.-H., Husemann M. (Schering Aktiengesellschaft), Int. PCT Pub. No. WO 2006108695, 2006;
- 17e. Luecking U., Nguyen D., von Bonin A., von Ahsen O., Krueger M., Briem H., Kettschau G., Prien O., Mengel A., Krolikiewicz K., Boemer U., Bothe U., Hartung I. (Schering Aktiengesellschaft), Int. PCT Pub. No. WO 2007071455, 2007;
- 17f. Lücking U., Siemeister G., Bader B. (Schering Aktiengesellschaft), Int. PCT Pub. No. WO 2007079982, 2007;
- 17g. Luecking U., Siemeister G., Jautelat R. (Bayer Schering Pharma Aktiengesellschaft), Int. PCT Pub. No. WO 2008025556, 2008;
- 17h. Prien O., Eis K., Lücking U., Guenther J., Zopf D. (Bayer Schering Pharma Aktiengesellschaft), DE 102007024470, 2008;
- 17i. Hartung I., Bothe U., Kettschau G., Luecking U., Mengel A., Krueger M., Thierauch K.-H., Lienau P., Boemer U. (Bayer Schering Pharma Aktiengesellschaft), Int. PCT Pub. No. WO 2008155140, 2008;
- 17j. Nguyen D., Von Bonin A., Haerter M., Schirok H., Mengel A., Von Ahsen O. (Bayer Schering Pharma Aktiengesellschaft), Int. PCT Pub. No. WO 2009089851, 2009;
- 17k. Härter M., Beck H., Ellinghaus P., Berhoerster K., Greschat S., Thierauch K.-H., Süssmeier F. (Bayer Schering Pharma Aktiengesellschaft), Int. PCT Pub. No. WO 2010054763, 2010;
- 17l. von Nussbaum F., Karthaus D., Anlauf S., Delbeck M., Li V. M.-J., Meibom D., Lustig K., Schneider D. (Bayer Schering Pharma Aktiengesellschaft), Int. PCT Pub. No. WO 2010115548, 2010;
- 17m. Schwede W., Klar U., Möller C., Rotgeri A., Bone W. (Bayer Schering Pharma Aktiengesellschaft), Int. PCT Pub. No. WO 2011009531, 2011.
- 18. Okamura H., Bolm C., Org. Lett. 2004, 6, 1305. [DOI] [PubMed] [Google Scholar]
- 19.For an alternative synthesis based on a palladium-catalyzed α-arylation, see: Sirvent J. A., Bierer D., Webster R., Lücking U., Synthesis 2017, 49, 1024. [Google Scholar]
- 20. Ertl P., Rohde B., Selzer P., J. Med. Chem. 2000, 43, 3714. [DOI] [PubMed] [Google Scholar]
- 21. Bradner J. E., Hnisz D., Young R. A., Cell 2017, 168, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a.A. Scholz, T. Oellerich, A. Hussain, S. Lindner, U. Luecking, A. O. Walter, P. Ellinghaus, R. Valencia, F. von Nussbaum, D. Mumberg, M. Brands, S. Ince, H. Serve, K. Ziegelbauer, Cancer Res 2016, 76, Abstract No. 3022; https://doi.org/10.1158/1538-7445.AM2016-3022;
- 22b.A. Scholz, U. Luecking, G. Siemeister, P. Lienau, U. Boemer, P. Ellinghaus, A. O. Walter, R. Valencia, S. Ince, F. von Nussbaum, D. Mumberg, M. Brands, K. Ziegelbauer, Cancer Res 2015, 75, Abstract No. DDT02-02; https://doi.org/10.1158/1538-7445.AM2015-DDT02-02;
- 22c.U. Luecking, A. Scholz, P. Lienau, G. Siemeister, D. Kosemund, R. Bohlmann, K. Eis, M. Gnoth, I. Terebesi, K. Meyer, K. Prelle, R. Valencia, S. Ince, F. von Nussbaum, D. Mumberg, K. Ziegelbauer, B. Klebl, A. Choidas, P. Nussbaumer, M. Baumann, C. Schultz-Fademrecht, G. Ruehter, J. Eickhoff, M. Brands, Cancer Res 2015, 75, Abstract No. 2828; https://doi.org/10.1158/1538-7445.AM2015-2828.
- 23. Kueng W., Silber E., Eppenberger U., Anal. Biochem. 1989, 182, 16. [DOI] [PubMed] [Google Scholar]
- 24.See, for example: Kerns E. H., Di L., Drug-like Properties: Concepts, Structure Design and Methods, Elsevier Inc./Academic Press, Burlington, MA (USA), 2008, pp. 276–286. [Google Scholar]
- 25. Ahmad A. B., Bennett P. N., Rowland M., J. Pharm. Pharmacol. 1983, 35, 219. [DOI] [PubMed] [Google Scholar]
- 26.Glide, version 5.7, Schrödinger, LLC, New York, NY, 2011.
- 27. Baumli S., Endicott J. A., Johnson L. N., Chem. Biol. 2010, 17, 931. [DOI] [PubMed] [Google Scholar]
- 28.LigPrep, version 2.5, Schrödinger, LLC, New York, NY, 2011.
- 29.APEX II, version 2011.8-0, Bruker AXS Inc., Madison, WI, 2011.
- 30. Krause L., Herbst-Irmer R., Sheldrick G. M., Stalke D., J. Appl. Crystallogr. 2015, 48, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.SHELXTL, version 6.14, Bruker AXS Inc., Madison, WI, 2003.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary