

Abstract
We report herein that 4‐alkyl‐1,4‐dihydropyridines (alkyl‐DHPs) can directly reach an electronically excited state upon light absorption and trigger the generation of C(sp3)‐centered radicals without the need for an external photocatalyst. Selective excitation with a violet‐light‐emitting diode turns alkyl‐DHPs into strong reducing agents that can activate reagents through single‐electron transfer manifolds while undergoing homolytic cleavage to generate radicals. We used this photochemical dual‐reactivity profile to trigger radical‐based carbon–carbon bond‐forming processes, including nickel‐catalyzed cross‐coupling reactions.
Keywords: cross-coupling, dihydropyridines, nickel catalysis, photochemistry, synthetic methods
The chemical reactivity of electronically excited molecules differs fundamentally from that in the ground state. This is the underlying reactivity concept of photochemistry,1 which has traditionally allowed the development of unique chemical transformations that are not achievable through conventional ground‐state pathways.2 For example, an excited‐state molecule is both a better electron donor (i.e., a better reductant) and a better electron acceptor (i.e., a better oxidant) than in the ground state.3 This explains why the light excitation of organic molecules can unlock unconventional reactivity manifolds. In this context, our laboratory has been exploring the potential of some chiral organocatalytic intermediates to directly reach an electronically excited state upon visible‐light absorption to then switch on novel catalytic functions that are unavailable to ground‐state organocatalysis.4 For example, we showed that electron‐rich enamines I, which act as nucleophiles in the ground state, could become strong reductants upon light excitation and trigger the formation of radicals through single‐electron transfer (SET) reduction of electron‐poor organic halides (Figure 1 a).4a,4b In the same vein, 1,4‐dihydropyridine derivatives II (H‐DHP, Figure 1 b) are primarily understood as hydride (H−) sources in their ground state.5 Other researchers recently demonstrated that exciting them with visible light affords strong photoreductants that can productively engage in SET manifolds.6
Figure 1.
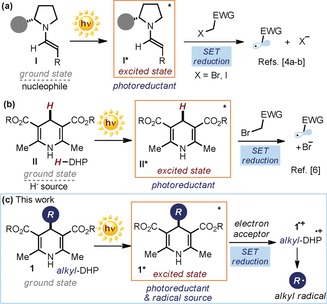
Radical generation strategies enabled by the excited‐state reactivity of organic molecules. a, b) Previous studies demonstrating that light excitation turns enamines (a) and 1,4‐dihydropyridines II (H‐DHP; b) into strong reductants and the resulting SET‐based radical generation mechanisms. c) The proposed strategy exploits the ability of 4‐alkyl‐1,4‐dihydropyridines (alkyl‐DHPs, 1) to become both photoreductants and precursors of alkyl radicals upon excitation. EWG= electron‐withdrawing group; SET=single‐electron transfer; filled dark blue circle represents a bulky substituent on the chiral amine.
These findings led us to wonder whether 4‐alkyl‐1,4‐dihydropyridines (alkyl‐DHPs, 1), which closely resemble the structure of H‐DHP II, might unveil a similar reactivity behavior upon excitation, becoming photoreductants in the excited state (Figure 1 c). If so, the ensuing SET event with a suitable electron acceptor would generate intermediate 1.+. The radical cation 1.+, which is generally accessed by SET oxidation of 1 using an external photoredox catalyst7 or a stoichiometric oxidant,8 is prone to a fast homolytic cleavage, eventually serving as a radical precursor.7, 8 Overall, the proposed photochemical mechanism would generate C(sp3)‐centered radicals, which could then trigger carbon–carbon bond‐forming processes under mild conditions and only relying upon the photochemistry of readily available alkyl‐DHPs 1, which are prepared from aldehydes in one step.8, 9 Herein, we demonstrate how this idea was translated into experimental reality.
To test the feasibility of our photochemical plan, we initially studied the photophysical behavior of diethyl 4‐benzyl‐1,4‐dihydropyridine‐3,5‐dicarboxylate (Bn‐DHP, 1 a), which was selected as the model substrate. The optical absorption spectrum of 1 a dissolved in CH3CN confirmed its ability to absorb in the visible frequency region, up to 420 nm (Figure 2 a, blue line). We found that 1 a was fluorescent when excited at 405 nm, which allowed us to record the emission spectrum (red dotted line in Figure 2 a, λ max=470 nm) and to measure the fluorescence lifetime in CH3CN using a picosecond pulsed laser.10 The reconvolution fitting shows a double exponential decay (Figure 2 b), affording the following values: τ 0=0.28 ns (below the instrument's detection limit) and τ 1=4.1 ns. Interestingly, the excited‐state lifetime of Bn‐DHP is comparable with the lifetime of unsubstituted 1,4‐dihydropyridine of type II (H‐DHP in Figure 1 b), which was measured to be 320 ps.6b Considering the ability of H‐DHP to act as a strong reducing agent in the excited state,6 these photophysical studies encouraged us to evaluate the photochemistry of the excited Bn‐DHP (1 a*).
Figure 2.
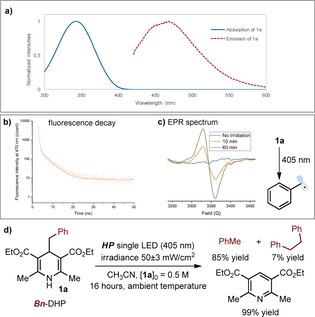
a) Absorption of 1 a (blue line, λ max=343 nm) and its emission (excitation at 405 nm, red dotted line, λ max=470 nm) in CH3CN. b) Fluorescence decay trace of 600 μm 1 a in CH3CN after picosecond photoexcitation at 405 nm. c) EPR spectrum of the benzyl radical generated from 1 a at 77 K after 10 minutes (orange line) and 60 minutes (green line) of light irradiation at 405 nm. d) Photochemical behavior of a solution of 1 a in CH3CN under 405 nm irradiation. Yield measured by 1H NMR analysis using trichloroethylene as the internal standard.
We conducted our experiments in CH3CN under irradiation by a high‐power single light‐emitting diode (HP single LED, λ max=405 nm) with an irradiance of 50 mW cm−2, as controlled by an external power supply (full details of the illumination set‐up are reported in the Supporting Information). Direct illumination of a CH3CN solution of 1 a led to the formation of toluene, 1,2‐diphenylethane, and the corresponding pyridine (Figure 2 d). On repeating the reaction in the presence of 2,2,6,6‐tetramethyl‐1‐piperidinyloxy (TEMPO, 1 equiv), a radical scavenger, we detected the formation of the benzyl‐TEMPO adduct in 55 % yield (details in the Supporting Information). Overall, these experiments indicate that the simple photoexcitation of 1 a can trigger the formation of benzyl radicals. This notion was further corroborated by EPR studies of a solution of 1 a in MeCN, conducted at 77 K and with the same illumination system used in the experiments in Figure 2 c. The EPR spectrum showed a strong isotropic X‐band absorption and afforded a g‐value of 2.0035 (Figure 2 c), which is consistent with reported data for the characterization of benzyl radicals.11
After establishing the ability of 1 a to generate C(sp3)‐centered radicals upon excitation, we evaluated its propensity to act as a strong reducing agent in the excited state. To test this possibility, we selected the aromatic ipso‐substitution of cyanoarenes as the model reaction.7a, 12 Indeed, this process classically proceeds through a radical coupling mechanism that requires the concomitant formation of a C(sp3)‐centered radical and a persistent cyanoarene radical anion III (Scheme 1 a). The ensuing C−C bond formation affords a cyclohexadienyl anion IV that is prone to rapid rearomatization through the elimination of cyanide to afford the arene alkylation product. When mixing either 4‐cyanopyridine (2 a, Scheme 1 b) or tetracyanobenzene (2 b, Scheme 1 c) with 1 a under light irradiation, the corresponding products 3 a and 3 b were generated in fairly good yields. These results are consonant with the excited state of 1 a acting both as a strong reducing agent, affording the cyanoarene radical anion of type III upon SET reduction of substrates 2, and as a source of a benzyl radical, generated upon homolytic cleavage of the radical cation 1 a.+ that emerges from the SET. The reduction potential of the excited Bn‐DHP 1 a [E red* (1 a*/1 a. +)] was estimated as −2.0 V vs. Ag/Ag+ in CH3CN on the basis of electrochemical and spectroscopic measurements. This establishes the thermodynamic feasibility of the SET reduction of both 2 a (E red=−1.87 V vs. SCE in CH3CN)13a and 2 b (E red=−0.64 V vs. SCE in CH3CN).13b
Scheme 1.
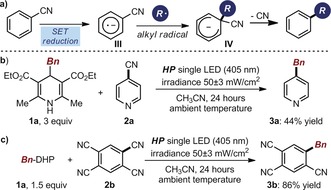
a) Mechanism of the aromatic ipso‐substitution of cyanoarenes, governed by the coupling of an alkyl radical and the persistent cyanoarene radical anion III. Benzylation of 4‐cyanopyridine 2 a (b) and tetracyanobenzene 2 b (c) enabled by the direct photoexcitation of 1 a.
These studies demonstrate that alkyl‐DHPs 1 possess a dual reactivity profile, since excitation with a violet‐light‐emitting diode turns them into strong reducing agents that are able to activate reagents through SET manifolds while undergoing homolytic cleavage to directly generate alkyl radicals. We reasoned that this unique reactivity behavior could be fully exploited in the framework of a photochemical nickel‐catalyzed cross‐coupling method. Merging nickel catalysis14 and photoredox catalysis15 has recently emerged as a versatile platform for developing highly enabling cross‐coupling methods.16 The success of this approach relies on the ability of Ni0 complexes to undergo facile oxidative addition with aromatic halides while engaging in radical‐capture mechanisms. This sequence results in the formation of NiIII complexes, which afford the cross‐coupled products through rapid reductive elimination. At the same time, the chemistry relies on the reactivity of visible‐light‐activated photocatalysts that, upon excitation, can generate alkyl radicals through SET activation of non‐traditional cross‐coupling nucleophiles. Crucially, the photoredox catalyst is also involved in modulating the oxidation state of nickel complexes by SET reduction, an essential step to restore the original Ni0 catalyst. Since alkyl‐DHPs 1 can act as both photoreductants and precursors of alkyl radicals upon excitation, we surmised that their photochemistry could enable a nickel‐catalyzed C(sp2)−C(sp3) cross‐coupling process without the need for any external photoredox catalyst.17 The proposed mechanism, depicted in Scheme 2, would start with the photoexcitation of alkyl‐DHPs 1. In the first catalytic cycle, the resulting excited‐state intermediate 1* (E red* (1 a*/1 a. +)=−2.0 V vs. Ag/Ag+ in CH3CN) would reduce the NiII precatalyst through two discrete SET events to afford the active Ni0 intermediate (E red NiII/Ni0=−1.2 V versus SCE in DMF).18 The concomitant formation of the radical cation 1.+ would secure the formation of the alkyl radical, which is then intercepted by the Ni0 complex. The emerging NiI organometallic species would then undergo oxidative addition with the aryl bromide 4. This would lead to the NiIII intermediate, which, after reductive elimination, forges the desired C(sp2)−C(sp3) bond in the cross‐coupled product 5. Finally, SET reduction of the generated NiI complex by the excited alkyl‐DHPs 1* would complete the nickel catalytic cycle while regenerating the C(sp3) radical.
Scheme 2.
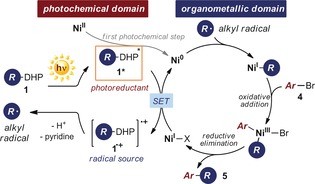
Proposed reaction mechanism for the nickel‐catalyzed C(sp2)−C(sp3) cross‐coupling driven by the photochemical activity of alkyl‐DHPs 1.
Pleasingly, the proposed light‐triggered cross‐coupling process turned out to be feasible at ambient temperature in CH3CN using NiCl2⋅DME as the metal catalyst (5 mol %), bipyridine (10 mol %) and 2,6‐lutidine as the base (1 equiv). Control experiments established the necessity of light irradiation (using high‐powered LED,19 λ max=405 nm, with an irradiance of 50 mW cm−2), with the process being completely inhibited in the dark. As depicted in Figure 3, a variety of aryl bromides 4 were successfully coupled with Bn‐DHP 1 a to afford the corresponding benzylated products 5 a–g in good to high yields. The process tolerated a wide range of functional groups, including cyano, aldehyde, ester, and chloride moieties. Moreover, a ortho substituent did not hinder the reaction, yielding the coupling product 5 f in good yield. To glean further insight into this process, we measured the quantum yield (Φ) of the photochemical cross‐coupling reaction leading to adduct 5 a. A value of 0.0034 (λ=405 nm in CH3CN, average of three runs) was determined, which is consonant with the mechanism proposed in Scheme 2.
Figure 3.
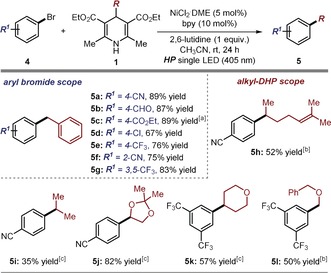
Survey of the aryl bromides 4 and the 4‐alkyl‐1,4‐dihydropyridines 1 that can participate in the nickel‐catalyzed cross‐coupling process enabled by the photochemical activity of 1. Reactions performed on a 0.5 mmol scale using 1.5 equiv of 1 and an irradiance of 50 mW cm−2. Yields refer to isolated products. [a] The NMR yield is given, since the compound was isolated together with 10 % of a byproduct. [b] Reaction time 72 hours. [c] 0.1 mmol scale reaction performed over 16 hours (average of two runs).
We then evaluated the possibility of using alkyl‐DHP substrates 1 bearing different alkyl fragments to benzyl at the C4‐position. Both linear and cyclic fragments could be successfully coupled with aryl bromides (adducts 5 h–l). An alkene, an acetal moiety, and an oxygen‐based heterocycle were all well‐tolerated, leading to the corresponding products 5 h, 5 j and 5 k, respectively. In addition, a primary radical successfully engaged in this coupling when adorned with a stabilizing α‐oxygen atom (adduct 5 l). These results are mechanistically relevant because they demonstrate that different 4‐alkyl‐1,4‐dihydropyridines 1 can be excited upon visible‐light irradiation and their photochemical activity successfully integrated into a nickel catalytic cycle to enable C(sp2)−C(sp3) cross‐coupling processes.
We then wondered whether the same photochemical method could be successfully translated to the nickel‐mediated cross‐coupling of 1 with acyl chlorides 6.20 As shown in Figure 4, an array of dialkyl and aryl/alkyl ketones 7 could be efficiently prepared under mild conditions and relying only on the visible‐light excitation of alkyl‐DHP substrates 1.
Figure 4.
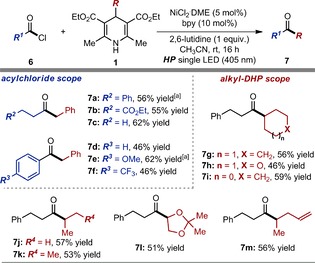
Survey of the acyl chlorides 6 and the 4‐alkyl‐1,4‐dihydropyridines 1 that can participate in the nickel‐catalyzed cross‐coupling process to afford ketones 7. Reactions performed on a 0.1 mmol scale using 1.5 equiv of 1 and an irradiance of 50 mW cm−2. Yields refer to isolated products (average of two runs). [a] 0.5 mmol scale reaction performed over 24 hours.
In summary, we have demonstrated that 4‐alkyl‐1,4‐dihydropyridines can unlock rich photochemistry upon light excitation. Selective absorption of violet light turns them into strong reducing agents that activate reagents through single‐electron transfer manifolds while undergoing homolytic cleavage to generate C(sp3)‐centered radicals. This light‐triggered dual‐reactivity profile was integrated into a nickel catalytic cycle to enable C(sp2)−C(sp3) cross‐coupling reactions without the need for an external photoredox catalyst. We are conducting ongoing studies to further exploit the photochemical activity of alkyl‐DHPs in other radical‐based carbon–carbon bond‐forming processes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the Generalitat de Catalunya (CERCA Program), MINECO (CTQ2016‐75520‐P), and the European Research Council (ERC‐2015‐CoG 681840—CATA‐LUX). L.B. thanks MINECO for a predoctoral fellowship (CTQ2013‐45938‐P). The authors thank Dr. Fernando Bozoglian for assistance with photophysical studies and Dr. Maria Dolores Gonzalez Candela for assistance with EPR experiments.
L. Buzzetti, A. Prieto, S. R. Roy, P. Melchiorre, Angew. Chem. Int. Ed. 2017, 56, 15039.
Contributor Information
Luca Buzzetti, http://www.iciq.org/research/research_group/prof‐paolo‐melchiorre/.
Prof. Dr. Paolo Melchiorre, Email: pmelchiorre@iciq.es.
References
- 1.
- 1a. Balzani V., Ceroni P., Juris A., in Photochemistry and Photophysics, Wiley-VCH, Weinheim, 2014; [Google Scholar]
- 1b. Klán P., Jacob J., in Photochemistry of Organic Compounds, Wiley, Hoboken, 2010. [Google Scholar]
- 2. Albini A., Fagnoni M., in Handbook of Synthetic Photochemistry, Wiley-VCH, Weinheim, 2010. [Google Scholar]
- 3. Turro N. J., Ramamurthy V., Scaiano J. C., in Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, CA, 2010, Chapter 7. [Google Scholar]
- 4.
- 4a. Silvi M., Arceo E., Jurberg I. D., Cassani C., Melchiorre P., J. Am. Chem. Soc. 2015, 137, 6120–6123; [DOI] [PubMed] [Google Scholar]
- 4b. Filippini G., Silvi M., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 4447–4451; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4518–4522; [Google Scholar]
- 4c. Silvi M., Verrier C., Rey Y. P., Buzzetti L., Melchiorre P., Nat. Chem. 2017, 9, 868–873; [DOI] [PubMed] [Google Scholar]
- 4d. Filippini G., Nappi M., Melchiorre P., Tetrahedron 2015, 71, 4535–4542. [Google Scholar]
- 5. Zheng C., You S.-L., Chem. Soc. Rev. 2012, 41, 2498–2518. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Fukuzumi S., Hironaka K., Tanaka T., J. Am. Chem. Soc. 1983, 105, 4722–4727; [Google Scholar]
- 6b. Jung J., Kim J., Park G., You Y., Cho E. J., Adv. Synth. Catal. 2016, 358, 74–80; [Google Scholar]
- 6c. Emmanuel M. A., Greenberg N. R., Oblinsky D. G., Hyster T. K., Nature 2016, 540, 414–417; [DOI] [PubMed] [Google Scholar]
- 6d. Panferova L. I., Tsymbal A. V., Levin V. V., Struchkova M. I., Dilman A. D., Org. Lett. 2016, 18, 996–999; [DOI] [PubMed] [Google Scholar]
- 6e. Chen W., Tao H., Huang W., Wang G., Li S., Cheng X., Li G., Chem. Eur. J. 2016, 22, 9546–9550. [DOI] [PubMed] [Google Scholar]
- 7.Examples of 4-alkyl-1,4-dihydropyridines serving as radical precursors under the action of external photoredox catalysts:
- 7a. Nakajima K., Nojima S., Sakata K., Nishibayashi Y., ChemCatChem 2016, 8, 1028–1032; [Google Scholar]
- 7b. Gutiérrez-Bonet Á., Tellis J. C., Matsui J. K., Vara B. A., Molander G. A., ACS Catal. 2016, 6, 8004–8008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Chen W., Liu Z., Tian J., Ma J., Cheng X., Li G., J. Am. Chem. Soc. 2016, 138, 12312–12315; [DOI] [PubMed] [Google Scholar]
- 7d. Nakajima K., Nojima S., Nishibayashi Y., Angew. Chem. Int. Ed. 2016, 55, 14106–14110; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14312–14316; For a review, see: [Google Scholar]
- 7e. Huang W., Cheng X., Synlett 2017, 28, 148–158. [Google Scholar]
- 8. Gutiérrez-Bonet Á., Remeur C., Matsui J. K., Molander G. A., J. Am. Chem. Soc. 2017, 139, 12251–12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tewari N., Dwivedi N., Tripathi R., Tetrahedron Lett. 2004, 45, 9011–9014. [Google Scholar]
- 10.Our attempts to perform Stern–Volmer quenching studies using different electron acceptors, including the electron-poor cyano substrate 2 a, met with failure. For all the tested 4-alkyl-1,4-dihydropyridines, including 1 a, we observed an increase in fluorescence intensity upon multiple irradiation of the substrate alone. This phenomenon is likely ascribable to the formation of a strongly emitting compound, which is generated upon photodegradation of 1. This emission behavior, which is further detailed in the Supporting Information (Section B4, Figure S6), precluded a reliable quenching measurement.
- 11. Kim K. H., Bai X., Brown R. C., J. Anal. Appl. Pyrolysis 2014, 110, 254–263. [Google Scholar]
- 12.For selected examples, see:
- 12a. Pac C., Nakasone A., Sakurai H., J. Am. Chem. Soc. 1977, 99, 5806–5808; [Google Scholar]
- 12b. Borg R. M., Arnold D. R., Cameron T. S., Can. J. Chem. 1984, 62, 1785–1802; [Google Scholar]
- 12c. Mella M., Fagnoni M., Albini A., J. Org. Chem. 1994, 59, 5614–5622; [Google Scholar]
- 12d. McNally A., Prier C. K., MacMillan D. W. C., Science 2011, 334, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. McDevitt P., Vittimberga B. M., J. Heterocycl. Chem. 1990, 27, 1903–1908; [Google Scholar]
- 13b. Bagnato J. D., Shum W. W., Strohmeier M., Grant D. M., Arif A. M., Miller J. S., Angew. Chem. Int. Ed. 2006, 45, 5322–5326; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5448–5452. [Google Scholar]
- 14. Tasker S. Z., Standley E. A., Jamison T. F., Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shaw M. H., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For a review:
- 16a. Twilton J., Le C., Zhang P., Shaw M. H., Evans R. W., MacMillan D. W. C., Nat. Rev. Chem. 2017, 1, 0052; Selected examples of synergistic nickel/photoredox catalysis: [Google Scholar]
- 16b. Tellis J. C., Primer D. N., Molander G. A., Science 2014, 345, 433–436; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c. Zuo Z., Ahneman D. T., Chu L., Terrett J. A., Doyle A. G., MacMillan D. W. C., Science 2014, 345, 437–440; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Corcé V., Chamoreau L.-M., Derat E., Goddard J.-P., Ollivier C., Fensterbank L., Angew. Chem. Int. Ed. 2015, 54, 11414–11418; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11576–11580; [Google Scholar]
- 16e. Jouffroy M., Primer D. N., Molander G. A., J. Am. Chem. Soc. 2016, 138, 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.4-Alkyl-1,4-dihydropyridines were recently used as alkyl radical precursors in nickel-catalyzed cross-coupling reactions; the reported methods, however, require the use of an external photoredox catalyst; see Refs. [7b,d].
- 18.
- 18a. Johnston P., Smith R. T., Allmendinger S., MacMillan D. W. C., Nature 2016, 536, 322–325; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Durandetti M., Devaud M., Perichon J., New J. Chem. 1996, 20, 659–667. [Google Scholar]
- 19.The use of an LED strip (λ max=405 nm) severely reduced the reactivity of the nickel cross-coupling reaction. Details of the importance of the light intensity and the use of HP LEDs are reported in Section D2 in the Supporting Information.
- 20.Previous methods required the use of an external photoredox catalyst and alkyltrifluoroborates as radical precursors:
- 20a. Amani J., Sodagar E., Molander G. A., Org. Lett. 2016, 18, 732–735; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Amani J., Molander G. A., J. Org. Chem. 2017, 82, 1856–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary