

Abstract
The Na+‐taurocholate cotransporting polypeptide (NTCP/SLC10A1) is believed to be pivotal for hepatic uptake of conjugated bile acids. However, plasma bile acid levels are normal in a subset of NTCP knockout mice and in mice treated with myrcludex B, a specific NTCP inhibitor. Here, we elucidated which transport proteins mediate the hepatic uptake of conjugated bile acids and demonstrated intestinal sensing of elevated bile acid levels in plasma in mice. Mice or healthy volunteers were treated with myrcludex B. Hepatic bile acid uptake kinetics were determined in wild‐type (WT), organic anion transporting polypeptide (OATP) knockout mice (lacking Slco1a/1b isoforms), and human OATP1B1‐transgenic mice. Effects of fibroblast growth factor 19 (FGF19) on hepatic transporter mRNA levels were assessed in rat hepatoma cells and in mice by peptide injection or adeno‐associated virus–mediated overexpression. NTCP inhibition using myrcludex B had only moderate effects on bile acid kinetics in WT mice, but completely inhibited active transport of conjugated bile acid species in OATP knockout mice. Cholesterol 7α‐hydroxylase Cyp7a1 expression was strongly down‐regulated upon prolonged inhibition of hepatic uptake of conjugated bile acids. Fgf15 (mouse counterpart of FGF19) expression was induced in hypercholanemic OATP and NTCP knockout mice, as well as in myrcludex B–treated cholestatic mice, whereas plasma FGF19 was not induced in humans treated with myrcludex B. Fgf15/FGF19 expression was induced in polarized human enterocyte‐models and mouse organoids by basolateral incubation with a high concentration (1 mM) of conjugated bile acids. Conclusion: NTCP and OATPs contribute to hepatic uptake of conjugated bile acids in mice, whereas the predominant uptake in humans is NTCP mediated. Enterocytes sense highly elevated levels of (conjugated) bile acids in the systemic circulation to induce FGF15/19, which modulates hepatic bile acid synthesis and uptake. (Hepatology 2017;66:1631–1643).
Abbreviations
- AAV
adeno‐associated virus
- Asbt
apical sodium‐dependent bile acid transporter
- BDL
bile duct ligation
- BW
body weight
- C4
7α‐hydroxy‐4‐cholesten‐3‐one
- CA
cholate
- Cyp7a1
cholesterol 7α‐hydroxylase
- FGF15/19
fibroblast growth factor 15/19
- Fgfr4
fibroblast growth factor receptor 4
- FRET
Förster resonance energy transfer
- FXR
farnesoid X receptor
- GCA
glycocholic acid
- GCDCA
glycochenodeoxycholic acid
- GFP
green fluorescent protein
- HBV
hepatitis B virus
- HBV/HDV
hepatitis B/D virus
- hFGF19
human FGF19
- HPLC
high‐pressure liquid chromatography
- Ibabp
ileal bile acid binding protein
- Mrp2
multidrug resistance‐associated protein 2
- Mrp4
multidrug resistance‐associated protein 4
- NTCP
sodium taurocholate cotransporting polypeptide
- OATP
organic anion transporting polypeptide
- Ostα/β
organic solute transporter alpha/beta
- Shp
small heterodimer partner
- TβMCA
tauro‐beta‐muricholic acid
- TCA
taurocholic acid
- TCDCA
taurochenodeoxycholic acid
- TGR5
transmembrane G protein‐coupled receptor 5
- v.g.
vector genomes
- WT
wild type
Bile acids are amphipathic molecules that solubilize lipids in the intestinal tract, and are potent systemic signaling hormones, acting through farnesoid X receptor (FXR) and transmembrane G protein‐coupled receptor 5 (TGR5) in various organs.1 Bile acids undergo effective (re)uptake from the portal blood, limiting escape of bile acids to the systemic circulation. The sodium‐dependent taurocholate cotransporting polypeptide (Slc10a1/NTCP) extracts the majority of conjugated bile acids at the basolateral membrane of the liver.2 In 2012, NTCP was found to be the main receptor for the hepatitis B virus (HBV).3 The hepatitis B/D virus (HBV/HDV) entry inhibitor, myrcludex B, interferes with viral entry into the hepatocyte4, 5 and is currently being tested in HBV/HDV clinical trials.6, 7 Notably, myrcludex B is a specific NTCP inhibitor in vivo,8 and administration of myrcludex B in healthy volunteers and mutations in the SLC10A1 gene result in increased plasma conjugated bile acid levels (A. Blank, personal communication9, 10).
The role of NTCP as the major hepatic uptake transporter for conjugated bile acids in vivo was further corroborated in NTCP knockout mice.8 More than half of the adult NTCP knockout mice maintain normal levels of (conjugated) bile acids in plasma, although hepatic bile acid uptake is delayed. NTCP knockout mice that show elevated plasma levels of conjugated bile acids display altered hepatic expression of members of the sodium‐independent organic anion transporting polypeptide (Slco/OATP) family. OATP1A4 was strongly up‐regulated and OATP1A1 was completely down‐regulated. It was previously demonstrated that members of the OATP family are essential for efficient hepatic uptake of unconjugated bile acids. Total Slco1a/1b‐family knockout mice showed a marked conjugated hyperbilirubinemia and elevated unconjugated bile acid levels in blood,11 and the latter phenotype was also present in Slco1b2 knockout mice, suggesting that this isoform is most critical for unconjugated bile acid uptake.12 The three most abundant hepatic OATP isoforms in rodents are OATP1B2, OATP1A1, and OATP1A4.13 Here, we investigated whether expression of OATP isoforms is sufficient to maintain hepatic uptake of conjugated bile acids in adult mice, even in the absence of functional NTCP. To this end, we used Slco1a/1b knockout mice injected with myrcludex B to inhibit NTCP. We also investigated whether functional expression of human OATP1B1 in mice compensates for loss of mouse OATPs and NTCP regarding transport of conjugated bile acids. Finally, while searching for an explanation for the two distinct phenotypical presentations of NTCP‐deficient mice, we found that enterocytes can sense highly elevated basolateral levels of bile acids, to induce fibroblast growth factor (FGF) 15/FGF19, which eventually modifies bile acid synthesis as well as the hepatic bile acid uptake machinery in mice.
Materials and Methods
Detailed information on materials and methods can be found in the Supporting Information.
MOUSE STUDIES
Male Slc10a1 knockout mice (C57Bl6/J background) were housed and bred in the Academic Medical Center, Amsterdam.8 Control wild‐type (WT) mice were purchased from Envigo, Horst, the Netherlands. Male Slco1a/1b knockouts mice (lacking Slco1b2 and Slco1a1 to Slco1a5) and Slco1a/1b knockouts reconstituted with liver‐specific expression of human OATP1B1 (FVB/N background) and WT littermates were housed and bred in the Netherlands Cancer Institute, Amsterdam11 or obtained from Taconic, Silkeborg, Denmark. A single dose of myrcludex B (intravenously, 5 μg/g body weight [BW]) was administered in acute taurocholate (TCA) clearance studies to block NTCP‐mediated TCA uptake. Male WT mice were subjected to common bile duct ligation (BDL) and cholecystectomy, as described.14 Treatment with myrcludex B (5‐day subcutaneously, 2.5 μg/g BW) was performed in male (OATP1B1‐humanized) Slco1a/1b knockouts and in BDL mice. To investigate acute regulation of hepatic bile acid uptake transporters, male WT and Fgf15 knockout mice (3‐4 months of age, described in an earlier work15) were injected intraperitoneally with recombinant human FGF19 (hFGF19; 1 mg/kg) or saline at 3 am. Between 8:00 and 9:00 am, mice were killed by isofluorane overdosage. Db/db male mice (11‐week‐old, #000642, BKS background) were purchased from The Jackson Laboratory (Ben Harbor, ME) and received a single intravenous dose of 3 × 1011 vector genomes (v.g.) of adeno‐associated virus (AAV) encoding either FGF19 or a control protein (GFP). Db/db mice were euthanized 24 weeks post‐AAV administration to examine long‐term effects of FGF19. The study design and animal care and handling were approved by the Institutional Animal Care and Use Committee of the University of Amsterdam (Amsterdam, The Netherlands) and the University of Texas Southwestern Medical Center of Dallas (Dallas, TX).
HUMAN STUDY
The Clinical Research Unit of the Department of Clinical Pharmacology and Pharmacoepidemiology at the Heidelberg University Hospital (EN ISO‐standard 9001) conducted a clinical study, which followed the guideline of good clinical practice, the ethical principles expressed in the Declaration of Helsinki, and all legal requirements for clinical studies in Germany. The responsible ethics committee of the Medical Faculty of Heidelberg University (Heidelberg, Germany) and the competent national authority approved the study (BfArM, Germany, EudraCT: 2014‐003289‐26). Each participant provided written informed consent before any study‐related procedure was conducted. Further details are described in Blank et al. (2017; manuscript submitted).
STATISTICAL ANALYSIS
Data are provided as the mean ± SEM. Differences between groups were analyzed using the Mann‐Whitney U test or Kruskal‐Wallis test. Statistical significance was considered at P < 0.05, and calculations and graphs were generated using GraphPad Prism software (version 6.0; GraphPad Software Inc.).
Results
TCA TRANSPORT IN Slco1a/1b KNOCKOUT MICE AFTER MYRCLUDEX B
Despite delayed bile acid uptake, only a subset of adult NTCP knockout mice showed elevated plasma bile acid levels.8 In these mice, OATP1A1 was completely absent in the liver (Fig. 1A). In contrast, OATP1A4 expression was up‐regulated, in particular in the periportal area (Fig. 1B). Expression of both OATP isoforms was unaffected in normocholanemic NTCP knockout mice (data not shown). To investigate the contribution of the OATP isoforms to hepatic uptake of conjugated bile acids, TCA clearance was determined in WT and Slco1a/1b knockout mice. Although plasma TCA elimination of WT mice injected with myrcludex B was slightly reduced during the first 5 minutes of clearance compared to vehicle injections, eventually all TCA was effectively cleared, suggesting significant NTCP‐independent TCA uptake in mouse liver (Fig. 1C; red line, open dots). Plasma TCA clearance was not reduced in Slco1a/1b knockout mice treated with vehicle compared to WT mice. Strikingly, plasma TCA clearance in Slco1a/1b knockout mice injected with myrcludex B (Fig. 1C, red line, open squares) was completely abrogated.
Figure 1.
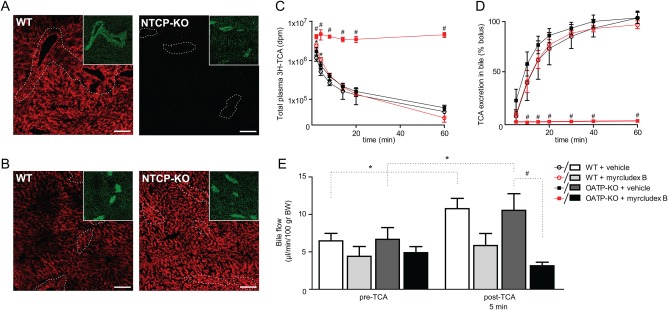
TCA kinetics in WT and Slco1a/1b (OATP1A/1B) knockout mice. (A,B) Immunofluorescent visualization of mouse Oatp1a1 (A) or –Oatp1a4 (B) in male WT and Slc10a1 knockout (NTCP‐KO) mice with high plasma bile acid levels. Pericentral hepatocytes were visualized by costaining of glutamine synthetase (green, as insert). Scale bar is 40 μm. (C) Plasma TCA clearance in WT and OATP1A/1B knockout mice after myrcludex B or vehicle administration. TCA with tracer amounts of [3H]TCA was injected into the tail vein of mice and tritium activity (dpm) was measured in plasma (C) or bile (D) at indicated time points. (E) Hepatic bile flow pre‐TCA and post‐TCA injection in WT and OATP1A/1B knockout mice injected with myrcludex B or vehicle. (C‐E) N = 5/6 mice per group. Asterisk indicates significant difference between vehicle and myrcludex B in WT mice, and hash indicates significant difference between vehicle and myrcludex B in OATP1A/1B knockout mice (P < 0.05; Mann‐Whitney U test).
Biliary TCA excretion matched these results, because labeled TCA was hardly detectable (<1%) in bile of Slco1a/1b knockout mice after myrcludex B treatment, even after 60 minutes. In contrast, 38% and 56% of the labeled TCA was recovered over a 10‐minute period in bile of vehicle‐injected WT or Slco1a/1b knockout mice versus 41% recovery in bile of WT mice injected with myrcludex B (Fig. 1D). This shows that NTCP and OATPs together mediate all hepatic uptake of conjugated bile acid.
To ensure that these effects were not attributed to defective apical bile acid excretion, hepatic bile flow was determined after myrcludex B injections. WT mice showed similar bile flow as Slco1a/1b knockout mice, 10 minutes after vehicle injections (Fig. 1E). Myrcludex B treatment did not block bile flow, although there was a tendency to a reduction in both genotypes, even before TCA administration (∼30%; not significant). Upon intravenous administration of TCA, bile flow transiently increased in WT and Slco1a/1b knockout mice. This increase was less prominent in WT mice after myrcludex B injections, and bile flow did not increase at all upon TCA administration in mice that functionally lack both uptake pathways.
ELEVATED LEVELS OF CONJUGATED BILE ACIDS IN Slco1a/1b KNOCKOUT MICE INJECTED WITH MYRCLUDEX B
The TCA kinetics data raised the question as to whether myrcludex B causes elevated conjugated bile acid levels in blood of Slco1a/1b knockout mice. Measurement of individual plasma bile acid species by high‐performance liquid chromatography (HPLC) showed increased conjugated bile acid levels 3 hours after the last myrcludex B injection in Slco1a/1b knockout mice, reaching a concentration of ∼1 mM and mainly composed of TCA (904.4 ± 108.6 μM) and tauro‐β‐muricholic acid (TβMCA; 181.6 ± 18.7 μM; Fig. 2A). Plasma unconjugated bile acids, such as cholate (CA), were modestly elevated as well (Fig. 2B). Similar results were obtained with myrcludex B treatment in Slco1a/1b knockout mice that express the human OATP1B1 specifically in the liver.16
Figure 2.
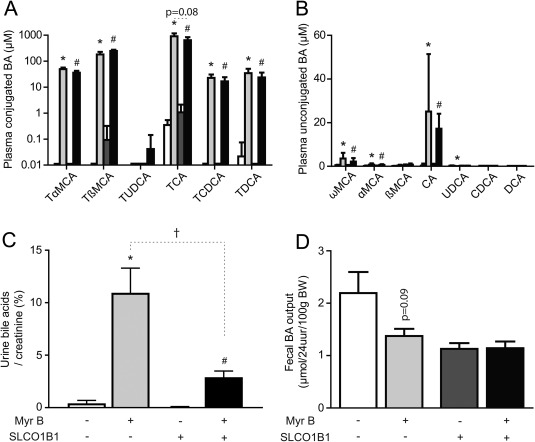
Myrcludex B increases bile acid levels in plasma and urine, but not in feces, of (OATP1B1‐humanized) Slco1a/1b knockout mice. Myrcludex B or vehicle was injected for 5 days. Plasma composition of (A) conjugated and (B) unconjugated bile acid species as quantified by HPLC. Concentrations (μM) are given on a 10log scale for 6 mice/group. (C) Urinary bile acid concentrations corrected for creatinine concentrations in each mouse (μmol/mmol). (D) Total fecal bile acid excretion (μmol/24 hours/100 g BW). White and light‐gray bars indicate OATP‐KO mice, injected with vehicle or myrcludex B, respectively. Dark‐gray or black bars indicate OATP1B1‐humanized OATP‐KO mice, injected with vehicle or mycludex B, respectively. Asterisk indicates significant change for OATP‐KO mice, and hash indicates significant change for OATP1B1‐humanized OATP‐KO mice. Dagger indicates significant change between genotypes upon myrcludex B treatment (P < 0.05; Mann‐Whitney U test). Abbreviations: BA, bile acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; KO, knockout; MCA, muricholic acid; Myr B, myrcludex B; TDCA, taurodeoxycholic acid; TUDCA, tauroursodeoxycholic acid.
Total plasma bile acid concentrations were virtually normalized in both mouse models 24 hours after myrcludex B injection (Supporting Fig. S1A), indicating only transiently inhibited hepatic bile acid uptake upon daily myrcludex B injection in mice. This is consistent with studies of myrcludex B in humans, rats, and dogs, reporting a rapid distribution to the liver (maximum hepatic binding 4 hours postinjection) and an elimination half‐life of ∼12 hours.7, 17
Renal bile acid elimination mirrored plasma bile acid levels, because urinary concentrations in Slco1a/1b knockout and OATP1B1‐humanized Slco1a/1b knockout mice were low after vehicle injections and massively increased 3 hours after myrcludex B in both genotypes (Fig. 2C). OATP1B1‐humanized Slco1a/1b knockout mice had reduced renal bile acid excretion compared to Slco1a/1b knockout mice. Also, plasma TCA levels showed a trend to decrease in OATP1B1‐humanized Slco1a/1b knockouts after myrcludex B administration. Functionality of human OATP1B1 was demonstrated by normalization of plasma bilirubin levels, which were elevated in Slco1a/1b knockout mice (Supporting Table S2). The renal bile acid transporter, organic solute transporter alpha/beta (Ostα/β), was induced by myrcludex B treatment (269% in Slco1a/1b knockout and 178% in OATP1B1‐humanized Slco1a/1b knockout mice; Supporting Fig. S1B). The same figure shows that expression of apical sodium‐dependent bile acid transporter (Asbt), multidrug resistance‐associated protein 2 (Mrp2), and multidrug resistance‐associated protein 4 (Mrp4) was unaffected by myrcludex B. Fecal bile acid excretion was not significantly affected in Slco1a/1b knockouts upon myrcludex B treatment (Fig. 2D). In both Slco1a/1b models, we observed no signs of hepatobiliary damage after myrcludex B treatment, because plasma aspartate aminotransferase/alanine aminotransferase and alkaline phosphatase were comparable and not elevated in any group (Supporting Table S2). Conjugated bilirubin levels were higher in Slco1a/1b knockout mice compared to OATP1B1‐humanized Slco1a/1b knockout mice, in line with OATP‐mediated bilirubin transport,18 and myrcludex B treatment did not affect plasma levels of (conjugated) bilirubin in either genotype (Supporting Table S2). Histological examination of hematoxylin and eosin–stained liver sections showed no abnormalities in (OATP1B1‐humanized) Slco1a/1b knockouts after 5‐day myrcludex B administration (Supporting Fig. S2).
THREE MURINE HEPATIC OATP ISOFORMS TRANSPORT CONJUGATED BILE ACIDS IN VITRO
Bile acid uptake experiments were performed in vitro to investigate whether the three liver‐specific OATPs can mediate conjugated bile acid transport. All three OATP isoforms were capable of significant TCA uptake compared to untransfected U2OS cells (mock; Fig. 3A). In line with a previous study of Leuthold et al.,19 mouse OATP1A1 had the highest conjugated TCA uptake velocity (after NTCP, the positive control of this experiment). OATP‐mediated uptake of conjugated bile acids was confirmed using the Förster resonance energy transfer (FRET)‐based bile acid sensor we previously developed to assess real‐time intracellular bile acid uptake dynamics.20 FRET ratio increased upon addition of taurochenodeoxycholic acid (TCDCA) when OATP1B2, OATP1A1, OATP1A4, or NTCP were transiently expressed in U2OS cells, whereas mock‐transfected U2OS cells did not show bile acid uptake (Fig. 3B). Expression of OATP1B2, OATP1A1, and OATP1A4 at the plasma membrane was confirmed by cell‐surface biotinylation and western blotting analysis (Fig. 3C).
Figure 3.
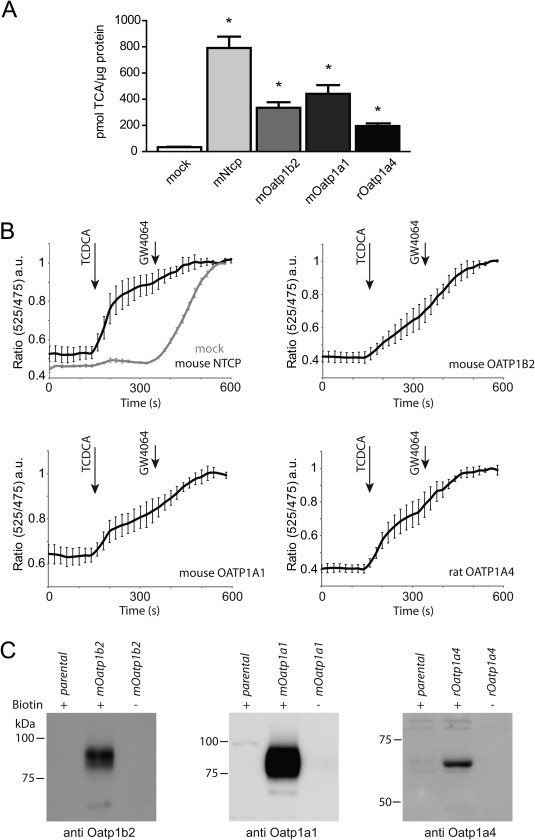
OATP isoforms mediate uptake of conjugated bile acids in vitro. (A) [3H]TCA uptake in U2OS transiently overexpressing mouse Ntcp, Oatp1b2, Oatp1a1, and rat Oatp1a4. N = 6 wells/group from two independent experiments. NTCP‐ and OATP‐mediated transport was compared to parental cells (mock), and asterisk indicates significant difference (P < 0.05). (B) FRET‐based sensing of NTCP‐ and OATP‐mediated TCDCA influx. The nuclear FRET sensor for bile acids (NucleoBAS) was cotransfected with empty plasmid (mock) or each individual bile acid uptake transporter in U2OS cells. The emission ratio of citrin (525 nm)/cerulean (475 nm) is plotted in time. Compound addition is indicated by arrows. To fully activate NucleoBAS, each experiment ended with addition of 5 μM of GW4064. N = 6 cells/group, from two independent experiments. (C) Evident plasma membrane expression of mouse OATP1B2, OATP1A1, and rat OATP1A4 was demonstrated by cell surface biotinylation. (+) indicates with biotin. (–) indicates without biotin.
OATP1A1 DOWN‐REGULATION IS NOT ATTRIBUTED TO INFLAMMATION
We aimed to uncover the mechanism underlying the reduced OATP1A1 expression, associated with strongly elevated levels of conjugated bile acids in NTCP knockout mice. Inflammation has previously been described to down‐regulate Oatp expression in a cytokine‐mediated manner.21 We could indeed confirm that inflammation significantly reduces hepatic expression of Oatp1a1 in a sterile inflammation model (T‐cell transfer; Supporting Fig. S3A). However, cytokine levels in the blood of Slco1a/1b and Slc10a1 knockout mice were below detection levels (Supporting Fig. S3B), even when bile acid levels were highly elevated. Given that histology also did not show any hepatic recruitment of leukocytes (Supporting Fig. S2), this suggests that inflammation does not underlie the observed changes in Oatp expression. Fortunately, analysis of Cyp7a1 transcriptional dynamics pointed us to an intestine‐derived signal that may modulate expression of hepatic bile acid transporters, including OATPs.
CHOLESTEROL 7α‐HYDROXYLASE DOWN‐REGULATION POINTS TO INDUCTION OF ILEAL FGF15 WHEN BILE ACIDS LEVELS ARE ELEVATED
Cholesterol 7α‐hydroxylase (CYP7A1) mediates the first and rate‐limiting step in bile acid production. We first determined hepatic Cyp7a1 mRNA expression in mice that received myrcludex B once 1.5 hours before sacrifice. Cyp7a1 increased in Slco1a/1b knockout mice, but not in WT mice (Fig. 4A). This matches the previously described CYP7A1 induction in human liver chimeric mice upon myrcludex B treatment.22 Remarkably, and in contrast to this acute upregulation, 5‐day myrcludex B treatment caused significant down‐regulation of Cyp7a1 mRNA levels, in both Slco1a/1b knockout mice and OATP1B1‐humanized Slco1a/1b knockout mice (Fig. 4B). Similarly, reduced bile acid synthesis was functionally demonstrated in NTCP knockout mice with elevated bile acid levels, because 7α‐hydroxy‐4‐cholesten‐3‐one (C4) levels were significantly lower in plasma (Fig. 4C). Down‐regulation of Cyp7a1 could indicate increased intestinal FXR‐FGF15 signaling.23, 24 Several ileal FXR target genes were tested to see whether intestinal FXR is activated by 5‐day myrcludex B treatment in Slco1a/1b knockout mice (Fig. 5A). Both small heterodimer partner (Shp) and Fgf15 mRNA strongly increased in Slco1a/1b knockout mice and in OATP1B1‐humanized Slco1a/1b knockout mice after myrcludex B treatment. Ileal bile acid binding protein (Ibabp) mRNA expression moderately increased, but only in total Slco1a/1b knockout mice. To confirm induction of the ileal FXR‐FGF15 axis when bile acid levels are elevated in the systemic circulation, we also analyzed ileal mRNA expression in WT and NTCP knockout mice with high plasma bile acid levels (Fig. 5B). NTCP knockout mice display clearly increased Shp and Fgf15 mRNA levels.
Figure 4.
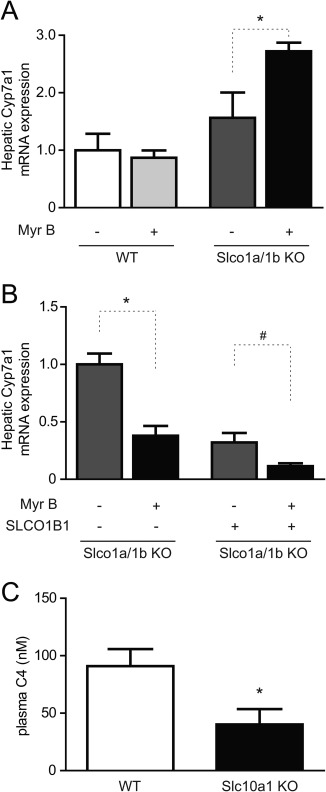
Cyp7a1 expression after acute or prolonged myrcludex B treatment. (A) Hepatic mRNA expression of the rate‐limiting bile acid synthesis enzyme, Cyp7a1, shortly (1.5 hours) after vehicle or myrcludex B treatment in WT or Slco1a/1b knockout (KO) mice. Values are normalized to vehicle‐treated WT mice. Asterisk indicates significant increase (P < 0.05) between vehicle and myrcludex B injection in Slco1a/1b knockout mice. (B) Hepatic Cyp7a1 mRNA expression after 5 daily injections with vehicle or myrcludex B in (OATP1B1‐humanized) Slco1a/1b knockout mice. Asterisk or hash symbols indicate significant decrease (P < 0.05; 5‐6 fasted male mice/group) compared to vehicle controls. Data are calculated using the geometric mean of reference genes Rplp0 and Tbp and normalized to vehicle‐treated Slco1a/1b knockout mice. (C) C4 levels, a marker for bile acid synthesis, were reduced in plasma of Slc10a1 knockout compared to WT mice. Data from 5‐6 fasted female mice/group. Abbreviations: KO, knockout; Myr B, myrcludex B.
Figure 5.
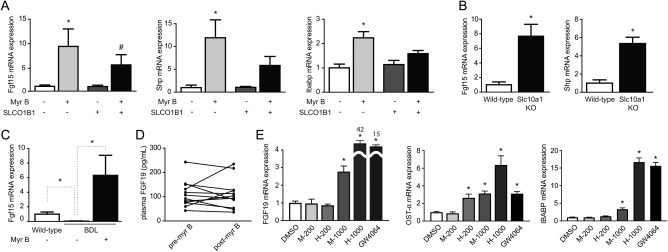
Millimolar concentrations of conjugated bile acids, present in blood after myrcludex B treatment, induce intestinal FXR‐FGF15/19 signaling. (A) Relative mRNA expression of FXR target genes Shp and Fgf15 in the distal ileum in (OATP1B1‐humanized) Slco1a/1b knockout mice after 5‐day injections with vehicle or myrcludex B. (B) Relative mRNA expression of Shp and Fgf15 in the distal ileum for WT and Slc10a1 knockout mice with high plasma bile acid levels. Data are calculated using the geometric mean of reference genes Hprt and Ppib and normalized to vehicle‐treated Slco1a/1b KO mice or Slc10a1 KO mice. Asterisk and hash symbols indicate significant changes (P < 0.05; 6 fasted male mice/group). (C) Relative Fgf15 mRNA expression in the distal ileum after 5‐day BDL (5‐6 fasted male mice/group), normalized to vehicle‐treated WT mice. (D) Effects of myrcludex B on human plasma FGF19 levels. Blood samples were drawn at 9:45 am (fasting), immediately before myrcludex B administration. After 3 hours, a consecutive blood sample was taken. FGF19 levels were measured by enzyme‐linked immunosorbent assay in 12 individuals. (E) Mouse and human conjugated bile acids (200 μM and 1 mM) were added to the basolateral compartment of filter‐grown T84 cells. mRNA expression of FXR target genes FGF19, OSTα, and IBABP was determined after 24‐hour incubation. GW4064 (5 μM) was used as a positive control. Data are calculated using the geometric mean of reference genes Ppib and Hprt and normalized to the vehicle‐treated condition. Asterisk indicates significant change (P < 0.05; 5‐6 replicates/group). Abbreviations: DMSO, dimethyl sulfoxide; KO, knockout; Myr B, myrcludex B.
ENTEROCYTES SENSE BASOLATERAL ELEVATIONS IN BILE ACID LEVELS TO INDUCE Fgf15/FGF19
To verify whether high concentrations of conjugated bile acids in plasma activate intestinal FXR‐FGF15 from the basolateral side of the enterocyte, mice underwent 5‐day BDL. In this model, bile acids are essentially absent from the intestinal lumen. Plasma bile acid concentrations were elevated in BDL mice, and this further increased (reaching 3‐4 mM) upon daily myrcludex B treatment. Plasma bile acids mostly consist of TCA (∼78%) during myrcludex B treatment, whereas plasma of vehicle‐treated BDL mice is mainly the more hydrophilic TβMCA (∼72%; Supporting Fig. S4A). Ileal Fgf15 mRNA expression significantly decreased in vehicle‐treated BDL mice compared to WTs. However, myrcludex B treatment in BDL mice strongly induced Fgf15 expression (Fig. 5C), supporting the notion that bile acids can enter enterocytes from the basolateral side at highly elevated concentrations (≥1 mM). Subsequently, we determined systemic FGF19 release in 12 healthy human volunteers with elevated bile acid levels in blood after myrcludex B treatment. All individuals displayed an increase of bile acid levels in blood 3 hours after myrcludex B injection (range 70‐550 μM; Supporting Fig. S4B). Plasma FGF19 levels did not increase after myrcludex B treatment (Fig. 5D). Additionally, we determined plasma FGF19 kinetics in 4 individuals and showed that diurnal variation of plasma FGF19 was not affected by myrcludex B treatment (Supporting Fig. S4C).
Murine bile acids (TCA > tauromuricholic acid > TCDCA) or human bile acids (glycocholic acid [GCA] = glycochenodeoxycholic acid [GCDCA] >> TCA) were added to the basolateral compartment of polarized cells in increasing concentrations, to investigate the discrepancy in FGF15/19 induction between mice and humans upon myrcludex B treatment. Of note, bile acid concentrations in myrcludex B–treated mice are substantially higher than in myrcludex B–treated humans (1,000 vs. 200 μM). One‐millimolar mouse bile acid mixture induced significantly more FGF19 compared to 200 μM of human bile acids in filter‐grown T84 cells (Fig. 5E). Human conjugated bile acid species are slightly more effective than murine ones in inducing FGF19 at 1,000 μM. OSTα and IBABP mRNA responded in similar fashion as FGF19, in line with potent FXR activation by conjugated bile acids. Cell viability and epithelial integrity were not affected by this treatment (Supporting Fig. S5A). A similar induction of Fgf15 mRNA was demonstrated in mouse organoids upon basolateral exposure to millimolar levels of conjugated bile acid, with preserved organoid morphology (Supporting Fig. S5B,C).
FGF19‐TREATMENT MODULATES HEPATIC BILE ACID UPTAKE TRANSPORTERS
We subsequently tested whether FGF15/19 induction affects the hepatic bile acid uptake machinery. Ntcp and Oatp1b2, Oatp1a1, and Oatp1a4 are all significantly down‐regulated by FGF19 in a rat hepatoma cell line (HPCT‐1e3), which expressed Fgfr4 and β‐klotho, but not Fgf15 mRNA (Fig. 6A). High concentrations of the bile acid, TCA (100 μM), did not significantly affect mRNA expression of Ntcp or Oatp isoforms, and similar lack of effect was found upon incubation with CA (data not shown). Subsequently, we investigated the regulation of Ntcp and Oatp 6 hours after administration of recombinant hFGF19 in mice. hFGF19 injections down‐regulated Ntcp mRNA by ∼50% in WT mice (Figure 6B). Furthermore, Oatp1b2 and Oatp1a4 were down‐regulated by FGF19 as well to a similar extent, indicating that acute FGF19‐mediated repression of hepatic bile acid uptake transporters also occurs in vivo. Similar results were obtained in FGF15 knockout mice (Supporting Fig. S6A). Finally, we investigated the effects of continuous FGF19 exposure on hepatic bile acid uptake transporters in vivo. FGF19 overexpression caused significant down‐regulation of Oatp1a1 and Oatp1b2, whereas Oatp1a4 showed a trend to increase (Fig. 6C). In diet‐induced obese mice, Oatp1a4 was significantly induced (Supporting Fig. S6B). Ntcp expression was up‐regulated in long‐term FGF19 overexpression mouse models.
Figure 6.
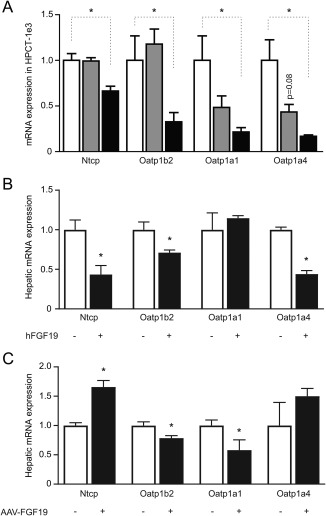
FGF19 modulates the hepatic bile acid uptake machinery in rat hepatocytes (HPCT‐1e3) and mice upon peptide injection or AAV‐mediated overexpression. (A) Effects of TCA (100 μM) or hFGF19 (40 ng/mL) on Ntcp, Oatp1b2, Oatp1a1, and Oatp1a4 expression in rat hepatoma cells (HPCT‐1e3) cells was determined by qRT‐PCR using the geometric mean of Rplp0 and Tbp as a reference. Data are normalized to vehicle (6 replicates/group). Asterisk indicates significant change (P < 0.05). (B) Relative hepatic mRNA expression of Ntcp and Oatp isoforms was determined 6 hours (acutely) after a single injection of hFGF19 (1 mg/kg intraperitoneally) or vehicle in male WT mice, using the housekeeping gene Rplp0 as a reference. Data are normalized to vehicle‐treated WT mice. Asterisk symbols indicate significant (P < 0.05; 5 fasted male mice/group). (C) Relative hepatic mRNA expression of Ntcp and Oatp isoforms after 24 weeks of AAV‐induced (3 × 1011 v.g.) FGF19 overexpression in db/db male mice, using the housekeeping gene, Rplp0, as a reference. Data are normalized to AAV‐GFP treatment. Asterisk indicates significant change (P < 0.05; 8‐10 fasted male mice/group).
Discussion
This study provides evidence that OATP1A/1B isoforms control hepatic conjugated bile acid uptake during (transient) NTCP deficiency in mice. Myrcludex B, the first specific NTCP inhibitor25 currently used in human HBV/HDV studies, totally blocks plasma TCA clearance and increases plasma conjugated bile acids in Slco1a/1b knockout, but not in WT mice. Therefore, the first major finding is that the activity of OATP isoforms establishes systemic bile acid clearance during NTCP deficiency, which explains the physiological systemic bile acid levels in many NTCP‐deficient mice.
Furthermore, our findings indicate that microsomal epoxide hydrolase 126 does not mediate conjugated bile acid transport, as supported by a previous study.27 The strongly reduced OATP1A1 expression in NTCP knockout mice appears a relevant causative factor for hypercholanemia. This isoform is not present in humans, which might contribute to the differential phenotype between humans and mice upon myrcludex B treatment. Similarly, we can refute other OATP proteins (e.g., OATP2B1) as hepatic transporters for conjugated bile acids in mice. Mice lacking only Slco1b2 or the total Slco1a/1b family display reduced uptake of unconjugated bile acids, and 10‐ to 13‐fold elevated levels of unconjugated bile acids in plasma,11, 12 whereas conjugated bile acid concentrations remain mostly unchanged. Systemic conjugated bile acid concentrations in the Slco1a/1b knockout mouse model treated with myrcludex B evidently increase, to an even higher level than in humans treated with myrcludex B.6, 7, 9 Humanizing the Slco1a/1b knockout mouse with OATP1B1 (one of the human isoforms) still results in high plasma‐conjugated bile acid levels, whereas plasma bilirubin levels normalize. These mice do show reduced urinary bile acid excretion upon myrcludex B treatment. Nevertheless, human OATP1B1 alone is unable to effectively clear circulating conjugated bile acids. Our study indicates that NTCP dominates hepatic uptake of conjugated bile acids in humans, whereas the activity of the OATP1A/1B family members can functionally complement NTCP in mice. However, loss of function of NTCP in mice results in an unstable bile acid balance that can easily tip toward hypercholanemia. Using mathematical simulation of bile acid transport, Cravetto et al. have demonstrated that the plasma bile acid concentration is very sensitive to isolated defects of hepatic bile acid uptake, but only at largely reduced fractional hepatic extraction (>70% reduced uptake rate).28 We have previously shown such a reduction in NTCP‐deficient normocholanemic mice, which need 3.5 times more time to clear a fixed amount of conjugated cholate.8
In the quest for cellular regulators of the hepatic bile acid uptake machinery, systemic inflammation was proven absent in Slc10a1 and Slco1a/1b knockout mice (undetectable cytokine levels, normal liver histology) and is therefore unlikely to regulate hepatic transporter expression. Remarkably, hepatic CYP7A1 levels, regulating the size of the bile acid pool,29 acutely increased after myrcludex B treatment in Slco1a/1b knockout mice. Immediate sensing of decreased hepatic bile acid uptake seems to occur, and this may increase bile acid synthesis. Such a process is in line with the study of Oehler et al.,22 describing increased CYP7A1 upon HBV/myrcludex B binding to NTCP in humanized mouse livers. No clear bile acid concentration differences among the different groups were found in this study. Here, we show that elevated levels of conjugated bile acids in blood (as observed in Slc10a1 and myrcludex B–treated Slco1a/1b knockout or BDL mice) reduce Cyp7a1 expression. This is likely explained by activation of the intestinal FXR‐FGF15/19‐hepatic FGFR4/β‐Klotho axis.23, 24 Previously, Tomiyama et al. demonstrated acute, Klotho‐dependent down‐regulation of Oatp1a1 upon FGF19 injection,30 in line with our in vitro results (Fig. 6A). However, in vivo, we detected Oatp1a1 down‐regulation (and Oatp1a4 up‐regulation) only upon prolonged FGF19 treatment, not following single FGF19 injections. Therefore, we cannot conclude whether the effects of FGF15/19 on expression of hepatic bile acid uptake transporters is direct or a consequence of FGF15/19 effects on bile acid pool size or composition.23
For intestinal FXR‐FGF15/19 signaling to occur, we hypothesize that OSTα/β mediates uptake of conjugated bile acid at the basolateral side of enterocytes. In physiological situations, the heterodimer, OSTα/β, is a bile acid export transporter in the small intestine.31 Initial studies described OSTα/β as a bile acid transporter that could mediate both efflux and uptake,32 and we could confirm this using the FRET sensor for bile acids.20 Our data provide mechanistic evidence for Fgf15/19 induction by systemic bile acids, shown by selective basolateral exposure of filter‐grown T84 cells and mouse organoids to (millimolar levels of) conjugated bile acids. Also, BDL‐mice treated with myrcludex B show highly increased (∼3‐4 mM) plasma bile acid concentrations (mostly TCA) and Fgf15, whereas vehicle‐injected BDL mice predominantly display TβMCA, a weak FXR agonist (or even antagonist),33 at around 300 μM. Low FGF15 levels in vehicle‐injected BDL mice could also be attributed to up‐regulation of cytosolic bile acids buffers or extrusion mechanisms pumping out bile acids sufficiently given that Ibabp and Mrp2/3 are increased upon BDL in mice.34 Plasma FGF19 levels were unaltered after myrcludex B treatment in (healthy) humans, suggesting that (local) intestinal bile acid levels are insufficient to induce FGF signaling, whereas this threshold is reached in mice. Notably, the first described NTCP‐deficient (p.R252H) individual displayed highly elevated plasma bile acid levels, accompanied by plasma FGF19 levels close to the upper limit of normal.10 Plasma C4 levels, a quantitative indicator of bile acid synthesis, were below the lower limit of normal in this patient (12.8 nmol/L; normal, 18‐134).35
In summary, we conclude that (1) NTCP and OATP isoforms cooperatively form a stable bile acid uptake machinery in mice, whereas NTCP is predominant in humans, and (2) intestinal sensing of highly elevated levels of conjugated bile acids in blood promotes FGF15/FGF19 signaling, reducing hepatic bile acid synthesis and modulating bile acid transporters.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29251/suppinfo.
Supporting Information
Acknowledgments
We thank Rashmi Dwarka, Karin Prummel, Arno van Cruchten, Lizette Haazen, and Els Wagenaar for technical assistance. Walter Honscha and Bruno Hagenbuch provided materials. We further acknowledge Bert Groen (plasma C4 measurements), David Mangelsdorf and Lei Ling (FGF19 study), Annette Eidam, Stephan Urban and Walter E. Haefeli (Clinical study myrcludex B).
Potential conflict of interest: Dr. Beuers consults and received lecture fees from Intercept and Novartis. He received lecture fees from Falk Foundation, Gilead, Shire, and Zarator. He received grants from Dr. Falk. Dr. Blank received grant money from DZIF and non‐financial support from Myr GmbH.
S.F.J.v.d.G. is supported by the Netherlands Organization for Scientific Research (Vidi; 91713319) and the European Research Council (Starting grant 337479). The clinical study was funded by the “Deutsches Zentrum für Infektionsforschung” (DZIF [German Center for Infection Research] DZIF TTU 05.901).
REFERENCES
- 1. Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile‐acid signalling for metabolic diseases. Nat Rev Drug Discov 2008;7:678‐693. [DOI] [PubMed] [Google Scholar]
- 2. Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res 2009;50:2340‐2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012;1:e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lutgehetmann M, Mancke LV, Volz T, Helbig M, Allweiss L, Bornscheuer T, et al. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology 2012;55:685‐694. [DOI] [PubMed] [Google Scholar]
- 5. Li W, Urban S. Entry of hepatitis B and hepatitis D virus into hepatocytes: basic insights and clinical implications. J Hepatol 2016;64(1 Suppl):S32‐S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bogomolov P, Alexandrov A, Voronkova N, Macievich M, Kokina K, Petrachenkova M, et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B—first results of a phase Ib/IIa study. J Hepatol 2016. ;65:490‐498. [DOI] [PubMed] [Google Scholar]
- 7. Blank A, Markert C, Hohmann N, Carls A, Mikus G, Lehr T, et al. First‐in‐human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J Hepatol 2016;65:483‐489. [DOI] [PubMed] [Google Scholar]
- 8. Slijepcevic D, Kaufman C, Wichers CG, Gilglioni EH, Lempp FA, Duijst S, et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1‐binding in na+‐taurocholate cotransporting polypeptide knockout mice. Hepatology 2015;62:207‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haag M, Hofmann U, Murdter TE, Heinkele G, Leuthold P, Blank A, et al. Quantitative bile acid profiling by liquid chromatography quadrupole time‐of‐flight mass spectrometry: monitoring hepatitis B therapy by a novel Na+‐taurocholate cotransporting polypeptide inhibitor. Anal Bioanal Chem 2015;407:6815‐6825. [DOI] [PubMed] [Google Scholar]
- 10. Vaz FM, Paulusma CC, Huidekoper H, de Ru M, Lim C, Koster J, et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2015;61:260‐267. [DOI] [PubMed] [Google Scholar]
- 11. van de Steeg E, Wagenaar E, van der Kruijssen CM, Burggraaff JE, de Waart DR, Elferink RP, et al. Organic anion transporting polypeptide 1a/1b‐knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J Clin Invest 2010;120:2942‐2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Csanaky IL, Lu H, Zhang Y, Ogura K, Choudhuri S, Klaassen CD. Organic anion‐transporting polypeptide 1b2 (Oatp1b2) is important for the hepatic uptake of unconjugated bile acids: Studies in Oatp1b2‐null mice. Hepatology 2011;53:272‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta 2003;1609:1‐18. [DOI] [PubMed] [Google Scholar]
- 14. Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct‐ligated mice. Gastroenterology 2003;125:825‐838. [DOI] [PubMed] [Google Scholar]
- 15. Wright TJ, Ladher R, McWhirter J, Murre C, Schoenwolf GC, Mansour SL. Mouse FGF15 is the ortholog of human and chick FGF19, but is not uniquely required for otic induction. Dev Biol 2004;269:264‐275. [DOI] [PubMed] [Google Scholar]
- 16. van de Steeg E, van der Kruijssen CM, Wagenaar E, Burggraaff JE, Mesman E, Kenworthy KE, Schinkel AH. Methotrexate pharmacokinetics in transgenic mice with liver‐specific expression of human organic anion‐transporting polypeptide 1B1 (SLCO1B1). Drug Metab Dispos 2009;37:277‐281. [DOI] [PubMed] [Google Scholar]
- 17. Schieck A, Schulze A, Gahler C, Muller T, Haberkorn U, Alexandrov A, et al. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology 2013;58:43‐53. [DOI] [PubMed] [Google Scholar]
- 18. van de Steeg E, Stranecky V, Hartmannova H, Noskova L, Hrebicek M, Wagenaar E, et al. Complete OATP1B1 and OATP1B3 deficiency causes human rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest 2012;122:519‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leuthold S, Hagenbuch B, Mohebbi N, Wagner CA, Meier PJ, Stieger B. Mechanisms of pH‐gradient driven transport mediated by organic anion polypeptide transporters. Am J Physiol Cell Physiol 2009;296:C570‐C582. [DOI] [PubMed] [Google Scholar]
- 20. van der Velden LM, Golynskiy MV, Bijsmans IT, van Mil SW, Klomp LW, Merkx M, van de Graaf SF. Monitoring bile acid transport in single living cells using a genetically encoded FRET sensor. Hepatology 2013;57:740‐752. [DOI] [PubMed] [Google Scholar]
- 21. Hartmann G, Cheung AK, Piquette‐Miller M. Inflammatory cytokines, but not bile acids, regulate expression of murine hepatic anion transporters in endotoxemia. J Pharmacol Exp Ther 2002;303:273‐281. [DOI] [PubMed] [Google Scholar]
- 22. Oehler N, Volz T, Bhadra OD, Kah J, Allweiss L, Giersch K, et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology 2014;60:1483‐1493. [DOI] [PubMed] [Google Scholar]
- 23. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell metabolism 2005;2:217‐225. [DOI] [PubMed] [Google Scholar]
- 24. Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 2007;48:2664‐2672. [DOI] [PubMed] [Google Scholar]
- 25. Schieck A, Muller T, Schulze A, Haberkorn U, Urban S, Mier W. Solid‐phase synthesis of the lipopeptide Myr‐HBVpreS/2‐78, a hepatitis B virus entry inhibitor. Molecules 2010;15:4773‐4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu QS, Xing W, Qian B, von Dippe P, Shneider BL, Fox VL, Levy D. Inhibition of human m‐epoxide hydrolase gene expression in a case of hypercholanemia. Biochim Biophys Acta 2003;1638:208‐216. [DOI] [PubMed] [Google Scholar]
- 27. Honscha W, Platte HD, Oesch F, Friedberg T. Relationship between the microsomal epoxide hydrolase and the hepatocellular transport of bile acids and xenobiotics. Biochem J 1995;311(Pt 3):975‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cravetto C, Molino G, Hofmann AF, Belforte G, Bona B. Computer simulation of portal venous shunting and other isolated hepatobiliary defects of the enterohepatic circulation of bile acids using a physiological pharmacokinetic model. Hepatology 1988;8:866‐878. [DOI] [PubMed] [Google Scholar]
- 29. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000;102:731‐744. [DOI] [PubMed] [Google Scholar]
- 30. Tomiyama K, Maeda R, Urakawa I, Yamazaki Y, Tanaka T, Ito S, et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc Natl Acad Sci USA 2010;107:1666‐1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, et al. OSTalpha‐OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005;42:1270‐1279. [DOI] [PubMed] [Google Scholar]
- 32. Seward DJ, Koh AS, Boyer JL, Ballatori N. Functional complementation between a novel mammalian polygenic transport complex and an evolutionarily ancient organic solute transporter, OSTalpha‐OSTbeta. J Biol Chem 2003;278:27473‐27482. [DOI] [PubMed] [Google Scholar]
- 33. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro‐beta‐muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013;17:225‐235. [DOI] [PubMed] [Google Scholar]
- 34. Slitt AL, Allen K, Morrone J, Aleksunes LM, Chen C, Maher JM, et al. Regulation of transporter expression in mouse liver, kidney, and intestine during extrahepatic cholestasis. Biochim Biophys Acta 2007;1768:637‐647. [DOI] [PubMed] [Google Scholar]
- 35. Freudenberg F, Gothe F, Beigel F, Rust C, Koletzko S. Serum 7‐alpha‐hydroxy‐4‐cholesten‐3‐one as a marker for bile acid loss in children. J Pediatr 2013;163:1367‐1371.e1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29251/suppinfo.
Supporting Information