

Abstract
Parathyroid carcinoma (PC) may occur as part of a complex hereditary syndrome or an isolated (i.e., non‐syndromic) non‐hereditary (i.e., sporadic) endocrinopathy. Studies of hereditary and syndromic forms of PC, which include the hyperparathyroidism‐jaw tumor syndrome (HPT‐JT), multiple endocrine neoplasia types 1 and 2 (MEN1 and MEN2), and familial isolated primary hyperparathyroidism (FIHP), have revealed some genetic mechanisms underlying PC. Thus, cell division cycle 73 (CDC73) germline mutations cause HPT‐JT, and CDC73 mutations occur in 70% of sporadic PC, but in only ∼2% of parathyroid adenomas. Moreover, CDC73 germline mutations occur in 20%–40% of patients with sporadic PC and may reveal unrecognized HPT‐JT. This indicates that CDC73 mutations are major driver mutations in the etiology of PCs. However, there is no genotype–phenotype correlation and some CDC73 mutations (e.g., c.679_680insAG) have been reported in patients with sporadic PC, HPT‐JT, or FIHP. Other genes involved in sporadic PC include germline MEN1 and rearranged during transfection (RET) mutations and somatic alterations of the retinoblastoma 1 (RB1) and tumor protein P53 (TP53) genes, as well as epigenetic modifications including DNA methylation and histone modifications, and microRNA misregulation. This review summarizes the genetics and epigenetics of the familial syndromic and non‐syndromic (sporadic) forms of PC.
Keywords: CDC73, familial isolated primary hyperparathyroidism, genetic syndromes, hyperparathyroidism‐jaw tumor syndrome, multiple endocrine neoplasia type 1
1. INTRODUCTION
Parathyroid carcinoma (PC) is a rare endocrine malignancy accounting for 0.005% of all cancers and <1% of primary hyperparathyroidism (pHPT) (Hundahl, Fleming, Fremgen, & Menck, 1999; Ruda, Hollenbeak, & Stack, 2005). Data from the Surveillance, Epidemiology, and End Results cancer registry showed a 60% increase in PC incidence from 1988 to 2003, which, in part, may be due to increased screening of serum calcium and an increased number of patients undergoing surgery for asymptomatic pHPT (Lee, Jarosek, Virnig, Evasovich, & Tuttle, 2007). PC was first reported in 1909 by the Swiss surgeon Fritz de Quervain in a 68‐year‐old man who presented with a large neck mass and died from local recurrence and pulmonary metastasis (Quervain, 1909). Most PCs secrete parathyroid hormone (PTH) resulting in hypercalcemia, however, approximately 40 PC cases have been reported in which there was no increase in PTH production and morbidity resulted from tumor invasion and spread (Wang et al., 2015). PC, parathyroid adenoma (PA), and atypical parathyroid adenoma (APA) cannot be reliably distinguished on the basis of plasma concentrations of calcium and PTH in individual patients, although plasma calcium and PTH concentrations are often higher in patients with PC than patients with PA. Thus, the diagnosis of PC relies on histological criteria, which require demonstration of either capsular invasion with growth into adjacent tissues, vascular and/or perineural tumor invasion, and/or metastasis (Bondeson, et al., 2004). Moreover, the presence of four or more associated features of malignancy that include: capsular invasion without extension to surrounding soft tissue; mitosis >5/10 high power fields; broad intratumoral fibrous bands; coagulative tumor necrosis; diffuse sheet‐like monotonous small cells with high nucleus:cytoplasmic ratio; diffuse cellular atypia; and presence of macronuclei in many tumor cells, qualifies for a diagnosis of PC, whereas the presence of only one to three of these features, qualifies for a diagnosis of APA, which is considered to have features of carcinomas that lack unequivocal evidence for invasive growth (Bondeson, et al., 2004; Chan, 2013; DeLellis, 2011; Kumari, Chaudhary, Pradhan, Agarwal, & Krishnani, 2016). Indeed, using such clinicopathological criteria only ∼15%–35% of the prospectively diagnosed PC cases will continue to behave in a malignant manner, whereas ≤50% of PCs will have an initial diagnosis of benign disease (Gill, 2014; Marsh, Hahn, Howell, & Gill, 2007). Prospective diagnosis of PC is important, as cure can only be achieved following complete surgical resection. Therefore, a broader understanding of the molecular and hereditary basis of PC would provide insight to improve pre‐ and post‐surgical diagnosis and staging.
The aim of this review is to summarize the current knowledge of the molecular and hereditary basis of PC. A PubMed and EMBASE literature search was undertaken on July 1, 2016 using the search term: “parathyroid carcinoma”, its free and controlled vocabulary EMTREE and MeSH synonyms, cross‐referenced with “genetics,” “epigenetics,” “mutations,” “CDC73,” “hyperparathyroidism‐jaw tumor,” “familial isolated hyperparathyroidism,” “MEN1,” “MEN2,” and its free and controlled vocabulary EMTREE and MeSH synonyms. There were no restrictions on language, publication type, or date. Additionally, reference lists from all major reviews were examined for citations that did not appear in the PubMed or EMBASE search.
2. CLINICAL FEATURES OF PARATHYROID CARCINOMA
PC may occur as part of a complex syndrome and hereditary disorder, or as a non‐hereditary (i.e., sporadic) and isolated (i.e., non‐syndromic) endocrinopathy (Table 1 and Figure 1). PC most commonly occurs as a sporadic non‐syndromic disorder. The hereditary syndromes associated with PC include the hyperparathyroidism‐jaw tumor (HPT‐JT) syndrome, the multiple endocrine neoplasia (MEN) type 1 (MEN1) and type 2 (MEN2) syndromes, and potentially the non‐syndromic familial isolated primary hyperparathyroidism (FIHP), which may be clinically difficult to distinguish from the MEN1 and HPT‐JT syndromes (Figure 1).
Table 1.
Syndromic and hereditary forms of parathyroid carcinoma
| Conditiona | Syndromic or isolated | Gene affected | Chromosomal locationb | Protein functionc | Inheritanced | pHPT featurese | Associated conditionsf |
|---|---|---|---|---|---|---|---|
| HPT‐JT | Syndromic | CDC73 | 1q31.2 | TS | AD | PA (cystic)/PC | Jaw, renal, and uterine tumors |
| FIHP | Isolated | CDC73 MEN1 | 1q31.2 11q13 | TS TS | ADg | PA/PC Hyperplasia/PA/PC | |
| MEN1 | Syndromic | MEN1 | 11q13 | TS | AD | Hyperplasia/PA/PC | Enteropancreatic tumors (75%), pituitary (50%), and adrenal hyperplasia (13%) or tumors (13%) |
| MEN2 | Syndromic | RET | 10q11.21 | Onco | AD | Hyperplasia/PA/PC | MTC (66%) and pheochromocytoma (33%) |
HPT‐JT, hyperparathyroidism‐jaw tumor; FIHP, familial isolated primary hyperparathyroidism; MEN1, multiple endocrine neoplasia type 1; MEN2, multiple endocrine neoplasia type 2.
Cytogenetic band according to HUGO Gene Nomenclature Committee.
TS, tumor suppressor; Onco, proto‐oncogene.
AD, autosomal dominant.
pHPT, primary hyperparathyroidism; PA, parathyroid adenoma; PC, parathyroid carcinoma.
MTC, medullary thyroid cancer.
Some families may show autosomal recessive inheritance.
Figure 1.
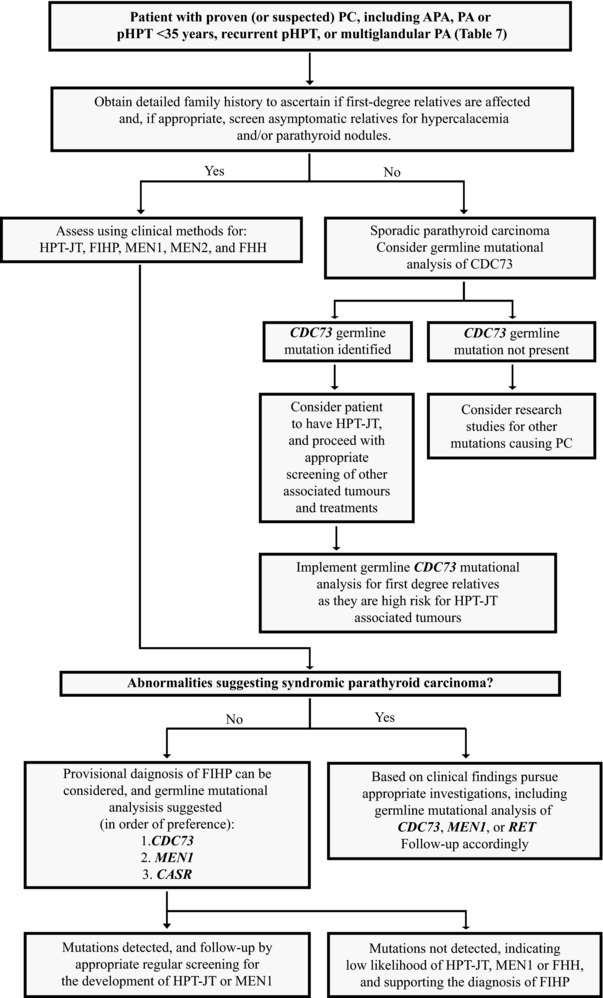
A genetic testing approach to patients with parathyroid carcinoma. PC, parathyroid carcinoma; APA, atypical parathyroid adenoma; PA, parathyroid adenoma; pHPT, primary hyperparathyroidism; HPT‐JT, hyperparathyroidism‐jaw tumor; FIHP, familial isolated primary hyperparathyroidism; MEN1, multiple endocrine neoplasia type 1; MEN2, multiple endocrine neoplasia type 2; FHH, familial hypocalciuric hypercalcemia
The clinical findings of PC are generally non‐specific and the diagnosis of PC is rarely made before surgery and histological examination of the tumor. Thus, distinguishing between benign and malignant disease is a challenge in the management of patients with pHPT. The most frequent symptoms of PC are those associated with hypercalcemia and are: fatigue, weakness, weight loss, anorexia, nausea, vomiting, abdominal pain, polyuria, and polydipsia. Other clinical features may include bone pain, fractures, anemia, nephrolithiasis, pancreatitis, and peptic ulcer disease (Busaidy et al., 2004; Chen et al., 2003; Hakaim & Esselstyn, 1993; Schantz & Castleman, 1973; Wynne, van Heerden, Carney, & Fitzpatrick, 1992). Renal and bone involvement is frequent and may coexist in >50% of PC patients (Wynne et al., 1992). Osteoporosis, osteitis fibrosa cystica, subperiosteal bone resorption, or salt‐and‐pepper skull lesions have been reported to occur in ∼40%–90% of PC patients, whereas bone disease occurs in <10% of patients with benign pHPT (Schantz & Castleman, 1973; Shane & Bilezikian, 1982; Wang & Gaz, 1985; Silverberg et al., 1990; Wynne et al., 1992). Renal involvement has been reported to occur in ∼30%–85% of PC patients with nephrocalcinosis occurring in ∼15%–55%, nephrolithiasis in ∼55%–70%, and renal insufficiency in ∼55%–85% of patients (Holmes, Morton, & Ketcham, 1969; Schantz & Castleman, 1973; Shane & Bilezikian, 1982; Wynne et al., 1992). Conversely, renal involvement in benign pHPT is considerably lower, affecting ∼20% of patients (Silverberg et al., 1990).
A palpable tumor is found in ∼50% of PC patients, whereas it is rarely identifiable in patients with benign pHPT (Holmes et al., 1969; Wynne et al., 1992). More than 90% of PC cases involve functioning tumors with plasma PTH concentrations 3–10 times higher than normal upper limit, whereas plasma PTH concentrations 2–3 times higher are typically found in benign pHPT (Holmes et al., 1969; Wynne et al., 1992). Recently, a population‐based study reported a positive predictive value of >80% for PTH levels ≥10 times higher than the upper normal limit (Schaapveld et al., 2011). Most PC patients have severe hypercalcemia at presentation (calcium >14 mg/dl, i.e., >3.50 mmol/l), whereas in benign pHPT calcium levels are generally 1–2 mg/dl (i.e., 0.25–0.50 mmol/l) above normal (Wang & Gaz, 1985; Wynne et al., 1992; Chen et al., 2003). Plasma alkaline phosphatase activity is more commonly elevated in patients with PC than benign pHPT as a result of bone involvement (Silverberg et al., 1990; Chen et al., 2003). However, there is considerable overlap of these elevations of plasma calcium and PTH concentrations and alkaline phosphatase activity in patients with PC and PA, thereby making it difficult to rely upon them for establishing an unequivocal diagnosis of PC. However, PC patients have been reported to have elevated levels of urinary human chorionic gonadotropin subunits, particularly the hyperglycosylated isoforms, which are associated with an increased risk of hip fracture and death, and this difference from patients with benign pHPT requires further study (Rubin, Bilezikian, Birken, & Silverberg, 2008).
3. SYNDROMIC AND HEREDITARY FORMS OF PARATHYROID CARCINOMA
The syndromic and hereditary forms of PC are associated with germline mutations of the cell division cycle 73 (CDC73) gene, also referred to as the hyperparathyroidism type 2 (HRPT2) gene, MEN type 1 (MEN1), and rearranged during transfection (RET) genes (Table 1). The RET mutations, which are activating, are dominant at the cellular level, and only one copy of the mutated gene is required for tumor development. However, for the MEN1 and CDC73 mutations, which are inactivating and recessive at the cellular level, two mutations are required for a tumor to develop: for the hereditary tumors, these two recessive mutations comprise one germline and one somatic mutation that may involve a chromosomal loss and be detected as loss of heterozygosity (LOH) in the tumor. Such tumors may also occur sporadically, that is, without a family history and without inheritance of the germline mutation, and in these patients, both the recessive mutations will have likely occurred as somatic mutations in the tumor. This genetic model of neoplasia involving two recessive mutations in the development of tumors is known as Knudson's two‐hit hypothesis. The genetic mechanisms involved in the etiology of the MEN1 and HPT‐JT syndromes due to MEN1 and CDC73 mutations are consistent with Knudson's two‐hit hypothesis (Knudson, 1971; Thakker, 1993).
3.1. Hyperparathyroidism‐jaw tumor (HPT‐JT)
HPT‐JT (MIM# 145001) is a rare syndrome characterized by pHPT, fibro‐osseous lesions (ossifying fibroma) of the mandible and maxilla, and tumors of the kidney and uterus (Jackson, 1958; Bradley et al., 2005b). Parathyroid tumors, of which 15% are carcinomas, are generally the first manifestation, and occur in >90% of HPT‐JT cases (Bradley & Thakker, 2006). pHPT is usually caused by a solitary parathyroid tumor, but multiglandular involvement may affect >15% of cases (Marx, 2000; Bradley & Thakker, 2006; Mehta et al., 2014).
3.1.1. CDC73
HPT‐JT is an autosomal dominant disease due to germline mutations of the CDC73 gene (Tables 1, 2, 3). CDC73, which is comprised of 17 exons (Figure 2A) and is located on chromosome 1q31.2, encodes the protein, parafibromin, which is associated in the polymerase associated factor (Paf1) complex (Figure 2B and C) with the proteins: PAF1; tryptophan‐aspartic acid dipeptide terminating repeat domain 61 (WDR61); and the RNA polymerase‐associated proteins—left open reading frame homolog (LEO1), cyclin three requiring homolog (CTR9), and restores TATA‐binding protein function homolog (RTF1). The Paf1 complex interacts with the RNA polymerase II subunit A (POLR2A), regulating genetic transcription, and with the histone methyltransferase complex, regulating histone modifications (Figure 2C) (Rozenblatt‐Rosen et al., 2005; Yart et al., 2005). Functions attributed to parafibromin include the downregulation of cyclin D1 expression and direct interaction with β‐catenin resulting in the activation of transcription of target genes (Figure 2C) (Woodard et al., 2005; Mosimann, Hausmann, & Basler, 2006; Zhang et al., 2006; Bradley et al., 2007). Parafibromin also has a role in embryonic development regulating genes involved in cell growth and survival (Figure 2C) (Wang et al., 2008).
Table 2.
Spectrum of diseases associated with CDC73 mutations
| Hyperparathyroidism‐jaw tumor |
|---|
Diagnosis may be established in individuals with:
|
| CDC73‐related familial isolated primary hyperparathyroidism |
|---|
Diagnosis may be established in individuals with:
|
| CDC73‐related parathyroid carcinoma |
|---|
Diagnosis may be established in individuals with:
|
pHPT, primary hyperparathyroidism; HPT‐JT, hyperparathyroidism‐jaw tumor; PC, parathyroid carcinoma; FIHP, familial isolated primary hyperparathyroidism.
Table 3.
Summary of CDC73 mutations associated with hyperparathyroidism‐jaw tumor
| Mutation a | Exon/intron | Codon b | Predicted effect c | Type d | Original designation | References |
|---|---|---|---|---|---|---|
| c.‐16_8del | Exon 1 | p.Met? | G | c.‐16:8del; p.Met1? | Bellido et al. (2016) | |
| c.3G > A | Exon 1 | 1 | p.Met? | G | 3G→A | Carpten et al. (2002)r |
| c.13_30del | Exon 1 | 5 | p.Leu5_Gln10del | Sp ,1 | 13_30delCTTAGCGTCCTGCGACAG | Moon et al. (2005) |
| c.18_46del | Exon 1 | 6 | p.Ser6ArgfsX50 | Gp | c.18_48del31 | Parfitt, Harris, Wright, and Kalamchi (2015) |
| c.14_17dup | Exon 1 | 7 | p.Val7X | Gp | c.14_17dupTTAG | Khadilkar et al. (2015)r , e |
| c.[24del;20T > C]q | Exon 1 | 7 | p.Val7AlafsX14 | G | nt20AGGACG→GGGAG | Aldred et al. (2006) |
| c.22del | Exon 1 | 8 | p.Leu8CysfsX13 | G | c.22delC | Carlson & Smith (2008) |
| c.25C > T | Exon 1 | 9 | p.Arg9X | G | 25C→T | Carpten et al. (2002)r |
| c.25C > T | Exon 1 | 9 | p.Arg9X | G | c.25 > T | Newey et al. (2010)f |
| c.25C > T | Exon 1 | 9 | p.Arg9X | ND | R9X | Schmidt, Bradrick, and Gabali (2009) |
| c.25C > T | Exon 1 | 9 | p.Arg9X | ND | p.Arg9Stop (R9X) | Mathews, Winchester, Alsaygh, Bartlett, and Luttrell (2016) |
| c.30del | Exon 1 | 10 | p.Gln10HisfsX11 | G | 30delG | Carpten et al. (2002)r |
| c.12_31dup | Exon 1 | 11 | p.Tyr11CysfsX17 | Gp | 41 bp duplication/insertion | Carpten et al. (2002)r |
| c.35_41del | Exon 1 | 12 | p.Asn12ArgfsX7 | G | 34delAACATCC | Carpten et al. (2002)r |
| c.40C > T | Exon 1 | 14 | p.Gln14X | G | c.40C > T | Khadilkar et al. (2015) |
| c.40del | Exon 1 | 14 | p.Gln14ArgfsX7 | Gp | 39delC | Carpten et al. (2002)r |
| c.40del | Exon 1 | 14 | p.Gln14ArgfsX7 | G | 39delC | Mizusawa et al. (2006) |
| c.40del | Exon 1 | 14 | p.Gln14ArgfsX7 | G | 39delC | Yamashita, Akiyama, Mizusawa, Yoshimoto, and Goto (2007) |
| c.70del | Exon 1 | 24 | p.Glu24LysfsX2 | Sp | c.70delG | Sriphrapradang et al. (2014) |
| c.76del | Exon 1 | 26 | p.Ile26SerfsX11 | G | c.76delA | Howell et al. (2003)g , h |
| c.76del | Exon 1 | 26 | p.Ile26SerfsX11 | Gp ,2 | c.76delA | Howell et al. (2003)g , h |
| c.76del | Exon 1 | 26 | p.Ile26SerfsX11 | Gp | c.76delA | Frank‐Raue et al. (2011) |
| c.85del | Exon 1 | 29 | p.Glu29SerfsX8 | Sp ,1 | 85delG | Moon et al. (2005) |
| c.85del | Exon 1 | 29 | p.Glu29SerfsX8 | G | 85del | Rekik et al. (2010) |
| c.85G > T | Exon 1 | 29 | p.Glu29X | G | c.93G > T exon 1 | Bricaire et al. (2013) |
| c.85G > T | Exon 1 | 29 | p.Glu29X | G | c.85G > T | Abdulla, O'Leary, Isorena, Diaz, and Yeh, (2013) |
| c.96G > A | Exon 1 | 32 | p.Trp32X | G | c.96G > A | Sarquis et al. (2008)h , i |
| c.96G > A | Exon 1 | 32 | p.Trp32X | NDp | c.96G > A | Kutcher et al. (2013) |
| c.131+1G > A | Intron 1 | splice [d]o | G | c.131+1G > A | Newey et al. (2010)f | |
| c.140_144del | Exon 2 | 47 | p.Lys47ArgfsX17 | G3 | c.136_144 del5 | Iacobone et al. (2009)h , j |
| c.165C > G | Exon 2 | 55 | p.Tyr55X | Gp | 165C‐G | Carpten et al. (2002)r |
| c.165C > A | Exon 2 | 55 | p.Tyr55X | NDp | c.165C > A | Veiguela, Isidro, Jorge, and Ruano (2010) |
| c.179T > A | Exon 2 | 60 | p.Ile60Asn | S3 | c.179T > A | Masi et al. (2014) |
| c.188T > C | Exon 2 | 63 | p.Leu63Pro | G | c.188T > C | Newey et al. (2010)f |
| c.188T > C | Exon 2 | 63 | p.Leu63Pro | G4 | c.188T > C | Iacobone et al. (2009)h , j |
| c.191T > C | Exon 2 | 64 | p.Leu64Pro | Gp | L64P | Hahn et al. (2010)g , h , k |
| c.205dup | Exon 2 | 69 | p.Leu69ProfsX13 | Gp | c.205dupC | Pichardo‐Lowden, Manni, Saunders and Baker (2011)l |
| c.226C > T | Exon 2 | 76 | p.Arg76X | G | c.226C > T | Newey et al. (2010)f |
| c.238‐1G > A | Intron 2 | splice [a]o | Gp ,1 | IVS2‐1G > A | Moon et al. (2005) | |
| c.284T > C | Exon 3 | 95 | p.Leu95Pro | G | L95P | Panicker, Zhang, Dagur, Gastinger and Simonds, (2010) |
| c.284T > C | Exon 3 | 95 | p.Leu95Pro | Sp ,5 | c.284T > C | Yu et al. (2015)h |
| c.306_307+13del | Exon 3 | 103 | p.Ser103AsnfsX5 | Gp | *306delGTgtgagtacttttt | Carpten et al. (2002)r |
| c.307+5G > T | Intron 3 | splice [vus] | G | c.307+5G > T | Frank‐Raue et al. (2011) | |
| c.356del | Exon 4 | 119 | p.Gln119ArgfsX14 | Gp ,5 | 356delA | Carpten et al. (2002)r , h |
| c.358C > T | Exon 4 | 120 | p.Arg120X | NDp | c.358C > T | Mele, Rolighed, Jespersen, Rejnmark and Christiansen (2016) |
| c.374_375dup | Exon 5 | 126 | p.Arg126AsnfsX8 | S4 | c.375_376insAA | Masi et al. (2008)h |
| c.406A > T | Exon 5 | 136 | p.Lys136X | G | 406A→T | Carpten et al. (2002)r |
| c.433_442delinsAGA | Exon 5 | 145 | p.Cys145ArgfsX55 | G | c.433_442delinsAGA | Iacobone et al. (2009) |
| c.424‐5T > C | Intron 5 | splice [vus] | G | c.424‐5T > C | Frank‐Raue et al. (2011) | |
| c.639del | Exon 7 | 213 | p.Phe213LeufsX6 | G | 636delT | Carpten et al. (2002)r |
| c.664C > T | Exon 7 | 222 | p.Arg222X | Gp | c.664C > T | Wang et al. (2012)r |
| c.668_669delinsG | Exon 7 | 223 | p.Asp223GlyfsX34 | Gp | 669delAT/insG | Bradley et al. (2005b)r , h |
| c.679_680insAG | Exon 7 | 227 | p.Arg227LysfsX31 | G | 679insAG | Bradley et al. (2005b)r , h |
| c.679_680insAG | Exon 7 | 227 | p.Arg227LysfsX31 | Gp | 679insAG | Carpten et al. (2002)r , h |
| c.686_689del | Exon 7 | 229 | p.Arg229AsnfsX27 | Sp ,2 | c.686delGAGT | Howell et al. (2003)g , h , m |
| c.687_688del | Exon 7 | 229 | p.Arg229SerfsX37 | G | c.679delAG | Howell et al. (2003)g , h |
| c.687_688del | Exon 7 | 229 | p.Arg229SerfsX37 | G | c.679delAG | Sarquis et al. (2008) |
| c.687_688del | Exon 7 | 229 | p.Arg229SerfsX37 | G | AGCACA^GAGAGagTATGGAGGACA | Teh et al. (2004)f , n |
| c.687_688del | Exon 7 | 229 | p.Arg229SerfsX37 | G | c.687_688del | Newey et al. (2010)f |
| c.700C > T | Exon 7 | 234 | p.Arg234X | G | 700C→T | Bradley et al. (2006) |
| c.700C > T | Exon 7 | 234 | p.Arg234X | Gp | R234X | Raue, Haag and Frank‐Raue (2007) |
| c.700C > T | Exon 7 | 234 | p.Arg234X | G | c.700C > T | Newey et al. (2010)f |
| c.745dup | Exon 8 | 249 | p.Ile249AsnfsX18 | G | c.745dupA | Newey et al. (2010)f |
| c.766_767del | Exon 8 | 256 | p.Val256LysfsX10 | G | 255delTG/256delGT | Cavaco et al. (2004) |
| c.1126_1127insTT | Exon 13 | 276 | p.Asn376IlefsX10 | G | 1126InsTT | Pimenta et al. (2006) |
| c.1135G > A | Exon 13 | 379 | p.Asp379Asn | G | 1135 G → A | Bradley et al. (2006) |
| c.1239del | Exon 14 | 413 | p.Gln413HisfsX15 | G | 1238delA | Carpten et al. (2002)r |
| c.1247del | Exon 14 | 416 | p.Gly416AlafsX12 | G | c.1247delG | Howell et al. (2009)f |
| c.1346del | Exon 15 | 449 | p.Gly449ValfsX30 | Gp | c.1346delG | Frank‐Raue et al. (2011) |
| c.1382del | Exon 15 | 461 | p.Leu461CysfsX18 | Gp | c.1379delT | Chiofalo et al. (2014) |
| c.1432_1433del | Exon 16 | 478 | p.Leu478GlufsX3 | G | c.1432_1433delCT | Frank‐Raue et al. (2011) |
| c.*12C > A | 3′‐UTR | Expression [vus] | G | c.*12C > A | Frank‐Raue et al. (2011) | |
| Gross deletion | G | c.307+?_513‐?del exons 4, 5, 6 | Kong et al. (2014) | |||
| Gross deletion | G | c.307+?_513‐?del exons 4, 5, 6 | Bricaire et al. (2013) | |||
| Gross deletion | Gp | 1q31,1–1q31,3 del | Bricaire et al. (2013)r | |||
| Gross deletion | G | Whole gene deletion | Cascon et al. (2011) | |||
| Gross deletion | Gp | Whole gene deletion | Bricaire et al. (2013)r |
Mutations are numbered in relation to the cell division cycle 73 (CDC73) cDNA reference sequence (GenBank accession number NM_024529.4) whereby nucleotide +1 corresponds to the A of the ATG‐translation initiation codon. All mutations were analyzed using the Leiden Open Variation Database (LOVD) Mutalyzer sequence variant nomenclature checker (https://www.lovd.nl/mutalyzer/) and annotated using the Human Genome Variation Society (HGVS) guidelines (https://www.hgvs.org/).
Codon numbering starts from initiation codon of CDC73 mRNA.
Predicted effect: splice, splice site mutation; [d] donor splice site; [a] acceptor splice site; [vus] variant of unknown significance; ? indicates unlikely translation of protein as initiator met is lost.
Mutation type: G, germline; S, somatic; ND, not defined. Equal superscript numbers represent germline and/or somatic mutations occurring in the same patient.
Criteria for diagnosis of PC were not reported, but the patient had persistent disease and clinical suspicion of thoracic metastasis.
Reported as HPT‐JT, but the authors did not provide details about the presence or absence of jaw tumors.
Additional clinical details about these kindreds are provided Bradley et al. (2005b).
Reported as HPT‐JT, but occurrence of jaw tumors, which may not always occur in HPT‐JT patients, was not detected in any family members.
Reported in other publication as a possible FIHP case, but the frequent recurrence, presence of APA and renal and uterine tumors favors the diagnosis of HPT‐JT (Silveira et al., 2008).
Initially reported as FIHP by Masi et al. (2008).
Initially reported as FIHP by Howell et al. (2003).
It is possible this is a case of HPT‐JT associated with PC since: the patient was diagnosed with three renal cysts, while “a maternal cousin had jaw pain and presumably bone destruction of the jaw, termed a ‘hole in the jaw’.” Furthermore, histological description of the proband's parathyroid gland was consistent with an APA (“…vascular and capsular invasion, but no definitive features of PC were identified”) and disease recurrence on the contra‐lateral side (again with diagnosis of APA) suggests a more malignant behavior.
Reported as a germline mutation in a later publication, but inconsistency between patients’ gender and age are observed (Sarquis et al., 2008).
Unclear if this kindred was included in the previous study of Howell et al. (2003).
For detailed information of the effect of CDC73 mutation on splicing please consult Hahn, McDonnell, and Marsh (2009).
Mutations identified in kindreds with case reports of PC.
Discordant codon/nucleotide change in the original report.
Criteria for diagnosis of PC not reported.
PC, parathyroid carcinoma; HPT‐JT, hyperparathyroidism‐jaw tumor; FIHP, familial isolated primary hyperparathyroidism; APA, atypical parathyroid adenoma.
Figure 2.
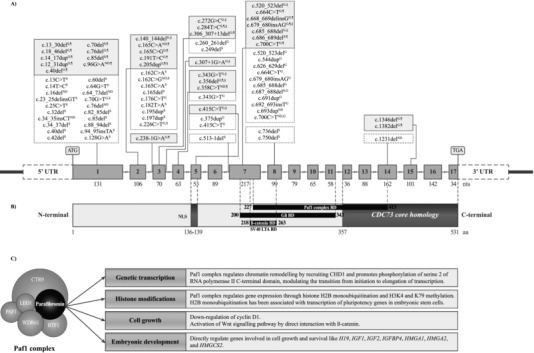
Schematic representation of the genomic organization of the human CDC73 gene, parafibromin protein, and its functions. (A) Upper panel, schematic representation of genomic structure of cell division cycle 73 (CDC73) comprising 17 exons. ATG and TGA represent the initiation and stop codons, respectively. Sites of CDC73 mutations associated with sporadic and familial parathyroid carcinoma (PC) are shown (S somatic mutation; G germline mutation; ND not defined; white, dotted line boxes, CDC73 mutations associated with sporadic PC; gray, full line boxes, CDC73 mutations associated with syndromic or hereditary forms of PC, where ¶ means hyperparathyroidism‐jaw tumor and § means familial isolated primary hyperparathyroidism). (B) Middle panel, schematic representation of parafibromin protein structure and known functional domains. CDC73 encodes a 531‐amino acid protein, whose C‐terminal domain shares 27% homology with the yeast CDC73 (CDC73 core homology domain). The nuclear localization signal (NLS) is encoded by exon 5, the evolutionary conserved polymerase‐associated factor 1 (Paf1) complex‐binding domain (Paf1 complex BD) by exons 7–14, the Gli binding domain (Gli BD) by exons 7–11, and the β‐catenin interaction binding domain (β‐catenin BD) and the SV40 large T antigen binding domain (SV40 LTA BD) by exons 7 and 8. (C) Lower panel, schematic representation of parafibromin functions. Parafibromin is a component of the Paf1 protein complex, which regulates chromatin remodeling and gene expression via histone modification. Parafibromin also regulates cell growth, via cyclin D1 and Wnt signaling, and embryonic development via genes involved in cell growth and survival. H19, H19 fetal liver mRNA; IGF1 and IGF2, insulin‐like growth factor 1 and 2; IGFBP4, insulin‐like growth factor binding protein 4; HMGA1 and HMGA2, high mobility AT‐hook 1 and 2; HMGCS2, 3‐hydroxy‐3‐methylglutaryl‐Coenzyme A synthase 2
About 75% of HPT‐JT patients will have germline CDC73 mutations within the coding region (Table 3 and Figure 2A and B), and the PCs will usually have LOH of CDC73 resulting in a loss of parafibromin expression. The ∼25% of HPT‐JT families, who do not harbor CDC73 mutations or deletions of the coding region or adjacent splice sites, may have abnormalities involving the CDC73 promoter regions, untranslated regions, uncharacterized alternate transcripts, whole exon or gene deletions that are not readily detected by polymerase chain reaction (PCR) and sequencing, mutations in unidentified genes, or epigenetic modifications (Carpten et al., 2002; Cetani et al., 2004; Bradley et al., 2005b; Bradley & Thakker, 2006). Approximately 55% of reported germline CDC73 mutations are associated with HPT‐JT, and these comprise: 60% frameshift, 26% nonsense, and 3% loss of initiator methionine mutations that are predicted to result in parafibromin truncation or loss of protein transcription; 5% missense; and 5% splice site mutations (Newey, Bowl, Cranston, & Thakker, 2010). Of the remaining 45% of germline CDC73 mutations, 21% are reported from patients with FIHP, and of these 50% are frameshift insertion/deletions, 29% are missense, and 21% are splice site mutations; 15% are reported from patients with sporadic PC, and of these 50% are frameshift insertion/deletions, 40% are nonsense, and 10% are missense mutations; 6% are reported from patients with sporadic adenomas, and of these 50% are missense and 50% are nonsense mutations; and 3% are reported from patients with sporadic ossifying fibromas of the jaw and all of these are frameshift insertion or deletions (Newey et al., 2010). Moreover, the same germline CDC73 mutations may be associated with HPT‐JT, FIHP, and sporadic PC in different patients; for example, the c.679_680insAG, p.Arg227LysfsX31 mutation has been reported to occur in patients with HPT‐JT (Figure 2 and Table 3), FIHP (Figure 2 and Table 4) and sporadic PC (Figure 2 and Table 5), and the c131+1G > A mutation has been reported to occur in patients with HPT‐JT and FIHP (Tables 3 and 4) (Carpten et al., 2002; Shattuck et al., 2003b; Cetani et al., 2004; Simonds et al., 2004; Bradley et al., 2005a; Newey et al., 2010). Thus, there is a lack of genotype–phenotype correlation and the underlying mechanisms for this variability remain to be elucidated (Howell et al., 2003; Shattuck et al., 2003b; Thakker, 2016).
Table 4.
Summary of CDC73, MEN1, and CASR mutations associated with familial isolated primary hyperparathyroidism
| Mutation a | Exon/intron | Codon b | Predicted effect c | Type d | Original designation | References |
|---|---|---|---|---|---|---|
| CDC73 | ||||||
| c.61_64del | Exon 1 | 21 | p.Lys21GlufsX4 | S1 | c.61_64del4 | Kelly et al. (2006) |
| c.62_66del | Exon 1 | 21 | p.Lys21ArgfsX43 | G | 62–66del | Mizusawa et al. (2006) |
| c.70_73del | Exon 1 | 24 | p.Glu24X | S2 | 70–73del | Mizusawa et al. (2006) |
| c.95_102del | Exon 1 | 32 | p.Trp32X | S2 | 95–102del | Mizusawa et al. (2006) |
| c.128G > A | Exon 1 | 43 | p.Trp43X | S | 128G→A | Carpten et al. (2002)q |
| c.131+1G > A | Intron 1 | splice [d]n | G | IVS1+1G > A | Cetani et al. (2004) | |
| c.131+1G > A | Intron 1 | splice [d]n | G | IVS1+1 g→a | Bradley et al. (2005a) | |
| c.140_144del | Exon 2 | 47 | p.Lys47ArgfsX17 | Go ,1 | c.140_144del5 | Kelly et al. (2006) |
| c.157G > T | Exon 2 | 53 | Glu53X | G | c.157G > T (Glu53X) | Kong et al. (2014) |
| c.188T > C | Exon 2 | 63 | p.Leu63Pro | G | c.188T > C | Newey et al. (2010) |
| c.191T > C | Exon 2 | 64 | p.Leu64Pro | G | 191T→C | Villablanca et al. (2004) |
| c.194dup | Exon 2 | 65 | p.Asn65LysfsX2 | G | 194dupA | Takeuchi et al. (2015) |
| c.205dup | Exon 2 | 69 | p.Leu69ProfsX13 | Go | c.205dupC | Pichardo‐Lowden et al. (2011)e |
| c.237+1G > C | Intron 2 | splice [d]n | G | IVS2+1G→C | Villablanca et al. (2004)n (rt) | |
| c.253_258del | Exon 3 | 85 | p.Val85_Val86del | G | c.252_257del6 | Pazienza et al. (2013) |
| c.272G > C | Exon 3 | 91 | p.Arg91Pro | Go | Arg91Pro | Zhang et al. (2012)f |
| c.284T > C | Exon 3 | 95 | p.Leu95Pro | So ,3 | c.284T > C | Yu et al. (2015)g |
| c.293T > C | Exon 3 | 98 | p.Leu98Pro | G | c.293T > C exon 3 | Bricaire et al. (2013)n (ut) |
| c.307+1G > A | Intron 3 | splice [d] | Go | IVS3+1 G > A | Kong et al. (2014) | |
| c.308–9T > A | Intron 3 | splice [vus] | G | c.308–9T > A intron 3 | Bricaire et al. (2013)n (rt,jt?) | |
| c.343G > T | Exon 4 | 115 | p.Glu115X | Go | c.343G > T | Guarnieri et al. (2008)h |
| c.356del | Exon 4 | 119 | p.Gln119ArgfsX14 | Go ,3 | 356delA | Bradley et al. (2006)g |
| c.415C > T | Exon 5 | 139 | p.Arg139X | Go | c.415C > T | Guarnieri et al. (2008)h , i , n (rt,ut) |
| c.483_486del | Exon 6 | 162 | p.Glu162GlyfsX39 | G | c.481_484delAAAG exon 6 | Bricaire et al. (2013)n (ut,jt?) |
| c.505C > T | Exon 6 | 169 | p. Gln169X | G | c.505C > T | Ghemigian et al. (2013) |
| c.520_523del | Exon 7 | 174 | p.Ser174LysfsX27 | Go | c.518_521delTCTC | Guarnieri et al. (2008)h,i,n(rt,ut) |
| c.520_523del | Exon 7 | 174 | p.Ser174LysfsX27 | G2 | 518–521del | Mizusawa et al. (2006) |
| c.664C > T | Exon 7 | 222 | p.Arg222X | G | R222X | Khadilkar et al. (2015) |
| c.664C > T | Exon 7 | 222 | p.Arg222X | G | c.664 C > T (Arg222X) | Kong et al. (2014) |
| c.679_680insAG | Exon 7 | 227 | p.Arg227LysfsX31 | Go | 679_680insAG | Simonds et al. (2004)n (lip) |
| c.685_688del | Exon 7 | 229 | p.Arg229TyrfsX27 | Go | 685delAGAG | Guarnieri et al. (2006)n (rt,ut) |
| c.745dup | Exon 8 | 249 | p.Ile249AsnfsX18 | G | 745 dup 1 bp | Bradley et al. (2006) |
| Gross deletion | G | c.237‐?_308‐?del exon 3 | Bricaire et al. (2013)n (rt,jt?) | |||
| Gross deletion | G | c.131 ?_308‐?del exons 2–3 | Bricaire et al. (2013) | |||
| Gross deletion | Go | Deletion exon 1–10 | Korpi‐Hyovalti et al. (2014)n (rt) | |||
| MEN1 | ||||||
| c.13_15delinsACGCT | Exon 2 | 5 | p.Ala5ThrfsX115 | G | 13insACGCTdelGCC | Cardinal et al. (2005)i |
| c.249_252del | Exon 2 | 85 | p.Ile85SerfsX33 | G | 249del4 | Karges et al. (2000) |
| c.255_256insCAGTGGCCGACCTGTCTAT | Exon 2 | 86 | p.Ile86GlnfsX37 | G | 2543ins18 | Bergman et al. (2000)i, j |
| c.255_256insCAGTGGCCGACCTGTCTAT | Exon 2 | 86 | p.Ile86GlnfsX37 | G | c.255_256insCAGTGGCCGACCTGTCTAT | Warner et al. (2004)i |
| c.255_256insCAGTGGCCGACCTGTCTAT | Exon 2 | 86 | p.Ile86GlnfsX37 | G | 255ins19 | Cardinal et al. (2005)i |
| c.334G > C | Exon 2 | 112 | p.Val112Leu | G | L112V | Villablanca et al. (2002) |
| c.458A > T | Exon 3 | 153 | Asp153Val | G | D153V | Pannett et al. (2003) |
| c.532_535del | Exon 3 | 178 | p.Ser178ArgfsX6 | G | codon 177–178(delGTCT) | Pannett et al. (2003) |
| c.551T > A | Exon 3 | 184 | p.Val184Glu | G | V184E | Fujimori et al. (1998) |
| c.590C > T | Exon 3 | 197 | p.Thr197Ile | G | 590C > T | Warner et al. (2004)i |
| c.590C > T | Exon 3 | 197 | p.Thr197Ile | G | 590C→T | Cardinal et al. (2005)i |
| c.600_601dup | Exon 3 | 201 | p.Lys201ThrfsX24 | G | 711dupCA | Wautot et al. (2002)k |
| c.654G > Tp | Exon 3 | 218 | G | codon 219 (CGG→CGT) | Dwarakanathan, Zwart and Oathus (2000) | |
| c.659G > T | Exon 4 | 220 | p.Trp220Leu | G | Trp220Leu | Hannan et al. (2008) |
| c.673G > A | Exon 4 | 225 | p.Gly225Arg | G | G225R (GGA→AGA) | Mizusawa et al. (2006) |
| c.722G > T | Exon 4 | 241 | p.Cys241Phe | G | C240F | Wautot et al. (2002)k |
| c.763G > A | Exon 4 | 255 | p.Glu255Lys | G | E255K | Teh et al. (1998) |
| c.779A > C | Exon 4 | 260 | p.Gln260Pro | G | Q260P | Kassem, Kruse, Wong, Larsson and Teh (2000) |
| c.784‐9G > A | Intron 4 | splice [vus] | G | IVS4 ‐9G→A | Cetani et al. (2006)n (rt,lip,tn) | |
| c.800T > C | Exon 5 | 267 | p.Leu267Pro | G | 910T→C | Poncin et al. (1999) |
| c.824G > T | Exon 5 | 275 | p.Arg275Met | G | c.824G > T | Nagamura et al. (2012) |
| c.824+1G > A | Intron 5 | splice [d] | G | IVS5 +1G→A | Cetani et al. (2006)n (lip) | |
| c.914G > A | Exon 7 | 305 | p.Gly305Asp | G | G305D | Honda et al. (2000) |
| c.1021T > C | Exon 7 | 341 | p.Trp341Arg | G | c.T1021C: p.W341R | Isakov et al. (2013) |
| c.1021T > C | Exon 7 | 341 | p.Trp341Arg | G | W341R | Wautot et al. (2002)k |
| c.1049+2_1049+5del | Intron 7 | splice [vus] | G | codon 350 (delGAgt) | Pannett et al. (2003) | |
| c.1051T > A | Exon 8 | 351 | p.Tyr351Asn | G | Tyr351Asn | Hannan et al. (2008) |
| c.1059C > A | Exon 8 | 353 | p.Tyr353X | G | Y353X | Shimizu et al. (1997) |
| c.1058_1060del | Exon 8 | 353 | p.Tyr353del | G | 1057‐1059delACT | Warner et al. (2004)i |
| c.1058_1060del | Exon 8 | 353 | p.Tyr353del | G | 1057‐1060delACT | Cardinal et al. (2005)i |
| c.1069G > C | Exon 8 | 357 | p.Asp357His | G | D357H | Wautot et al. (2002)k |
| c.1087_1089del | Exon 8 | 363 | p.Glu363del | G | E363del | Miedlich, Lohmann, Schneyer, Lamesch and Paschke (2001) |
| c.1096G > T | Exon 8 | 366 | p.Glu366X | G | Q366X | Takami et al. (2000) |
| c.1190_1193del | Exon 9 | 397 | p.Thr397ArgfsX47 | G | 1298del4 | Wautot et al. (2002)k |
| c.1231G > C | Exon 9 | 411 | p.Ala411Pro | G | A411P | Pannett et al. (2003) |
| c.1241_1243del | Exon 9 | 414 | p.Leu414del | G | 1350del3 | Sato et al. (1998) |
| c.1241_1243del | Exon 9 | 414 | p.Leu414del | G | 1350del3 | Ohye et al. (1998) |
| c.1252G > C | Exon 9 | 418 | p.Asp418His | G | 1252G > C | Warner et al. (2004)l |
| c.1252G > C | Exon 9 | 418 | p.Asp418His | G | D418H | Cetani et al. (2006)n (tn) |
| c.1343_1353del | Exon 9 | 448 | p.Glu448AlafsX79 | G | 1452deL11 | Wautot et al. (2002)k |
| c.1350+1G > A | Intron 9 | splice [d] | Go | IVS9 +1G > A | Carrasco et al. (2004)q | |
| c. 1373_1376del | Exon 10 | 458 | p.Val458AlafsX100 | G | 1483del4 | Takami et al. (2000) |
| c.1382_1404del | Exon 10 | 461 | p.Glu461GlyfsX62 | G | 1486del23 | Wautot et al. (2002)k |
| c.1546dup | Exon 10 | 516 | p.Arg516ProfsX15 | G | 1546‐1547insC | Warner et al. (2004) |
| c.1548del | Exon 10 | 516 | p.Lys517SerfsX42 | G | 1658delG | Villablanca et al. (2002) |
| c.1676del | Exon 10 | 559 | p.Lys559ArgfsX3 | G | 1785delA | Cetani et al. (2002) |
| Gross deletion | G | gross deletion | Cebrian et al. (2003) | |||
| CASR | ||||||
| c.299C > T | Exon 3 | 100 | p.Thr100Ile | G | T100I | Warner et al. (2004)n (hca) |
| c.476T > C | Exon 3 | 159 | p.Leu159Pro | G | L159P | Simonds et al. (2002)n (hca,uccr) |
| c.658C > T | Exon 4 | 220 | p.Arg220Trp | G | R220W | Simonds et al. (2002)n (hca,uccr) |
| c.748G > A | Exon 4 | 250 | p.Glu250Lys | G | E250K | Simonds et al. (2002) |
| c.802_812del | Exon 4 | 268 | p.Val268GlnfsX6 | G | V268del‐11 × 273 | Simonds et al. (2002)n (hca,hcu) |
| c.1006_1008del | Exon 4 | 336 | p.Lys336del | G | K336del | Warner et al. (2004)n (hca,hcu) |
| c.1949T > C | Exon 7 | 650 | p.Leu650Pro | G | L650P | Warner et al. (2004)n (hca) |
| c.2065G > A | Exon 7 | 689 | p.Val689Met | G | V689M | Warner et al. (2004)n (hca) |
| c.2641T > C | Exon 7 | 881 | p.Phe881Leu | G | F881L | Carling et al. (2000)n (hca) |
| c.2657G > C | Exon 7 | 886 | p.Arg886Pro | G | R886P | Simonds et al. (2002)n (hca,uccr) |
Mutations are numbered in relation to the cell division cycle 73 (CDC73), multiple endocrine neoplasia type 1 (MEN1), and calcium‐sensing receptor (CASR) cDNA reference sequences (GenBank accession number NM_024529.4, NM_130799.2, NM_000388.3, respectively) whereby nucleotide +1 corresponds to the A of the ATG‐translation initiation codon. All mutations were analyzed using the Leiden Open Variation Database (LOVD) Mutalyzer sequence variant nomenclature checker (https://www.lovd.nl/mutalyzer/) and annotated using the Human Genome Variation Society (HGVS) guidelines (https://www.hgvs.org/).
Codon numbering starts from initiation codon of CDC73, MEN1, and CASR mRNA.
Predicted effect: splice, splice site mutation; [d] donor splice site; [a] acceptor splice site; [vus] variant of unknown significance.
Mutation type: G, germline; S, somatic; ND, not defined. Equal superscript numbers represent germline and/or somatic mutations occurring in the same patient.
It is possible this is a case of HPT‐JT associated with PC since: the patient was diagnosed with three renal cysts, while “a maternal cousin had jaw pain and presumably bone destruction of the jaw, termed a ‘hole in the jaw’.” Furthermore, histological description of the proband's parathyroid gland was consistent with an APA (“…vascular and capsular invasion, but no definitive features of PC were identified”) and disease recurrence on the contra‐lateral side (again with diagnosis of APA) suggests a more malignant behavior.
All mutation carriers (n = 3) of this kindred developed PC.
Kindred originally reported by Williamson et al., and classified as HPT‐JT by Carpten et al. and FIHP by Bradley et al., and associated with PC by Carpten et al., Bradley et al., and Yu et al. (Williamson et al., 1999; Carpten et al., 2002; Bradley et al., 2005a; Bradley et al., 2006; Yu et al., 2015).
Reported as a FIHP family, but no information was provided on the pHPT status of the mutation carriers.
Additional clinical details about these kindreds are provided by Corbetta et al. (2010) and Vaira et al. (2012).
Studies reported by the same group, therefore it is not possible to exclude that equal mutations described in different publications are from the same proband/kindred.
Mutation was incorrectly reported in the original publication and was posteriorly updated by Warner et al. (2004) and Cardinal et al. (2005).
The authors collected 165 MEN1 mutations in patients with MEN1, but seven probands/kindreds exhibited FIHP phenotype (i.e., only pHPT) and were included here.
In a posterior publication, this mutation was identified by the same group in a kindred with MEN1 syndrome, and it is unclear if there were two different kindreds with the same mutation or if it was an update of the previous kindred (Cardinal et al., 2005).
Presence in the probands/kindreds of: rt renal cysts/lesions, and/or ut uterine tumors (if the presence of renal cysts or uterine tumors was unknown, one “?” was added next to the previous superscripts; hjt? was added if the absence of jaw tumors was unknown), and/or lip lipoma, and/or tn thyroid nodules, and/or hca hypercalcemia, and/or hcu hypercalciuria, and/or uccr urine calcium/creatinine clearance ratio < 0.010 in most of the affected individuals.
For detailed information of the effect of CDC73 mutation on splicing please consult Hahn et al. (2009).
Mutations identified in kindreds with case reports of PC.
Discordant codon/nucleotide number in the original report. There is no predicted change on the amino acid (p.Arg218 = ), but the authors reported altered RNA splicing caused by this nucleotide change.
Criteria for diagnosis of PC not reported.
HPT‐JT, hyperparathyroidism‐jaw tumor; PC, parathyroid carcinoma; APA, atypical parathyroid adenoma; FIHP, familial isolated primary hyperparathyroidism; MEN1, multiple endocrine neoplasia type 1.
Table 5.
Summary of CDC73 mutations associated with sporadic parathyroid carcinoma
| Mutation a | Exon/intron | Codon b | Predicted effect c | Type d | Original designation | References |
|---|---|---|---|---|---|---|
| c.13C > T | Exon 1 | 5 | p.Leu5Phe | S | 13C > T | Guarnieri et al. (2012) |
| c.14T > C | Exon 1 | 5 | p.Leu5Pro | S1 | c.14T > C | Cavaco et al. (2011) |
| c.16del | Exon 1 | 6 | p.Ser6AlafsX15 | ND2 | 16delA | Shattuck et al. (2003b) |
| c.23_25delinsGT | Exon 1 | 8 | p.Leu8ArgfsX13 | S | 23TGCG > GTG | Shattuck et al. (2003b) |
| c.25C > T | Exon 1 | 9 | p.Arg9X | S | R9X | Cetani et al. (2004) |
| c.32del | Exon 1 | 11 | p.Tyr11SerfsX10 | S3 | c.32delA | Domingues et al. (2012)e |
| c.34_35insCT | Exon 1 | 12 | p.Asn12ThrfsX10 | ND | c.34_35insCT | Wang et al. (2012)j |
| c.34_37del | Exon 1 | 12 | p.Asn12SerfsX8 | S | 34‐37 delAACA | Enomoto et al. (2010)j |
| c.40del | Exon 1 | 14 | p.Gln14ArgfsX7 | S | 39delC | Shattuck et al. (2003b) |
| c.42del | Exon 1 | 15 | p.Lys15ArgfsX6 | S4 | c.42delG | Guarnieri et al. (2012) |
| c.60del | Exon 1 | 21 | p.Lys21ArgfsX5 | S5 | c.60delG | Cetani et al. (2013) |
| c.64_73del | Exon 1 | 22 | p.Gly22X | ND6 | 60del10 | Shattuck et al. (2003b) |
| c.64G > T | Exon 1 | 22 | p.Gly22X | S7 | c.64G > T | Cetani et al. (2013) |
| c.70G > T | Exon 1 | 24 | p.Glu24X | S8 | 70G > T | Shattuck et al. (2003b) |
| c.70G > T | Exon 1 | 24 | p.Glu24X | S | E24X | Cetani et al. (2007) |
| c.70G > T | Exon 1 | 24 | p.Glu24X | G | c.70G > T | Serrano‐Gonzalez, Shay, Austin, Maceri and Pitukcheewanont (2016) |
| c.76del | Exon 1 | 26 | p.Ile26SerfsX11 | S | c.76delA | Howell et al. (2003) |
| c.82_85del | Exon 1 | 28 | p.Gly28SerfsX8 | S9 | 82del4 | Shattuck et al. (2003b) |
| c.85del | Exon 1 | 29 | p.Glu29SerfsX8 | S10 | c.85delG | Siu et al. (2011) |
| c.88_94del | Exon 1 | 30 | p.Phe30GlyfsX5 | S | c.88_94delTTCTCCT | Frank‐Raue et al. (2011)h (rt) |
| c.94_95insTA | Exon 1 | 32 | p.Trp32LeufsX6 | S | c.94insTA | Guarnieri et al. (2012) |
| c.128G > A | Exon 1 | 43 | p.Trp43X | S | c.128G > A | Haven et al. (2007)f |
| c.162C > G | Exon 2 | 54 | p.Tyr54X | S | c.162C > G (Y54X) | Howell et al. (2003) |
| c.162C > G | Exon 2 | 54 | p.Tyr54X | S11 | 162C > G | Shattuck et al. (2003b) |
| c.162C > G | Exon 2 | 54 | p.Tyr54X | ND | c.162C > G | Wang et al. (2012) |
| c.162C > A | Exon 2 | 54 | p.Tyr54X | S12 | c.162C > A | Cavaco et al. (2011)g |
| c.165C > A | Exon 2 | 55 | p.Tyr55X | S13 | c.165C > A | Howell et al. (2003) |
| c.165del | Exon 2 | 55 | p.Tyr55X | S | c.165delC | Howell et al. (2003) |
| c.165del | Exon 2 | 55 | p.Tyr55X | S | c.165delC | Haven et al. (2007)f |
| c.176C > T | Exon 2 | 59 | p.Ser59Phe | G | c.176C > T | Haven et al. (2007)f |
| c.182T > A | Exon 2 | 61 | p.Leu61X | S | 182T > A | Cetani et al. (2007) |
| c.195dup | Exon 2 | 66 | p.Asn66X | S | 195insT | Cetani et al. (2004) |
| c.197dup | Exon 2 | 66 | p.Asn66LysfsX16 | S | 195insA | Cetani et al. (2004) |
| c.226C > T | Exon 2 | 76 | p.Arg76X | S | c.226C > T | Shattuck et al. (2003b) |
| c.226C > T | Exon 2 | 76 | p.Arg76X | G1 | c.226C > T | Cavaco et al. (2011) |
| c.226C > T | Exon 2 | 76 | p.Arg76X | G10 | c.226C > T | Siu et al. (2011) |
| c.249del | Exon 3 | 84 | p.Pro84LeufsX25 | S5 | c.248delT | Cetani et al. (2013) |
| c.260_261del | Exon 3 | 87 | p.Arg87LysfsX3 | G | c.260_261delGA | Wang et al. (2012) |
| c.343G > T | Exon 4 | 115 | p.Glu115X | G | E115X | Cetani et al. (2013) |
| c.343G > T | Exon 4 | 115 | p.Glu115X | G7 | E115X | Cetani et al. (2013) |
| c.375dup | Exon 5 | 126 | p.Arg126ThrfsX5 | G | 373insA | Shattuck et al. (2003b) |
| c.415C > T | Exon 5 | 139 | p.Arg139X | G | 415C > T | Cetani et al. (2007) |
| c.415C > T | Exon 5 | 139 | p.Arg139X | G | c.415C > T exon 5 | Bricaire et al. (2013)j , h (ut?,rt?,jt?) |
| c.513‐1del | Intron 6 | splice [a]i | S13 | IVS6‐1delG | Howell et al. (2003) | |
| c.520_523del | Exon 7 | 174 | p.Ser174LysfsX27 | G12 | c.518_521delTGTC | Cavaco et al. (2011)g |
| c.544dup | Exon 7 | 182 | p.Ile182AsnfsX11 | G | c.539_544insA, p.Ile182AsnfsX10 | Yu et al. (2015) |
| c.626_629del | Exon 7 | 209 | p.Lys209ArgfsX9 | G | c.626_629delAACA | Wang et al. (2012)h(rt) |
| c.664C > T | Exon 7 | 222 | p.Arg222X | G | 664C > T | Shattuck et al. (2003b) |
| c.664C > T | Exon 7 | 222 | p.Arg222X | G | c.664C > T exon 7 | Bricaire et al. (2013)h (rt) |
| c.679_680insAG | Exon 7 | 227 | p.Arg227LysfsX31 | G11 | 679insAG | Shattuck et al. (2003b) |
| c.685_688del | Exon 7 | 229 | p.Arg229TyrfsX27 | G | c.679_682delAGAG | Corbetta et al. (2010) |
| c.687_688del | Exon 7 | 229 | p.Arg229SerfsX37 | S | c.679_680delAG | Corbetta et al. (2010) |
| c.687_688del | Exon 7 | 229 | p.Arg229SerfsX37 | G4 | c.679_680delAG | Guarnieri et al. (2012) |
| c.687_688del | Exon 7 | 229 | p.Arg229SerfsX37 | G | c.687_688delAG | Wang et al. (2012) |
| c.687_688del | Exon 7 | 229 | p.Arg229SerfsX37 | G | c.687_688delAG | Witteveen et al. (2011)f |
| c.691dup | Exon 7 | 231 | p.Trp231LeufsX36 | G | c.692_693insT | Haven et al. (2007)f |
| c.693dup | Exon 7 | 232 | p.Arg232GlufsX35 | ND | c.693_694insG | Haven et al. (2007) |
| c.700C > T | Exon 7 | 234 | p.Arg234X | ND6 | 700C > T | Shattuck et al. (2003b) |
| c.700C > T | Exon 7 | 234 | p.Arg234X | G | R234X | Cetani et al. (2004) |
| c.700C > T | Exon 7 | 234 | p.Arg234X | G | 234 CGA to TGA | Enomoto et al. (2010) |
| c.736del | Exon 8 | 246 | p.Ser246ProfsX11 | S9 | 732delT | Shattuck et al. (2003b) |
| c.750del | Exon 8 | 250 | p.Phe250LeufsX7 | S8 | 746delT | Shattuck et al. (2003b) |
| c.1231del | Exon 14 | 411 | p.Gln411ArgfsX17 | ND2 | 1230delC | Shattuck et al. (2003b) |
| Gross deletion | G | Whole gene deletion | Bricaire et al. (2013)h(ut) | |||
| Gross deletion | G | Whole gene deletion | Caron et al. (2011) | |||
| Gross deletion | G3 | Whole gene deletion | Domingues et al. (2012)e |
Mutations are numbered in relation to the cell division cycle 73 (CDC73) cDNA reference sequence (GenBank accession number NM_024529.4) whereby nucleotide +1 corresponds to the A of the ATG‐translation initiation codon. All mutations were analyzed using the Leiden Open Variation Database (LOVD) Mutalyzer sequence variant nomenclature checker (https://www.lovd.nl/mutalyzer/) and annotated using the Human Genome Variation Society (HGVS) guidelines (https://www.hgvs.org/).
Codon numbering starts from initiation codon of CDC73 mRNA.
Predicted effect: splice, splice site mutation; [d] donor splice site; [a] acceptor splice site.
Mutation type: G, germline; S, somatic; ND, not defined. Equal superscript numbers represent germline and/or somatic mutations occurring in the same patient.
Initially reported as a benign parathyroid adenoma, but later reclassified as PC by Yu et al. (2015).
In a posterior publication, most of this cohort was updated by Witteveen et al. (2011).
PC diagnosis disputable since: the tumor recurrence occurred with several cervical nodules of parathyroid tissue (fibrous septae, with low pleomorphism and high proliferative activity); however, during the first surgery, where a typical parathyroid adenoma was removed, the capsule was ruptured, thus raising the possibility of local seeding.
Presence in the affected patient of: rt renal cysts/lesions, and/or ut uterine tumors (if the presence of renal cysts or uterine tumors was unknown, one “?” was added next to the previous superscripts; hjt? was added if the absence of jaw tumors was unknown).
For detailed information of the effect of CDC73 mutation on splicing please consult Hahn et al. (2009).
Criteria for diagnosis of PC not reported.
PC, parathyroid carcinoma.
3.2. Multiple endocrine neoplasia type 1 (MEN1)
MEN1 (MIM# 131100), also known as Wermer's syndrome, is characterized by the occurrence of parathyroid, pancreatic islet, and anterior pituitary tumors (Thakker, 1998). Parathyroid tumors are often the first and the most frequent tumors, and occur in approximately 95% of MEN1 patients (Thakker et al., 2012; Thakker, 2014). Unlike HPT‐JT, many MEN1 patients with parathyroid tumors have multiglandular disease.
3.2.1. MEN1
MEN1 is an autosomal dominant disease, due to germline mutations of the MEN1 gene located on chromosome 11q13. MEN1 encodes the protein menin, which has roles in transcriptional regulation, genome stability, cell division, and proliferation. These roles have been identified by studying menin interactions with proteins. Thus, menin's roles in: transcriptional regulation involves interactions with Jun‐mediated transcriptional activation, nuclear factor‐kappaB (NF‐κB)‐mediated transcriptional activation, small body size homolog (sma, C. elegans), and mothers against decapentaplegic homolog (mad, Drosophila) (SMAD) family members to inhibit transforming growth factor‐b (TGF‐b) and bone morphogenetic protein‐2 (BMP‐2) signaling, and forkhead transcription factor checkpoint suppressor 1 (CHES1) in an S‐phase checkpoint pathway response to DNA damage; genome stability entails interactions with subunit of replication protein (RPA2) and Fanconi anemia complementation group D2 protein (FANCD2) that is involved in DNA repair; cell division includes interactions with nonmuscle myosin II‐A heavy chain (NMHC II‐A), glial fibrillary acidic protein (GFAP) and vimentin; and in proliferation, the reported interactions are with non‐metastatic cells 1 protein (NME1) and activator of S‐phase kinase (ASK) (Thakker, 2014). More than 90% of tumors from MEN1 patients will have LOH of MEN1, with loss of menin expression, consistent with a tumor suppressor role for MEN1 (Thakker et al., 1989; Lemos & Thakker, 2008). Approximately 10% of MEN1 patients harbor de novo mutations and 10%–15% may develop a non‐familial form (i.e., sporadic) (Trump et al., 1996; Bassett et al., 1998). To date >1,800 MEN1 mutations have been reported and ∼40% of these mutations are frameshift, followed by ∼25% nonsense, ∼20% missense mutations, and ∼10% splice site mutations (Lemos & Thakker, 2008; The Universal Mutation Database, 2017). Therefore, >70% of mutations are predicted to lead to truncated, and thus inactivated, forms of menin, with the majority of missense mutations resulting in the mutant menin being targeted to the proteasome, thereby reducing its ability to act as a tumor suppressor (Lemos & Thakker, 2008; Lemos et al., 2009). However, 5%–10% of MEN1 cases do not harbor mutations in the MEN1 gene (Bassett et al., 1998; Lemos & Thakker, 2008). MEN1 germline mutations have been reported in patients with hereditary and sporadic MEN1, and in FIHP and somatic MEN1 mutations are detected in approximately 20% of sporadic parathyroid tumors (Thakker, 2010).
PC rarely occurs in patients with MEN1. To date only 13 PC cases, of whom eight (> 60%) had local invasion or metastasis, have been reported in association with MEN1 (Table 6); one of these patients developed multiglandular PC, and in the remainder of patients, the PC was associated with multiple adenomatosis or hyperplasia. Four (30%) of these MEN1 patients presented with hypercalcemic crisis (median total calcium 15.7 mg/dl, that is, 3.9 mmol/l, and PTH 309.5 pg/ml) at a mean age of 50 years old. MEN1 germline mutations were reported in six (>45%) of these patients, and comprised one nonsense, three frameshifting with premature truncations, and two missense mutations (Sato et al., 2000; Clerici et al., 2001; Tham et al., 2007; Juodele et al., 2011; Christakis et al., 2016). Somatic genetic abnormalities in these PCs were not reported.
Table 6.
Parathyroid carcinoma in multiple endocrine neoplasia
| Gender Age | 1st manifestation | Calciuma (mg/dL) | PTHb (pg/mL) | Associated conditions | Mutationc | Predicted effect | Notes | References |
|---|---|---|---|---|---|---|---|---|
| MEN1 | ||||||||
|
Hypercalcaemic crisis | 16.4 | 154.3 | Pituitary adenoma | ND |
|
[Wu, et al., 1992] | |
|
|
10.7 | ND | c.734delCd , ¶ | p.Pro245LeufsX36 |
|
[Sato, et al., 2000] | |
|
Hypercalcaemic crisis | 15.7 | 1,888 |
|
NR |
|
[Dionisi, et al., 2002] | |
|
Hypercalcaemic crisis | 14.8 | 264 |
|
No |
|
[Agha, et al., 2007] | |
|
Hypercalcaemic crisis | 15.6 | 355 |
|
No |
|
[Agha, et al., 2007] | |
|
Moderate hypercalcaemia | 13.4 | 1,354 |
|
c.1406_1413dup8e , ¶ | p.Gly472SerfsX90 |
|
[Shih, et al., 2009] |
|
|
10.6 | 68 |
|
NR |
|
[Kalavalapalli, et al., 2010] | |
|
Moderate hypercalcaemia | ≈12 | 204 |
|
c.549G>Tf , ¶ | p.Trp183Cys |
|
[del Pozo, et al., 2011] |
|
Cervical mass | 13.4 | 323 |
|
c.129_130insAg , ¶ | p.Val44SerfsX73 |
|
[Juodele, et al., 2011] |
|
Moderate hypercalcaemia | 12.7 | 248.2 |
|
NR |
|
[Lee, et al., 2014] | |
|
Moderate hypercalcaemia | 12.4 | 127.3 |
|
ND |
|
[Singh Ospina, et al., 2016] | |
|
Moderate hypercalcaemia | 10.5 | 42 |
|
c.703G>Ah , ¶ | p.Glu235Lys |
|
[Christakis, et al., 2016] |
|
|
13.8 | 673.1 |
|
c.1378C>Ti , ¶ | p.Arg460X |
|
[Christakis, et al., 2016] |
| MEN2 | ||||||||
|
Moderate hypercalcaemia | 13.6 | 443 | Medullary thyroid carcinoma | c.1901G>Aj , ¶ | p.Cys634Tyr |
|
[Jenkins, et al., 1997] |
|
Severe hypercalcaemia Osteitis fibrosa cystica | 15.1 | 1,399 | Medullary thyroid carcinoma | No |
|
[Alfaro, et al., 2002] | |
|
Asymptomatic | 9.2 | 57.5 | Pheochromocytoma | c.1852T>Ck , ‡ | p.Cys618Arg |
|
[Posada‐Gonzalez, et al., 2014] |
aTotal serum calcium reference limits: 8.8–10.5 mg/dL (converted to commonly used units).
bParathyroid hormone (PTH) serum reference limits 10–65 pg/mL (converted to commonly used units).
cMutations are numbered in relation to the multiple endocrine neoplasia type 1 (MEN1) and rearranged during transfection (RET) cDNA reference sequences (GenBank accession number NM_130799.2 and NM_020975.4, respectively) whereby nucleotide +1 corresponds to the A of the ATG‐translation initiation codon. All mutations were analysed using the Leiden Open Variation Database (LOVD) Mutalyzer sequence variant nomenclature checker (http://www.lovd.nl/mutalyzer/) and annotated using the Human Genome Variation Society (HGVS) guidelines (http://www.hgvs.org/).
Reported originally as: d c.842delC, e c.1406_13dup8;fW183; g c.129insA; h c.703G>A; i c.1378C>T; j p.C634Y;k Cys618Arg.
¶Germline mutation;‡germline or somatic origin not defined (possibly germline, since its identification led to the prophylactic thyroidectomy, where the PC was incidentally found).
¥Diagnosis of PC based on capsular invasion, mitoses in parenchymal cells, and nuclear polymorphism. MEN1 mutation (c.734delC) was not identified in the 4 family members screened.
$Diagnosis of PC based on capsular invasion, fibrosis, cellular pleomorphism, dense fibrotic bands, and angulated parathyroid cell nests.
PC, parathyroid carcinoma; PA, parathyroid adenoma; pHPT, primary hyperparathyroidism; MEN1, multiple endocrine neoplasia type 1; MEN2, multiple endocrine neoplasia type 2; ND, not done; NR, not reported.
3.3. Multiple endocrine neoplasia type 2 (MEN2)
MEN2, also known as Sipple's syndrome, comprises three variants referred to as MEN2A (MIM# 171400), MEN2B (also called MEN3) (MIM# 162300), and medullary thyroid carcinoma (MTC) (MIM# 155240). MEN2A is characterized by occurrence of MTC, pheochromocytoma, and parathyroid tumors, which occur in >99%, ∼40%, and ∼30% of patients, respectively (Howe, Norton, & Wells, 1993). MEN2B is characterized by occurrence of MTC and pheochromocytoma in association with mucosal neuromas, medullated corneal fibers, intestinal autonomic ganglion dysfunction, and a Marfanoid habitus (Thakker, 1998). In patients with MTC‐only, MTC is the sole manifestation.
3.3.1. RET
MEN2A, MEN2B, and MTC‐only are due to activating mutations of the RET gene, located on chromosome 10q11.21 (Mathew et al., 1987; Simpson et al., 1987; Donis‐Keller et al., 1993; Mulligan, et al., 1993). The RET gene encodes a receptor tyrosine‐protein kinase involved in cell proliferation, neuronal navigation, cell migration, and cell differentiation following binding of glial cell‐derived neurotrophic factor ligands. RET signaling has critical roles in kidney organogenesis and formation of neural crest‐derived lineages, and RET can also modulate cell adhesion via caspase cleavage and cell migration in an integrin‐dependent manner. Moreover, in the absence of ligand, RET can also trigger apoptosis via intracellular caspase cleavage of the receptor (Mehlen & Thibert, 2004; Plaza‐Menacho, Mologni, & McDonald, 2014). There is a genotype–phenotype correlation between RET mutations and MEN2A, MEN2B, and MTC‐only, with: the majority of MEN2A patients having RET germline mutations involving codons 609, 611, 618, or 620 of exon 10, or codon 634 of exon 11; MEN2B patients having mutations of codon 918; and MTC‐only patients having mutations involving codons 618, 790, 791, or 804 (Raue & Frank‐Raue, 2012). To date three PC cases have been reported in association with MEN2A (Table 6). All patients were men and all had PC metastasis at diagnosis. RET mutations were identified in two of these patients, and these comprised a c.1852T > C, p.Cys618Arg mutation, whose germline or somatic origin was not defined, and a germline c.1901G > A, p.Cys634Tyr mutation. The metastatic PC from the patient with the RET Cys634Tyr mutation had additional somatic genetic abnormalities involving LOH at loci from chromosomes 1, 2, 3p, 13q, and 16p (Jenkins et al., 1997).
3.4. Familial isolated primary hyperparathyroidism (FIHP)
FIHP (MIM♯ 145000), is an autosomal dominant disorder, and to date >100 families with FIHP have been reported (Simonds et al., 2002; Pannett et al., 2003; Pontikides et al., 2014). The prevalence of FIHP has been estimated to be ∼1% of all pHPT cases, with an age at diagnosis of 40 years old (Simonds et al., 2002). Patients with FIHP more frequently present with severe hypercalcemia when compared with MEN1 patients or sporadic pHPT patients, and the provisional diagnosis of FIHP may, in ∼20% of patients, be reclassified as HPT‐JT, MEN1, or familial hypocalciuric hypercalcemia (FHH) following development of syndromic manifestations (Simonds et al., 2002; Pontikides et al., 2014). Furthermore, FIHP predisposition for PC is particularly high for CDC73 mutation carriers (Simonds et al., 2002; Pontikides et al., 2014).
3.4.1. CDC73
CDC73 mutations occur in 8% of FIHP patients (Pontikides et al., 2014). The majority of CDC73 germline mutations associated with FIHP are frameshift or nonsense, predicting premature truncation of parafibromin, thereby supporting a tumor suppressor function (Table 4 and Figure 2A and B).
3.4.2. MEN1
MEN1 mutations occur in 20% of FIHP patients (Pontikides et al., 2014). LOH, particularly at the 11q13 region, is a common finding in FIHP tumor samples. MEN1 germline mutations have been reported in 42 FIHP families, and ∼40% of these were missense, ∼30% were frameshift, and 5% were nonsense mutations (Table 4) (Lemos & Thakker, 2008). Interestingly, FIHP patients, in contrast to MEN1 patients, have a significantly lower prevalence of frameshift/nonsense MEN1 mutations (∼35% vs. ∼65%).
3.4.3. CASR
The calcium sensing receptor (CASR) gene, locate on chromosome 3q13.33, encodes a G‐protein coupled receptor that is predominantly expressed in the parathyroids and kidneys, where it respectively regulates PTH secretion and renal tubular calcium reabsorption appropriate to the prevailing calcium concentration (Thakker, 2004). The CaSR is also expressed in other tissues where its function remains to be elucidated (Thakker, 2004). CASR mutations occur in 2% of FIHP patients (Pontikides et al., 2014). To date 10 kindreds with CASR mutations associated with FIHP have been reported, and all of them had heterozygous CASR mutations that were predicted to be inactivating (Table 4). However, no PC case has been reported in any individual from these FIHP kindreds. FIHP patients with MEN1 and CASR mutations are generally younger and have multiglandular disease, whereas patients with CDC73 mutations have a disproportionally high prevalence of PC (Warner et al., 2004; Iacobone et al., 2007).
3.4.4. GCM2
Recently, activating mutations of the glial cells missing 2 (GCM2) gene, located on chromosome 6p24.2, have been reported in FIHP patients (Guan et al., 2016). GCM2 encodes a protein that acts as a transcription factor regulating parathyroid development and may also act to regulate the effect of calcium on PTH expression and secretion by parathyroid cells (Kamitani‐Kawamoto et al., 2011; Han, Tsunekage, & Kataoka, 2015).
3.4.5. Other Genes
MEN1, CDC73, CASR, and GCM2 mutations may not be found in over 60% of FIHP patients (Pontikides et al., 2014). Interestingly, one study has reported a 1.7 Mb interval of significant genetic linkage for FIHP on chromosome 2p13.3‐14, although conservative mutations involving the protein phosphatase 3 regulatory subunit B alpha (PPP3R1) and prokineticin receptor 1 (PROKR1) genes, which are in this interval, were not identified (Warner et al., 2006).
4. SPORADIC AND NON‐HEREDITARY PARATHYROID CARCINOMA
Sporadic and non‐hereditary PC may be associated with abnormalities of tumor suppressor genes and oncogenes, similarly to those causing hereditary syndromic forms of PC, and these include CDC73 and MEN1 mutations (Figure 3). However, sporadic and non‐hereditary PCs may be associated with abnormalities of other genes, which include retinoblastoma 1 (RB), tumor protein P53 (TP53), cyclin D1 (CCND1), enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), adenomatous polyposis coli (APC), glycogen synthase kinase 3 beta (GSK3B), and prune homolog 2 (PRUNE2). In addition, epigenetic abnormalities and microRNAs (miRNAs) may also be involved (Figure 3). These will be reviewed.
Figure 3.
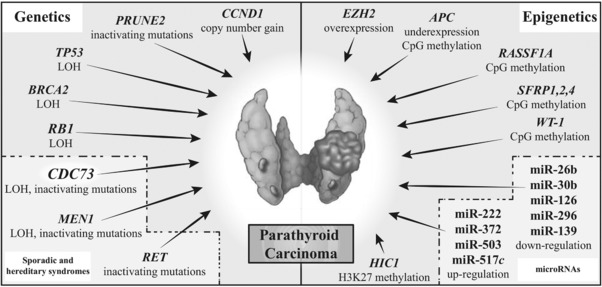
Molecular mechanisms of parathyroid carcinoma. LOH, loss of heterozygosity
4.1. CDC73
Approximately 40% of CDC73 mutations identified in patients with sporadic PC are germline mutations (Table 5 and Figures 2 and 3), and of these CDC73 mutations ∼65% occur in exons 1, 2, and 7, and the majority are frameshift or nonsense mutations, resulting in premature protein truncation and loss of protein function (Table 5) (Marsh et al., 2007; Newey et al., 2010). Moreover, a non‐random gain of mutated CDC73 alleles has been reported in PC, and this suggests that aberrant CDC73 expression may also be important in the pathogenesis of PC (Yu et al., 2015). For example, a recent study reported that 4 of 22 (∼20%) PCs had a 3–5 copy number gain of mutant alleles, with three of these four PCs also having loss of the wild‐type CDC73 allele through focal deletion or loss of the whole chromosome arm (Yu et al., 2015).
The identification of germline CDC73 mutations in patients with apparently sporadic PC is important, as it indicates that the patient and relatives are at risk of developing HPT‐JT‐associated tumors. Such germline CDC73 mutations are reported to occur in 20%–40% of patients with apparently sporadic PC, and somatic CDC73 mutations have been reported to occur in ∼40%–100% of apparently sporadic PCs (Table 5 and Figure 2) (Howell et al., 2003; Shattuck et al., 2003b; Cetani et al., 2004; Guarnieri et al., 2012). Moreover, LOH involving the CDC73 locus, on chromosome 1q31.2, is reported to occur in 50%–55% of sporadic PCs, and loss or reduced nuclear expression of parafibromin, detected by immunohistochemical (IHC) analysis, has been reported in >70% of PCs (Haven, van Puijenbroek, Karperien, Fleuren, & Morreau, 2004; Tan, et al., 2004; Cetani et al., 2007; Juhlin, et al., 2007; Yip et al., 2008). In contrast, germline CDC73 mutations were not found in patients with sporadic PAs, or in patients with hyperplastic parathyroids, and somatic CDC73 mutations and LOH of chromosome 1q have been reported to occur in <5% and <5%–10% of sporadic PAs, respectively (Carpten et al., 2002; Howell et al., 2003; Cetani et al., 2004; Krebs, Shattuck, & Arnold, 2005; Yip et al., 2008).
The absence of parafibromin nuclear staining, detected by IHC analysis, has been reported to occur in 15% of APAs, and <5%–20% of PAs, and it seems that the ability to distinguish between PC, APA, and PA using parafibromin IHC appears to be lower than CDC73 mutational analysis (Tan et al., 2004; Gill et al., 2006; Juhlin, et al., 2006; Cetani et al., 2007; Guarnieri et al., 2012; Cetani et al., 2013; Hu, Liao, Cao, Gao, & Zhao, 2016). However, one study has reported that the absence of parafibromin nuclear staining, detected by IHC analysis has a sensitivity of ∼70% and specificity of 95% for diagnosis of PCs (Hu et al., 2016). These findings indicate that CDC73 mutations are major driver mutations in the etiology of PCs.
4.2. MEN1
About 40%–50% of PCs have LOH of chromosome 11q, which is the location of the MEN1 gene, and >35% of PCs have combined LOH of 11q and 1q, which is the location of CDC73 (Figure 3). Combined LOH of 11q and 1q is rarely observed in PAs, and these findings suggest that MEN1 may be involved in PC pathogenesis (Dwight et al., 2000; Haven et al., 2004). In addition, somatic MEN1 mutations have been reported to occur in <15% PCs, in contrast to the higher frequencies of 35% and >45% of somatic MEN1 mutations and LOH involving chromosome 11 in sporadic PAs, respectively (Haven et al., 2007; Newey et al., 2012). Thus, the involvement of the MEN1 gene is likely to be a rare occurrence in PCs.
4.3. RB1
The retinoblastoma 1 (RB1) tumor suppressor gene, located on chromosome 13q14.2 encodes a protein (RB1) that is a negative regulator of the cell cycle. The active hypophosphorylated form of RB1 binds to the transcription factor E2 promoter binding factor 1 (E2F1) and leads to cell cycle arrest, whereas the phosphorylated form of RB1 allows dissociation from E2F1 and leads to transcription of E2F1 target genes that are involved in cell progression through G1 phase of the cell cycle (Asghar, Witkiewicz, Turner, & Knudsen, 2015). RB1 also maintains chromatin structure by stabilizing constitutive heterochromatin through stabilization of histone methylation (Gonzalo et al., 2005; Dyson, 2016). The RB1 gene has been implicated in the pathogenesis of PC, as allelic loss of RB1 has been observed in ∼30%–100% of PCs and decreased RB1 expression has been reported in >85% of PCs (Figure 3) (Cryns et al., 1994b; Dotzenrath et al., 1996; Szijan et al., 2000). This contrasts with the low rate (i.e., <5%) of RB1 allelic loss in PAs, and no loss of RB1 expression (Cryns et al., 1994b). However, no RB1 somatic mutations have been identified in PCs, although RB1 allelic loss has been reported to be associated with PC recurrence and aggressive PA (Pearce et al., 1996; Shattuck et al., 2003a).
4.4. TP53
Tumor protein P53 (TP53), is a tumor suppressor gene, which is located on chromosome 17p13.1 and encodes a protein (p53) that is a transcription factor whose level and post‐translational modification state are altered in response to cellular stress to induce growth arrest or apoptosis. Activated p53 suppresses cellular transformation by inducing growth arrest, apoptosis, DNA repair, and differentiation in damaged cells (Brosh & Rotter, 2009). TP53 allelic loss has been reported in 1 of 3 PCs studied (Figure 3), whereas TP53 overexpression has been observed in ∼10% of PAs (Cryns, Rubio, Thor, Louis, & Arnold, 1994a; Kishikawa et al., 1999). However, a somatic TP53 missense mutation (c.743G > A, p.Arg248Gln) has been reported in anaplastic PC cells, but not in differentiated PC cells, suggesting an association between this TP53 mutation and anaplastic transformation (Hakim & Levine, 1994; Tamura et al., 2009). The TP53 Arg248 residue is part of DNA binding domain (DBD) that interacts directly with the minor groove of DNA, and the p.Arg248Gln mutation is reported to result in the loss of DNA binding via the DBD (Ng et al., 2015). Interestingly, such TP53 mutations affecting Arg248 are reported to be present in ∼4% of all cancers (Petitjean et al., 2007).
4.5. CCND1
Cyclin D1 (CCND1), also known as parathyroid adenoma 1 (PRAD1), is an oncogene located on chromosome 11q13.3, that encodes cyclin D1, a 295‐amino acid protein that is a component of the cyclin D1‐cyclin‐dependent kinase 4 (CDK4) complex that phosphorylates RB1 and thus inhibits the actions of RB1 in regulating G1/S transition (Arnold et al., 1992). Overexpression of cyclin D1 occurs in ∼65%–90% of PCs, but in <40% of PAs and ∼60% of parathyroid hyperplasia (Hsi, Zukerberg, Yang, & Arnold, 1996; Vasef, Brynes, Sturm, Bromley, & Robinson, 1999; Haven et al., 2004). The overexpression of cyclin D1 is associated with PC cell proliferation and a Ki‐67 index of ≥5% (Haven et al., 2004). Overexpression of CCND1 gene may be associated with a 2–3 copy number gain of CCND1, which has been found to occur in five out of seven (∼70%) PCs (Figure 3), in contrast to the reported copy number gain of CCND1 in only three out of 14 (∼20%) PAs (Zhao et al., 2014). The increased CCND1 copy number in the PCs was associated with higher CCND1 mRNA levels and protein expression. However, the mechanisms linking CCND1 and PC tumorigenesis remain unknown. One hypothesis is that the potent inhibition of CCND1 expression by CDC73 may be lost after “two hits” on the CDC73 gene, which may then trigger CCDN1 disinhibition and tumorigenesis (Woodard et al., 2005).
4.6. EZH2
The enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) gene is located on chromosome 7q36.1, and encodes a 746‐amino acid histone methyltransferase enzyme that directly controls gene methylation and transcriptional repression (Vire et al., 2006). EZH2 mutations are rarely found in parathyroid tumors (Cromer et al., 2012; Sanpaolo et al., 2016). However, EZH2 copy number gain (four gene copies) has been reported to occur in ∼60% of PCs (Figure 3), ∼30% of PAs, and 50% of parathyroid hyperplasia (Svedlund et al., 2014). Furthermore, PC samples without gene copy number gain showed increased levels of EZH2 mRNA (Figure 3), suggesting the involvement of other indirect mechanisms (Svedlund et al., 2014). EZH2 may directly interact with β‐catenin inducing nuclear accumulation and activation of Wnt/β‐catenin signaling. EZH2 may also epigenetically repress Wnt antagonists like axis inhibition protein 2 (AXIN‐2), naked cuticle homolog 1 (NKD1), protein phosphatase 2 regulatory subunit B (PPP2R2B), prickle planar cell polarity protein 1 (PRICKLE1), and secreted frizzled related protein 5 (SFRP5), resulting in an increased activation of β‐catenin and increased expression of its target gene CCND1 (Bjorklund, Akerstrom, & Westin, 2007; Li et al., 2009; Cheng et al., 2011). EZH2 represses, through histone modification H3K27me2/3, the tumor suppressor gene hypermethylated in cancer 1 (HIC1), which is involved in controlling growth of parathyroid cells and is reported to be decreased in PCs and PAs (Svedlund et al., 2012).
4.7. APC
Adenomatous polyposis coli (APC) is a tumor suppressor gene located on chromosome 5q22.2, that encodes a 2,843‐amino acid protein, which inhibits canonical Wnt signaling by controlling β‐catenin ubiquitination and proteolysis. Loss of APC expression has been reported in PCs, although APC mutations and copy number changes have not been observed, thereby suggesting that APC may be involved in epigenetic mechanisms (Figure 3) (Juhlin et al., 2010; Svedlund et al., 2010; Andreasson et al., 2012; Newey et al., 2012; Yu et al., 2015). Thus, APC expression is reported to be lost in 75% of PCs (Figure 3), but maintained in 100% of PAs (Juhlin et al., 2009). Quantitative real time PCR (qRT‐PCR) and Western‐blot analysis has also revealed that APC mRNA is either undetectable or very low, and that APC protein expression is undetectable in PCs (Svedlund et al., 2010). These alterations in APC expression in PCs may involve hypermethylation of the APC promoter 1A, and indeed methylation levels of APC promoter 1A CpGs were found to be significantly higher in PCs (>85%) than normal parathyroids (>15%); this was associated with decreased APC expression and accumulation of active nonphosphorylated β‐catenin (Svedlund et al., 2010). Moreover, treatment of PC cultured cells with the DNA methylation inhibitor 5‐aza‐2′‐deoxycytidine (decitabine) resulted in re‐expression of APC mRNA, APC protein, and reduced cell viability, thereby suggesting that decitabine could be an additional option in the treatment of patients with recurrent or metastatic PC (Svedlund et al., 2010).
4.8. GSK3B
Glycogen synthase kinase 3 beta (GSK3B) protein expression has been reported to be lost in <35% of PCs and ∼5% of PAs (Juhlin et al., 2009). The GSK3B gene, located on chromosome 3q13.33, encodes a 420‐amino acid enzyme regulating glycogen synthesis, Wnt, and PI3‐kinase/AKT signaling pathways. However, loss of GSK3B expression was not associated with any increase of β‐catenin or cyclin D1 expression, thereby suggesting that GSK3B may act through a pathway different to the classical Wnt/β‐catenin pathway in the etiology of PC (Juhlin et al., 2009). This would be consistent with results from several studies that have reported that abnormal nuclear expression of β‐catenin is not a characteristic of PC (Semba, Kusumi, Moriya, & Sasano, 2000; Juhlin et al., 2009; Cetani et al., 2010).
4.9. PRUNE2
Prune homolog 2 (PRUNE2) germline and somatic mutations, comprising three missense mutations (one germline mutation in a PC without CDC73 mutations; two somatic mutations in two PCs without CDC73 mutations) and two nonsense somatic mutations (c.1609G > T, p.Glu537X and c.1420G > T, p.Glu474X in a single PC with a CDC73 mutation) have been reported to occur in four of 22 (∼20%) of PCs, but not PAs (Figure 3) (Yu et al., 2015). The PRUNE2 gene, located on chromosome 9q21.2, encodes a 3,088‐amino acid protein that regulates cell differentiation and survival by suppression of Ras homolog family member A (RhoA) activity. PRUNE2 has been reported to function as a tumor suppressor gene in prostate cancer, where prostate cancer antigen three (PCA3) regulates levels of PRUNE2 through formation of a PRUNE2/PCA3 double‐stranded RNA (Salameh et al., 2015).
4.10. Epigenetic mechanisms of parathyroid carcinoma
Epigenetic mechanisms, which may involve histone methylation modifications and DNA methylation, have been reported to occur in PCs (Figure 3), and these include overexpression of EZH2 and underexpression of HIC1, APC, and GSK3B as discussed above. Thus, the histone methyltransferases such as: EZH2, a H3‐lysine‐27‐methyltransferase enzyme; retinoblastoma protein‐interacting zinc finger gene 1 (RIZ1/PRDM2), a H3‐lysine‐9‐methyltransferase enzyme; and mixed lineage leukemia 2 (MLL2/KMT2D), a H3‐lysine‐4‐methyltransferase enzyme, have been reported to be involved in the pathogenesis of PC (Carling, Du, Fang, Correa, & Huang, 2003; Starker et al., 2011). Moreover, a somatic MLL2/KMT2D missense mutation (c.2522G > T, p.Cys841Phe) was reported in a PC, although in silico analysis has predicted that this is a likely tolerated/benign variant (Kasaian et al., 2013). Interestingly, menin and parafibromin, which are found to be mutated in such PCs, also interact with the histone methyltransferase SUV39H1 and function as transcription repressors by inducing H3K9 methylation (Rea et al., 2000; Yang et al., 2013). In addition, PCs have been reported to have promoter hypermethylation of: the Ras association domain family protein 1A (RASSF1A) gene, which encodes a Ras‐binding protein that down‐regulates cyclin D1 expression; and the secreted frizzled‐related protein 1 (SFRP1) gene, which was associated with epigenetic silencing and deregulated activation of the Wnt‐pathway (Figure 3). Hypermethylation of the promoters of the cyclin‐dependent kinase inhibitor 2A (CDKN2A), CDKN2B, Wilms tumor 1 (WT1), SFRP1, SFRP2, SFRP4, and RIZ1/PRDM2 genes, with reduced expression of the respective genes, have also have been reported in PCs, and expression of 5‐hydroxymethylcytosine (5hmC), an intermediate in DNA demethylation, was reported to be lower in PCs than in PAs (Starker et al., 2011; Sulaiman et al., 2013; Barazeghi et al., 2016). These results indicate that epigenetic mechanisms are likely involved in development of PC.
4.11. Role of microRNAs in parathyroid carcinoma
MicroRNAs (miRNAs) are small, 19–25 nucleotides long, non‐coding RNAs, that function as negative regulators of gene expression by decreasing translation or increasing degradation of the target mRNA. Studies of PCs have reported a global downregulation of approximately 60% miRNAs, when compared with normal parathyroid glands (Figure 3) (Corbetta et al., 2010; Rahbari et al., 2011). Downregulated miRNAs include miR‐26b, miR‐30b, miR‐126‐5p, miR‐296, and miR‐139 (Corbetta et al., 2010; Rahbari et al., 2011). Upregulated miRNAs include miR‐222, miR‐372, miR‐503, and miR‐517c (Corbetta et al., 2010; Vaira et al., 2012). These findings suggest that miRNA expression may contribute to development of PCs, and their role(s) remain to be elucidated.
5. GENETIC TESTING IN CLINICAL PRACTICE FOR PATIENTS WITH SUSPECTED PARATHYROID CARCINOMA
Distinguishing between PC (malignant disease) and PA (benign disease) on the basis of clinical and histological features is difficult and frequently not possible. This is because there is considerable overlap in the clinical features, including elevations of plasma calcium and PTH concentrations, and alkaline phosphatase activity, between patients with PC and PA (Wang & Gaz, 1985; Silverberg et al., 1990; Wynne et al., 1992; Chen et al., 2003). Moreover, histological examination may also not be able to reliably distinguish between PC, APA (an intermediate category between PC and PA), and PA (Bondeson, et al., 2004; DeLellis, 2011; Chan, 2013; Kumari et al., 2016). For example, it has been reported that use of pathological criteria has been associated with ≥50% of PCs, which actually behaved in a malignant manner being initially considered to be benign, and <15% of PCs were successfully diagnosed prospectively (Gill, 2014). However, CDC73 mutational analysis and parafibromin immunostaining has been reported to be more reliable, with: CDC73 mutations being identified in >75% of PCs but in <1% of PAs (Shattuck et al., 2003b; Krebs et al., 2005; Gill, 2014); and loss of nuclear parafibromin immunostaining occurring in >95% of PCs, but in <1% of PAs (Tan et al., 2004; Gill et al., 2006; Meyer‐Rochow et al., 2007). Moreover, even in the absence of a family history of PC or PAs, >30% of patients with PCs have a CDC73 germline mutation, thereby indicating they had an unrecognized HPT‐JT syndrome or FIHP (Shattuck et al., 2003b; Cetani et al., 2004; Gill, 2014). These observations indicate that parafibromin immunostaining is useful for diagnosis of PC, and that genetic testing for germline CDC73 mutations has an important role in the management of patients with proven or suspected PC, including APA, as these patients and their relatives are at risk of tumors associated with HPT‐JT. Identification of somatic mutations in some cancers is useful for targeting therapies, for example, epithelial growth factor receptor (EGFR) mutations for non‐small cell lung carcinoma (Paez et al., 2004; Gazdar, 2009); proto‐oncogene tyrosine‐protein kinase (KIT) mutations for chronic myeloid leukemia, gastrointestinal stromal tumors and melanoma (Heinrich, Blanke, Druker, & Corless, 2002; Willmore‐Payne, Holden, Tripp, & Layfield, 2005); or B‐Raf proto‐oncogene, serine/threonine kinase (BRAF) for melanoma and papillary thyroid cancer (Davies et al., 2002; Kimura et al., 2003). However, targeted therapies are not available for PC and at present genetic testing for somatic CDC73 mutations using parathyroid tumor DNA may not be clinically useful for establishing the diagnosis or staging, especially as such tumors may contain multiple mutations. For example, whole exome sequence analysis of PCs and PAs have reported that the number of somatic mutations in these tumors vary between 3–176 and 2–110, respectively, and that <50% of these tumors may have MEN1 mutations (Cromer et al., 2012; Newey et al., 2012; Yu et al., 2015). Moreover, our analysis, that has compared the frequency of somatic CDC73 mutations in the catalogue of somatic mutations in cancer (COSMIC) database with the frequency of germline CDC73 mutations in the exome aggregation consortium (ExAC) database, has revealed that there are ∼65‐fold more somatic non‐synonymous mutations than germline CDC73 mutations. This increased frequency of somatic CDC73 mutations is similar to that occurring for the neurofibromin 1 (NF1) gene, another tumor suppressor, in which somatic non‐synonymous mutations were ∼80‐fold more frequent than germline mutations. Recent studies have raised doubts about the pathogenicity of such mutations (Check Hayden, 2016; Lek, et al., 2016; Minikel, et al., 2016; Walsh et al., 2017), and while the clinical significance of such somatic mutations remains unknown, it would seem prudent at present, to reserve their investigation for research purposes only.
Genetic testing for germline CDC73 mutations may be helpful in clinical practice in several ways including: (1) confirmation of the high risk for developing PC and associated syndromic and hereditary forms of PC, so that appropriate screening for associated tumors (e.g., PC, uterine tumors, renal tumors) can be undertaken; (2) implementation of appropriate treatment (e.g., early parathyroidectomy for patients with HPT‐JT because of increased risk of PC); (3) identification of family members who may be asymptomatic but harbor the mutation and therefore require screening for tumor detection and early treatment; and (4) identification of the 50% of family members who do not harbor the familial germline mutation and can therefore be relieved of the anxiety burden of developing tumors, while reducing the cost to the individuals and their children, and also to the health services in not having to undertake unnecessary biochemical and radiological investigations (Newey & Thakker, 2011; Thakker et al., 2012; Eastell et al., 2014; Thakker, 2016).
A genetic testing approach in a patient with a proven or suspected PC (Figure 1) could be as follows. The indications for undertaking such genetic testing in a patient are occurrence of: a proven or suspected sporadic PC or APA; a PA in association with an ossifying fibroma, early‐onset uterine tumor, renal cysts or tumor, or endocrine tumor (e.g., pancreatic neuroendocrine or pituitary tumor); PA or pHPT occurring <35 years of age; recurrent pHPT, multiglandular parathyroid disease or hyperplasia or FIHP (Figure 1 and Table 7). A detailed family history for the occurrence of hypercalcemia, pHPT, MEN1, MEN2, HPT‐JT, FIHP, and FHH should be obtained as the presence of these disorders in a relative will help to guide decisions for appropriate germline mutational analysis of the MEN1, RET, CDC73, or CASR genes, with the results of the tests further guiding clinical management and treatments (Figure 1). In the absence of a family history, the diagnosis of sporadic PC should be considered, and germline mutational analysis of CDC73 undertaken as >30% of such patients will have a germline CDC73 mutation, and will therefore be at high risk of developing HPT‐JT‐associated tumors (Figure 1) (Thakker et al., 2012; Eastell et al., 2014; Wells et al., 2015; Thakker, 2016). The first degree relatives of these patients, even if asymptomatic, should also be offered tests for germline CDC73 mutations as these will help to identify if they have inherited the CDC73 mutation and are therefore at high risk of developing HPT‐JT‐associated tumors (Figure 1), or not inherited the CDC73 mutation in which case they can be reassured and have the burden of anxiety of developing PC and HPT‐JT‐associated tumors removed. Patients with sporadic PC who do not have CDC73 mutations, are likely to have another etiology for their disease and should be offered the opportunity of participating in research studies to elucidate the genetic abnormalities causing this rare disorder (Thakker et al., 2012; Eastell et al., 2014; Wells et al., 2015; Thakker, 2016). Thus, identification of germline mutations would be helpful in the clinical management of PC patients and their families.
Table 7.
Indications for CDC73 mutational analysis
| Sporadic PC |
| APA |
| Parathyroid tumor plus ossifying fibroma |
| Sporadic ossifying fibroma of the jaw |
| FIHP (MEN1 and CASR mutations excluded) |
| PA or pHPT < 35 years (MEN1 mutation excluded) |
| Recurrent pHPT (MEN1 mutation excluded) |
| Multiglandular PA/hyperplasia (MEN1 mutation excluded) |
| PA plus one or more of: |
| Early‐onset uterine lesion |
| Renal cysts/tumor |
| Pancreatic tumor |
| Thyroid tumor |
PC, parathyroid carcinoma; APA, atypical parathyroid adenoma; FIHP, familial isolated primary hyperparathyroidism; MEN1, multiple endocrine neoplasia type 1; CASR, calcium sensing receptor; PA, parathyroid adenoma; pHPT, primary hyperparathyroidism.
Modified from Newey et al. (2010).
6. CONCLUSIONS
PC is a rare endocrine cancer, presenting typically with symptoms of hypercalcemia and predisposition to recurrence and metastasis. A definitive diagnosis of PC, which is usually based on histological analysis, is often only made retrospectively. Improvements in predicting the predisposition to PC and in diagnosis of PC are required, to facilitate improvements in patient care. To this end, molecular genetic studies have helped in identifying the underlying causes of PC and a genetic approach (Figure 1) can be helpful for the management of patients. Molecular genetic studies have revealed CDC73 mutations to be major driver mutations in the etiology of PCs and defining and implementing clinical indications (Table 7) for CDC73 mutation analysis will aid in future management and counseling of patients at risk from PC and PC‐associated syndromes such as HPT‐JT. The genetic etiology causing PC involves other genes, which include MEN1, RET, and PRUNE2, as well as epigenetic mechanisms, alterations in miRNA expression and potentially as yet unidentified genes. PC is a rare neoplasm, and it is therefore essential that collaborative efforts that pool scarce tumor material and increase sample size are pursued to facilitate the identification of the genetic etiology of PC by next generation sequencing methodologies. These approaches are likely to yield important insights into the causative mechanisms for PC and to improved methods at detecting and diagnosing PCs, whose translation into the clinic are likely to lead to improved treatments and outcomes for patients.
Disclosure statement
The authors declare no conflict of interest.
Cardoso L, Stevenson M, Thakker RV. Molecular genetics of syndromic and non‐syndromic forms of parathyroid carcinoma. Human Mutation. 2017;38:1621–1648. https://doi.org/10.1002/humu.23337
Funding information Contract grant sponsors: United Kingdom Medical Research Council (MRC) (G9825289, G1000467); Wellcome Trust; National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme.
Communicated by Arupa Ganguly
REFERENCES
- Abdulla, A. , O'Leary, E. M. , Isorena, J. P. , Diaz, M. F. , & Yeh, M. W. (2013). Recurrent hyperparathyroidism and a novel nonsense mutation in a patient with hyperparathyriodism‐jaw tumor syndrome. Endocrine Practice, 19(6), e134–e137. [DOI] [PubMed] [Google Scholar]
- Agha, A. , Carpenter, R. , Bhattacharya, S. , Edmonson, S. J. , Carlsen, E. , & Monson, J. P. (2007). Parathyroid carcinoma in multiple endocrine neoplasia type 1 (MEN1) syndrome: Two case reports of an unrecognised entity. Journal of Endocrinological Investigation, 30(2), 145–149. [DOI] [PubMed] [Google Scholar]
- Aldred, M. J. , Talacko, A. A. , Savarirayan, R. , Murdolo, V. , Mills, A. E. , Radden, B. G. , … Larsson, C. (2006). Dental findings in a family with hyperparathyroidism‐jaw tumor syndrome and a novel HRPT2 gene mutation. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontology, 101(2), 212–218. [DOI] [PubMed] [Google Scholar]
- Alfaro, J. J. , Lamas, C. , Estrada, J. , & Lucas, T. (2002). MEN‐2A syndrome and pulmonary metastasis. Postgraduate Medicine Journal, 78(915), 51–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson, A. , Sulaiman, L. , do Vale, S. , Martins, J. M. , Ferreira, F. , Miltenberger‐Miltenyi, G. , … Juhlin, C. C. (2012). Molecular characterization of parathyroid tumors from two patients with hereditary colorectal cancer syndromes. Familial Cancer, 11(3), 355–362. [DOI] [PubMed] [Google Scholar]
- Arnold, A. , Motokura, T. , Bloom, T. , Rosenberg, C. , Bale, A. , Kronenberg, H. , … Kim, H. G. (1992). PRAD1 (cyclin D1): A parathyroid neoplasia gene on 11q13. Henry Ford Hospital Medicine Journal, 40(3–4), 177–180. [PubMed] [Google Scholar]
- Asghar, U. , Witkiewicz, A. K. , Turner, N. C. , & Knudsen, E. S. (2015). The history and future of targeting cyclin‐dependent kinases in cancer therapy. Nature Reviews Drug Discovery, 14(2), 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barazeghi, E. , Gill, A. J. , Sidhu, S. , Norlen, O. , Dina, R. , Palazzo, F. F. , … Westin, G. (2016). 5‐Hydroxymethylcytosine discriminates between parathyroid adenoma and carcinoma. Clinical Epigenetics, 8, 31 https://doi.org/10.1186/s13148-016-0197-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett, J. H. , Forbes, S. A. , Pannett, A. A. , Lloyd, S. E. , Christie, P. T. , Wooding, C. , … Thakker, R. V. (1998). Characterization of mutations in patients with multiple endocrine neoplasia type 1. American Journal of Human Genetics, 62(2), 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellido, V. , Larranaga, I. , Guimon, M. , Martinez‐Conde, R. , Eguia, A. , Perez de Nanclares, G. , … Gaztambide, S. (2016). A novel mutation in a patient with hyperparathyroidism‐jaw tumour syndrome. Endocrine Pathology, 27(2), 142–146. [DOI] [PubMed] [Google Scholar]
- Bergman, L. , Teh, B. , Cardinal, J. , Palmer, J. , Walters, M. , Shepherd, J. , … Hayward, N. (2000). Identification of MEN1 gene mutations in families with MEN 1 and related disorders. British Journal of Cancer, 83(8), 1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund, P. , Akerstrom, G. , & Westin, G. (2007). Activated β‐catenin in the novel human parathyroid tumor cell line sHPT‐1. Biochemical and Biophysical Research Communications, 352(2), 532–536. [DOI] [PubMed] [Google Scholar]
- Bondeson, L. , Grimelius, L. , DeLellis, R. A. , Lloyd, R. , Akerstrom, G. , Larsson, C. , … Bilezekian, J. P. (2004). Parathyroid carcinoma. In DeLellis R. A., Lloyd R. V., Heitz P. U., & Eng C. (Eds.), World Health Organization classification of tumors, pathology & genetics tumors of endocrine organs (3rd ed., pp. 124–127) Lyon: International Agency for Research on Cancer. [Google Scholar]
- Bradley, K. J. , Cavaco, B. M. , Bowl, M. R. , Harding, B. , Young, A. , & Thakker, R. V. (2005a). Utilisation of a cryptic non‐canonical donor splice site of the gene encoding PARAFIBROMIN is associated with familial isolated primary hyperparathyroidism. Journal of Medical Genetics, 42(8), e51 https://doi.org/10.1136/jmg.2005.032201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, K. J. , Hobbs, M. R. , Buley, I. D. , Carpten, J. D. , Cavaco, B. M. , Fares, J. E. , … Thakker, R. V. (2005b). Uterine tumours are a phenotypic manifestation of the hyperparathyroidism‐jaw tumour syndrome. Journal of Internal Medicine, 257(1), 18–26. [DOI] [PubMed] [Google Scholar]
- Bradley, K. J. , Cavaco, B. M. , Bowl, M. R. , Harding, B. , Cranston, T. , Fratter, C. , … Thakker, R. V. (2006). Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumours. Clinical Endocrinology (Oxford), 64(3), 299–306. [DOI] [PubMed] [Google Scholar]
- Bradley, K. J. , & Thakker, R. V. (2006). The hyperparathyroidism‐jaw tumour (HPT‐JT) syndrome. Clinical Cases in Mineral and Bone Metabolism, 3(2), 167–174. [Google Scholar]
- Bradley, K. J. , Bowl, M. R. , Williams, S. E. , Ahmad, B. N. , Partridge, C. J. , Patmanidi, A. L. , … Thakker, R. V. (2007). Parafibromin is a nuclear protein with a functional monopartite nuclear localization signal. Oncogene, 26(8), 1213–1221. [DOI] [PubMed] [Google Scholar]
- Bricaire, L. , Odou, M. F. , Cardot‐Bauters, C. , Delemer, B. , North, M. O. , Salenave, S. , … Group, G. T. E. (2013). Frequent large germline HRPT2 deletions in a French national cohort of patients with primary hyperparathyroidism. Journal of Clinical Endocrinology and Metabolism, 98(2), E403–408. [DOI] [PubMed] [Google Scholar]
- Brosh, R. , & Rotter, V. (2009). When mutants gain new powers: News from the mutant p53 field. Nature Reviews Cancer, 9(10), 701–713. [DOI] [PubMed] [Google Scholar]
- Busaidy, N. L. , Jimenez, C. , Habra, M. A. , Schultz, P. N. , El‐Naggar, A. K. , Clayman, G. L. , … Vassilopoulou‐Sellin, R. (2004). Parathyroid carcinoma: A 22‐year experience. Head and Neck, 26(8), 716–726. [DOI] [PubMed] [Google Scholar]
- Cardinal, J. W. , Bergman, L. , Hayward, N. , Sweet, A. , Warner, J. , Marks, L. , … Prins, J. B. (2005). A report of a national mutation testing service for the MEN1 gene: Clinical presentations and implications for mutation testing. Journal of Medical Genetics, 42(1), 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling, T. , Szabo, E. , Bai, M. , Ridefelt, P. , Westin, G. , Gustavsson, P. , … Rastad, J. (2000). Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. Journal of Clinical Endocrinology and Metabolism, 85(5), 2042–2047. [DOI] [PubMed] [Google Scholar]
- Carling, T. , Du, Y. , Fang, W. , Correa, P. , & Huang, S. (2003). Intragenic allelic loss and promoter hypermethylation of the RIZ1 tumor suppressor gene in parathyroid tumors and pheochromocytomas. Surgery, 134(6), 932–939. [DOI] [PubMed] [Google Scholar]
- Carlson, A. L. , & Smith, C. L. (2008). Primary hyperparathyroidism and jaw tumor syndrome: A novel mutation of the HRPT2 gene. Endocrine Practice, 14(6), 743–747. [DOI] [PubMed] [Google Scholar]
- Caron, P. , Simonds, W. F. , Maiza, J. C. , Rubin, M. , Cantor, T. , Rousseau, L. , … D'Amour, P. (2011). Nontruncated amino‐terminal parathyroid hormone overproduction in two patients with parathyroid carcinoma: A possible link to HRPT2 gene inactivation. Clinical Endocrinology (Oxford), 74(6), 694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpten, J. D. , Robbins, C. M. , Villablanca, A. , Forsberg, L. , Presciuttini, S. , Bailey‐Wilson, J. , … Hobbs M. R., (2002). HRPT2, encoding parafibromin, is mutated in hyperparathyroidism‐jaw tumor syndrome. Nature Genetics, 32(4), 676–680. [DOI] [PubMed] [Google Scholar]
- Carrasco, C. A. , Gonzalez, A. A. , Carvajal, C. A. , Campusano, C. , Oestreicher, E. , Arteaga, E. , … Fardella, C. E. (2004). Novel intronic mutation of MEN1 gene causing familial isolated primary hyperparathyroidism. Journal of Clinical Endocrinology and Metabolism, 89(8), 4124–4129. [DOI] [PubMed] [Google Scholar]
- Cascon, A. , Huarte‐Mendicoa, C. V. , Javier Leandro‐Garcia, L. , Leton, R. , Suela, J. , Santana, A. , … Robledo, M. (2011). Detection of the first gross CDC73 germline deletion in an HPT‐JT syndrome family. Genes, Chromosomes and Cancer, 50(11), 922–929. [DOI] [PubMed] [Google Scholar]
- Cavaco, B. M. , Guerra, L. , Bradley, K. J. , Carvalho, D. , Harding, B. , Oliveira, A. , … Leite, V. (2004). Hyperparathyroidism‐jaw tumor syndrome in Roma families from Portugal is due to a founder mutation of the HRPT2 gene. Journal of Clinical Endocrinology and Metabolism, 89(4), 1747–1752. [DOI] [PubMed] [Google Scholar]
- Cavaco, B. M. , Santos, R. , Felix, A. , Carvalho, D. , Lopes, J. M. , Domingues, R. , … Leite, V. (2011). Identification of de novo germline mutations in the HRPT2 gene in two apparently sporadic cases with challenging parathyroid tumor diagnoses. Endocrine Pathology, 22(1), 44–52. [DOI] [PubMed] [Google Scholar]
- Cebrian, A. , Ruiz‐Llorente, S. , Cascon, A. , Pollan, M. , Diez, J. J. , Pico, A. , … Robledo, M. (2003). Mutational and gross deletion study of the MEN1 gene and correlation with clinical features in Spanish patients. Journal of Medical Genetics, 40(5), e72 https://doi.org/10.1136/jmg.40.5.e72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetani, F. , Pardi, E. , Giovannetti, A. , Vignali, E. , Borsari, S. , Golia, F. , … Marcocci, C. (2002). Genetic analysis of the MEN1 gene and HPRT2 locus in two Italian kindreds with familial isolated hyperparathyroidism. Clinical Endocrinology (Oxford), 56(4), 457–464. [DOI] [PubMed] [Google Scholar]
- Cetani, F. , Pardi, E. , Borsari, S. , Viacava, P. , Dipollina, G. , Cianferotti, L. , … Marcocci, C. (2004). Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: Germline and somatic mutations in familial and sporadic parathyroid tumors. Journal of Clinical Endocrinology and Metabolism, 89(11), 5583–5591. [DOI] [PubMed] [Google Scholar]
- Cetani, F. , Pardi, E. , Ambrogini, E. , Lemmi, M. , Borsari, S. , Cianferotti, L. , … Marcocci, C. (2006). Genetic analyses in familial isolated hyperparathyroidism: Implication for clinical assessment and surgical management. Clinical Endocrinology (Oxford), 64(2), 146–152. [DOI] [PubMed] [Google Scholar]
- Cetani, F. , Ambrogini, E. , Viacava, P. , Pardi, E. , Fanelli, G. , Naccarato, A. G. , … Marcocci, C. (2007). Should parafibromin staining replace HRTP2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma? European Journal of Endocrinology, 156(5), 547–554. [DOI] [PubMed] [Google Scholar]
- Cetani, F. , Pardi, E. , Banti, C. , Collecchi, P. , Viacava, P. , Borsari, S. , … Marcocci, C. (2010). β‐catenin activation is not involved in sporadic parathyroid carcinomas and adenomas. Endocrine‐Related Cancer, 17(1), 1–6. [DOI] [PubMed] [Google Scholar]
- Cetani, F. , Banti, C. , Pardi, E. , Borsari, S. , Viacava, P. , Miccoli, P. , … Marcocci, C. (2013). CDC73 mutational status and loss of parafibromin in the outcome of parathyroid cancer. Endocrine Connections, 2(4), 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J. K. C. (2013). Tumors of thyroid and parathyroid glands In Fletcher C. D. M. (Ed.), Diagnostic histopathology of tumors (4th ed., pp. 1273–1293). Philadelphia: Elsevier Saunders. [Google Scholar]
- Check Hayden, E. (2016). A radical revision of human genetics. Nature, 538(7624), 154–157. [DOI] [PubMed] [Google Scholar]
- Chen, Q. , Kaji, H. , Nomura, R. , Sowa, H. , Yamauchi, M. , Tsukamoto, T. , … Chihara, K. (2003). Trial to predict malignancy of affected parathyroid glands in primary hyperparathyroidism. Endocrine Journal, 50(5), 527–534. [DOI] [PubMed] [Google Scholar]
- Cheng, A. S. , Lau, S. S. , Chen, Y. , Kondo, Y. , Li, M. S. , Feng, H. , … Sung, J. J. (2011). EZH2‐mediated concordant repression of Wnt antagonists promotes β‐catenin‐dependent hepatocarcinogenesis. Cancer Research, 71(11), 4028–4039. [DOI] [PubMed] [Google Scholar]
- Chiofalo, M. G. , Sparaneo, A. , Chetta, M. , Franco, R. , Baorda, F. , Cinque, L. , … Guarnieri, V. (2014). A novel CDC73 gene mutation in an Italian family with hyperparathyroidism‐jaw tumour (HPT‐JT) syndrome. Cellular Oncology (Dordrecht), 37(4), 281–288. [DOI] [PubMed] [Google Scholar]
- Christakis, I. , Busaidy, N. L. , Cote, G. J. , Williams, M. D. , Hyde, S. M. , Silva Figueroa, A. M. , … Perrier, N. D. (2016). Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; A clinico‐pathologic challenge. The MD Anderson case series and review of the literature. International Journal of Surgery, 31, 10–16. [DOI] [PubMed] [Google Scholar]
- Clerici, T. , Schmid, C. , Komminoth, Lange J., Spinas, G. A. , & Brandle, M. (2001). 10 Swiss kindreds with multiple endocrine neoplasia type 1: Assessment of screening methods. Swiss Medical Weekly, 131(25‐26), 381–386. [DOI] [PubMed] [Google Scholar]
- Corbetta, S. , Vaira, V. , Guarnieri, V. , Scillitani, A. , Eller‐Vainicher, C. , Ferrero, S. , … Spada, A. (2010). Differential expression of microRNAs in human parathyroid carcinomas compared with normal parathyroid tissue. Endocrine‐Related Cancer, 17(1), 135–146. [DOI] [PubMed] [Google Scholar]
- Cromer, M. K. , Starker, L. F. , Choi, M. , Udelsman, R. , Nelson‐Williams, C. , Lifton, R. P. , & Carling, T. (2012). Identification of somatic mutations in parathyroid tumors using whole‐exome sequencing. Journal of Clinical Endocrinology and Metabolism, 97(9), E1774–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns, V. L. , Rubio, M. P. , Thor, A. D. , Louis, D. N. , & Arnold, A. (1994a). p53 abnormalities in human parathyroid carcinoma. Journal of Clinical Endocrinology and Metabolism, 78(6), 1320–1324. [DOI] [PubMed] [Google Scholar]
- Cryns, V. L. , Thor, A. , Xu, H. J. , Hu, S. X. , Wierman, M. E. , Vickery, A. L., Jr. , … Arnold, A. (1994b). Loss of the retinoblastoma tumor‐suppressor gene in parathyroid carcinoma. New England Journal of Medicine, 330(11), 757–761. [DOI] [PubMed] [Google Scholar]
- Davies, H. , Bignell, G. R. , Cox, C. , Stephens, P. , Edkins, S. , Clegg, S. , … Futreal, P. A. (2002). Mutations of the BRAF gene in human cancer. Nature, 417(6892), 949–954. [DOI] [PubMed] [Google Scholar]
- del Pozo, C. , Garcia‐Pascual, L. , Balsells, M. , Barahona, M. J. , Veloso, E. , Gonzalez, C. , & Anglada‐Barcelo, J. (2011). Parathyroid carcinoma in multiple endocrine neoplasia type 1. Case report and review of the literature. Hormones (Athens), 10(4), 326–331. [DOI] [PubMed] [Google Scholar]
- DeLellis, R. A. (2011). Parathyroid tumors and related disorders. Modern Pathology, 24(Suppl 2), S78–93. [DOI] [PubMed] [Google Scholar]
- Dionisi, S. , Minisola, S. , Pepe, J. , De Geronimo, S. , Paglia, F. , Memeo, L. , & Fitzpatrick, L. A. (2002). Concurrent parathyroid adenomas and carcinoma in the setting of multiple endocrine neoplasia type 1: Presentation as hypercalcemic crisis. Mayo Clinic Proceedings, 77(8), 866–869. [DOI] [PubMed] [Google Scholar]
- Domingues, R. , Tomaz, R. A. , Martins, C. , Nunes, C. , Bugalho, M. J. , & Cavaco, B. M. (2012). Identification of the first germline HRPT2 whole‐gene deletion in a patient with primary hyperparathyroidism. Clinical Endocrinology (Oxford), 76(1), 33–38. [DOI] [PubMed] [Google Scholar]
- Donis‐Keller, H. , Dou, S. , Chi, D. , Carlson, K. M. , Toshima, K. , Lairmore, T. C. , … Wells, S. A., Jr. (1993). Mutations in the RET proto‐oncogene are associated with MEN 2A and FMTC. Human Molecular Genetics, 2(7), 851–856. [DOI] [PubMed] [Google Scholar]
- Dotzenrath, C. , Teh, B. T. , Farnebo, F. , Cupisti, K. , Svensson, A. , Toell, A. , … Larsson, C. (1996). Allelic loss of the retinoblastoma tumor suppressor gene: A marker for aggressive parathyroid tumors? Journal of Clinical Endocrinology and Metabolism, 81(9), 3194–3196. [DOI] [PubMed] [Google Scholar]
- Dwarakanathan, A. A. , Zwart, S. , & Oathus, R. C. (2000). Isolated familial hyperparathyroidism with a novel mutation of the MEN1 gene. Endocrine Practice, 6(3), 268–270. [DOI] [PubMed] [Google Scholar]
- Dwight, T. , Twigg, S. , Delbridge, L. , Wong, F. K. , Farnebo, F. , Richardson, A. L. , … Robinson, B. (2000). Loss of heterozygosity in sporadic parathyroid tumours: Involvement of chromosome 1 and the MEN1 gene locus in 11q13. Clinical Endocrinology (Oxford), 53(1), 85–92. [DOI] [PubMed] [Google Scholar]
- Dyson, N. J. (2016). RB1: A prototype tumor suppressor and an enigma. Genes and Development, 30(13), 1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastell, R. , Brandi, M. L. , Costa, A. G. , D'Amour, P. , Shoback, D. M. , & Thakker, R. V. (2014). Diagnosis of asymptomatic primary hyperparathyroidism: Proceedings of the Fourth International Workshop. Journal of Clinical Endocrinology and Metabolism, 99(10), 3570–3579. [DOI] [PubMed] [Google Scholar]
- Enomoto, K. , Uchino, S. , Ito, A. , Watanabe, S. , Shibuya, H. , Enomoto, Y. , & Noguchi, S. (2010). The surgical strategy and the molecular analysis of patients with parathyroid cancer. World Journal of Surgery, 34(11), 2604–2610. [DOI] [PubMed] [Google Scholar]
- Frank‐Raue, K. , Haag, C. , Schulze, E. , Keuser, R. , Raue, F. , Dralle, H. , & Lorenz, K. (2011). CDC73‐related hereditary hyperparathyroidism: Five new mutations and the clinical spectrum. European Journal of Endocrinology, 165(3), 477–483. [DOI] [PubMed] [Google Scholar]
- Fujimori, M. , Shirahama, S. , Sakurai, A. , Hashizume, K. , Hama, Y. , Ito, K. , … Fukushima, Y. (1998). Novel V184E MEN1 germline mutation in a Japanese kindred with familial hyperparathyroidism. American Journal of Medical Genetics, 80(3), 221–222. [DOI] [PubMed] [Google Scholar]
- Gazdar, A. F. (2009). Activating and resistance mutations of EGFR in non‐small‐cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 28 Suppl, 1, S24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghemigian, A. , Ghemigian, M. , Popescu, I. , Vija, L. , Petrova, E. , Dumitru, N. , & Dumitru, I. (2013). Familial isolated primary hyperparathyroidism due to HRPT2 mutation. Hormones (Athens), 12(3), 454–460. [DOI] [PubMed] [Google Scholar]
- Gill, A. J. , Clarkson, A. , Gimm, O. , Keil, J. , Dralle, H. , Howell, V. M. , & Marsh, D. J. (2006). Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidism‐jaw tumor (HPT‐JT) syndrome‐related adenomas from sporadic parathyroid adenomas and hyperplasias. American Journal of Surgical Pathology, 30(9), 1140–1149. [DOI] [PubMed] [Google Scholar]
- Gill, A. J. (2014). Understanding the genetic basis of parathyroid carcinoma. Endocrine Pathology, 25(1), 30–34. [DOI] [PubMed] [Google Scholar]
- Gonzalo, S. , Garcia‐Cao, M. , Fraga, M. F. , Schotta, G. , Peters, A. H. , Cotter, S. E. , … Blasco, M. A. (2005). Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nature Cell Biology, 7(4), 420–428. [DOI] [PubMed] [Google Scholar]
- Guan, B. , Welch, J. M. , Sapp, J. C. , Ling, H. , Li, Y. , Johnston, J. J. , … Agarwal, S. K. (2016). GCM2‐activating mutations in familial isolated hyperparathyroidism. American Journal of Human Genetics, 99(5), 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnieri, V. , Scillitani, A. , Muscarella, L. A. , Battista, C. , Bonfitto, N. , Bisceglia, M. , … Cole, D. E. (2006). Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: Implications for cancer surveillance. Journal of Clinical Endocrinology and Metabolism, 91(8), 2827–2832. [DOI] [PubMed] [Google Scholar]
- Guarnieri, V. , Bisceglia, M. , Bonfitto, N. , Cetani, F. , Marcocci, C. , Minisola, S. , … Scillitani, A. (2008). Re: Familial hyperparathyroidism: Surgical outcome after 30 years of follow‐up in three families with germline HRPT2 mutations. Surgery, 144(5), 839–840. [DOI] [PubMed] [Google Scholar]
- Guarnieri, V. , Battista, C. , Muscarella, L. A. , Bisceglia, M. , de Martino, D. , Baorda, F. , … Scillitani, A. (2012). CDC73 mutations and parafibromin immunohistochemistry in parathyroid tumors: Clinical correlations in a single‐centre patient cohort. Cellular Oncology (Dordrecht), 35(6), 411–422. [DOI] [PubMed] [Google Scholar]
- Hahn, M. A. , McDonnell, J. , & Marsh, D. J. (2009). The effect of disease‐associated HRPT2 mutations on splicing. Journal of Endocrinology, 201(3), 387–396. [DOI] [PubMed] [Google Scholar]
- Hahn, M. A. , Howell, V. M. , Gill, A. J. , Clarkson, A. , Weaire‐Buchanan, G. , Robinson, B. G. , … Marsh, D. J. (2010). CDC73/HRPT2 CpG island hypermethylation and mutation of 5'‐untranslated sequence are uncommon mechanisms of silencing parafibromin in parathyroid tumors. Endocrine‐Related Cancer, 17(1), 273–282. [DOI] [PubMed] [Google Scholar]
- Hakaim, A. G. , & Esselstyn, C. B., Jr. (1993). Parathyroid carcinoma: 50‐year experience at the Cleveland Clinic Foundation. Cleveland Clinical Journal of Medicine, 60(4), 331–335. [DOI] [PubMed] [Google Scholar]
- Hakim, J. P. , & Levine, M. A. (1994). Absence of p53 point mutations in parathyroid adenoma and carcinoma. Journal of Clinical Endocrinology and Metabolism, 78(1), 103–106. [DOI] [PubMed] [Google Scholar]
- Han, S. I. , Tsunekage, Y. , & Kataoka, K. (2015). Gata3 cooperates with Gcm2 and MafB to activate parathyroid hormone gene expression by interacting with SP1. Molecular and Cellular Endocrinology, 411, 113–120. [DOI] [PubMed] [Google Scholar]
- Hannan, F. M. , Nesbit, M. A. , Christie, P. T. , Fratter, C. , Dudley, N. E. , Sadler, G. P. , & Thakker, R. V. (2008). Familial isolated primary hyperparathyroidism caused by mutations of the MEN1 gene. Nature Clinical Practice. Endocrinology and Metabolism, 4(1), 53–58. [DOI] [PubMed] [Google Scholar]
- Haven, C. J. , van Puijenbroek, M. , Karperien, M. , Fleuren, G. J. , & Morreau, H. (2004). Differential expression of the calcium sensing receptor and combined loss of chromosomes 1q and 11q in parathyroid carcinoma. Journal of Pathology, 202(1), 86–94. [DOI] [PubMed] [Google Scholar]
- Haven, C. J. , van Puijenbroek, M. , Tan, M. H. , Teh, B. T. , Fleuren, G. J. , van Wezel, T. , & Morreau, H. (2007). Identification of MEN1 and HRPT2 somatic mutations in paraffin‐embedded (sporadic) parathyroid carcinomas. Clinical Endocrinology (Oxford), 67(3), 370–376. [DOI] [PubMed] [Google Scholar]
- Heinrich, M. C. , Blanke, C. D. , Druker, B. J. , & Corless, C. L. (2002). Inhibition of KIT tyrosine kinase activity: A novel molecular approach to the treatment of KIT‐positive malignancies. Journal of Clinical Oncology, 20(6), 1692–1703. [DOI] [PubMed] [Google Scholar]
- Holmes, E. C. , Morton, D. L. , & Ketcham, A. S. (1969). Parathyroid carcinoma: A collective review. Annals Surgery, 169(4), 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda, M. , Tsukada, T. , Tanaka, H. , Maruyama, K. , Yamaguchi, K. , Obara, T. , … Ishibashi, M. (2000). A novel mutation of the MEN1 gene in a Japanese kindred with familial isolated primary hyperparathyroidism. European Journal of Endocrinology, 142(2), 138–143. [DOI] [PubMed] [Google Scholar]
- Howe, J. R. , Norton, J. A. , & Wells, S. A., Jr. (1993). Prevalence of pheochromocytoma and hyperparathyroidism in multiple endocrine neoplasia type 2A: Results of long‐term follow‐up. Surgery, 114(6), 1070–1077. [PubMed] [Google Scholar]
- Howell, V. M. , Haven, C. J. , Kahnoski, K. , Khoo, S. K. , Petillo, D. , Chen, J. , … Teh, B. T. (2003). HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. Journal of Medical Genetics, 40(9), 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, V. M. , Gill, A. , Clarkson, A. , Nelson, A. E. , Dunne, R. , Delbridge, L. W. , … Marsh, D. J. (2009). Accuracy of combined protein gene product 9.5 and parafibromin markers for immunohistochemical diagnosis of parathyroid carcinoma. Journal of Clinical Endocrinology and Metabolism, 94(2), 434–441. [DOI] [PubMed] [Google Scholar]
- Hsi, E. D. , Zukerberg, L. R. , Yang, W. I. , & Arnold, A. (1996). Cyclin D1/PRAD1 expression in parathyroid adenomas: An immunohistochemical study. Journal of Clinical Endocrinology and Metabolism, 81(5), 1736–1739. [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Liao, Q. , Cao, S. , Gao, X. , & Zhao, Y. (2016). Diagnostic performance of parafibromin immunohistochemical staining for sporadic parathyroid carcinoma: A meta‐analysis. Endocrine, 54(3), 612–619. [DOI] [PubMed] [Google Scholar]
- Hundahl, S. A. , Fleming, I. D. , Fremgen, A. M. , & Menck, H. R. (1999). Two hundred eighty‐six cases of parathyroid carcinoma treated in the U.S. between 1985–1995: A National Cancer Data Base Report. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer, 86(3), 538–544. [DOI] [PubMed] [Google Scholar]
- Iacobone, M. , Barzon, L. , Porzionato, A. , Masi, G. , Macchi, V. , Marino, F. , … Favia, G. (2007). Parafibromin expression, single‐gland involvement, and limited parathyroidectomy in familial isolated hyperparathyroidism. Surgery, 142(6), 984–991. [DOI] [PubMed] [Google Scholar]
- Iacobone, M. , Masi, G. , Barzon, L. , Porzionato, A. , Macchi, V. , Ciarleglio, F. A. , … Favia, G. (2009). Hyperparathyroidism‐jaw tumor syndrome: A report of three large kindred. Langenbeck's Archives of Surgery, 394(5), 817–825. [DOI] [PubMed] [Google Scholar]
- Isakov, O. , Rinella, E. S. , Olchovsky, D. , Shimon, I. , Ostrer, H. , Shomron, N. , & Friedman, E. (2013). Missense mutation in the MEN1 gene discovered through whole exome sequencing co‐segregates with familial hyperparathyroidism. Genetics Research (Cambridge), 95(4), 114–120. [DOI] [PubMed] [Google Scholar]
- Jackson, C. E. (1958). Hereditary hyperparathyroidism associated with recurrent pancreatitis. Annals of Internal Medicine, 49(4), 829–836. [DOI] [PubMed] [Google Scholar]
- Jenkins, P. J. , Satta, M. A. , Simmgen, M. , Drake, W. M. , Williamson, C. , Lowe, D. G. , … Besser, G. M. (1997). Metastatic parathyroid carcinoma in the MEN2A syndrome. Clinical Endocrinology (Oxford), 47(6), 747–751. [DOI] [PubMed] [Google Scholar]
- Juhlin, C. , Larsson, C. , Yakoleva, T. , Leibiger, I. , Leibiger, B. , Alimov, A. , … Villablanca, A. (2006). Loss of parafibromin expression in a subset of parathyroid adenomas. Endocrine‐Related Cancer, 13(2), 509–523. [DOI] [PubMed] [Google Scholar]
- Juhlin, C. C. , Villablanca, A. , Sandelin, K. , Haglund, F. , Nordenstrom, J. , Forsberg, L. , … Hoog, A. (2007). Parafibromin immunoreactivity: Its use as an additional diagnostic marker for parathyroid tumor classification. Endocrine‐Related Cancer, 14(2), 501–512. [DOI] [PubMed] [Google Scholar]
- Juhlin, C. C. , Haglund, F. , Villablanca, A. , Forsberg, L. , Sandelin, K. , Branstrom, R. , … Hoog, A. (2009). Loss of expression for the Wnt pathway components adenomatous polyposis coli and glycogen synthase kinase 3‐β in parathyroid carcinomas. International Journal of Oncology, 34(2), 481–492. [PubMed] [Google Scholar]
- Juhlin, C. C. , Nilsson, I. L. , Johansson, K. , Haglund, F. , Villablanca, A. , Hoog, A. , & Larsson, C. (2010). Parafibromin and APC as screening markers for malignant potential in atypical parathyroid adenomas. Endocrine Pathology, 21(3), 166–177. [DOI] [PubMed] [Google Scholar]
- Juodele, L. , Serapinas, D. , Sabaliauskas, G. , Krasauskiene, A. , Krasauskas, V. , Verkauskiene, R. , … Juozaityte, E. (2011). Carcinoma of two parathyroid glands caused by a novel MEN1 gene mutation—A rare feature of the MEN 1 syndrome. Medicina (Kaunas), 47(11), 635–639. [PubMed] [Google Scholar]
- Kalavalapalli, S. , Talapatra, I. , & Connell, I. (2010). A complex case of multiple endocrine neoplasia type 1 with metastatic parathyroid carcinoma. Open Medicine, 5(1), 53–58. [Google Scholar]
- Kamitani‐Kawamoto, A. , Hamada, M. , Moriguchi, T. , Miyai, M. , Saji, F. , Hatamura, I. , … Kataoka, K. (2011). MafB interacts with Gcm2 and regulates parathyroid hormone expression and parathyroid development. Journal of Bone and Mineral Research, 26(10), 2463–2472. [DOI] [PubMed] [Google Scholar]
- Karges, W. , Jostarndt, K. , Maier, S. , Flemming, A. , Weitz, M. , Wissmann, A. , … Boehm, B. O. (2000). Multiple endocrine neoplasia type 1 (MEN1) gene mutations in a subset of patients with sporadic and familial primary hyperparathyroidism target the coding sequence but spare the promoter region. Journal of Endocrinology, 166(1), 1–9. [DOI] [PubMed] [Google Scholar]
- Kasaian, K. , Wiseman, S. M. , Thiessen, N. , Mungall, K. L. , Corbett, R. D. , Qian, J. Q. , … Jones, S. J. (2013). Complete genomic landscape of a recurring sporadic parathyroid carcinoma. Journal of Pathology, 230(3), 249–260. [DOI] [PubMed] [Google Scholar]
- Kassem, M. , Kruse, T. A. , Wong, F. K. , Larsson, C. , & Teh, B. T. (2000). Familial isolated hyperparathyroidism as a variant of multiple endocrine neoplasia type 1 in a large Danish pedigree. Journal of Clinical Endocrinology and Metabolism, 85(1), 165–167. [DOI] [PubMed] [Google Scholar]
- Kelly, T. G. , Shattuck, T. M. , Reyes‐Mugica, M. , Stewart, A. F. , Simonds, W. F. , Udelsman, R. , … Carpenter, T. O. (2006). Surveillance for early detection of aggressive parathyroid disease: Carcinoma and atypical adenoma in familial isolated hyperparathyroidism associated with a germline HRPT2 mutation. Journal of Bone and Mineral Research, 21(10), 1666–1671. [DOI] [PubMed] [Google Scholar]
- Khadilkar, K. S. , Budyal, S. R. , Kasliwal, R. , Lila, A. R. , Bandgar, T. , & Shah, N. S. (2015). Hrpt2‐ (Cdc73) related hereditary hyperparathyroidism: A case series from western India. Endocrine Practice, 21(9), 1010–1016. [DOI] [PubMed] [Google Scholar]
- Kimura, E. T. , Nikiforova, M. N. , Zhu, Z. , Knauf, J. A. , Nikiforov, Y. E. , & Fagin, J. A. (2003). High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC‐RAS‐BRAF signaling pathway in papillary thyroid carcinoma. Cancer Research, 63(7), 1454–1457. [PubMed] [Google Scholar]
- Kishikawa, S. , Shan, L. , Ogihara, K. , Utsunomiya, H. , Nakamura, M. , Nakamura, Y. , … Kakudo, K. (1999). Overexpression and genetic abnormality of p53 in parathyroid adenomas. Pathology International, 49(10), 853–857. [DOI] [PubMed] [Google Scholar]
- Knudson, A. G., Jr. (1971). Mutation and cancer: Statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America, 68(4), 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, J. , Wang, O. , Nie, M. , Shi, J. , Hu, Y. , Jiang, Y. , … Xing, X. (2014). Familial isolated primary hyperparathyroidism/hyperparathyroidism‐jaw tumour syndrome caused by germline gross deletion or point mutations of CDC73 gene in Chinese. Clinical Endocrinology (Oxford), 81(2), 222–230. [DOI] [PubMed] [Google Scholar]
- Korpi‐Hyovalti, E. , Cranston, T. , Ryhanen, E. , Arola, J. , Aittomaki, K. , Sane, T. , … Schalin‐Jantti, C. (2014). CDC73 intragenic deletion in familial primary hyperparathyroidism associated with parathyroid carcinoma. Journal of Clinical Endocrinology and Metabolism, 99(9), 3044–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs, L. J. , Shattuck, T. M. , & Arnold, A. (2005). HRPT2 mutational analysis of typical sporadic parathyroid adenomas. Journal of Clinical Endocrinology and Metabolism, 90(9), 5015–5017. [DOI] [PubMed] [Google Scholar]
- Kumari, N. , Chaudhary, N. , Pradhan, R. , Agarwal, A. , & Krishnani, N. (2016). Role of histological criteria and immunohistochemical markers in predicting risk of malignancy in parathyroid neoplasms. Endocrine Pathology, 27(2), 87–96. [DOI] [PubMed] [Google Scholar]
- Kutcher, M. R. , Rigby, M. H. , Bullock, M. , Trites, J. , Taylor, S. M. , & Hart, R. D. (2013). Hyperparathyroidism‐jaw tumor syndrome. Head and Neck, 35(6), E175–177. [DOI] [PubMed] [Google Scholar]
- Lee, K. M. , Kim, E. J. , Choi, W. S. , Park, W. S. , & Kim, S. W. (2014). Intrathyroidal parathyroid carcinoma mimicking a thyroid nodule in a MEN type 1 patient. Journal of Clinical Ultrasound, 42(4), 212–214. [DOI] [PubMed] [Google Scholar]
- Lee, P. K. , Jarosek, S. L. , Virnig, B. A. , Evasovich, M. , & Tuttle, T. M. (2007). Trends in the incidence and treatment of parathyroid cancer in the United States. Cancer, 109(9), 1736–1741. [DOI] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … Exome Aggregation, C. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos, M. C. , & Thakker, R. V. (2008). Multiple endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Human Mutation, 29(1), 22–32. [DOI] [PubMed] [Google Scholar]
- Lemos, M. C. , Harding, B. , Reed, A. A. , Jeyabalan, J. , Walls, G. V. , Bowl, M. R. , … Thakker, R. V. (2009). Genetic background influences embryonic lethality and the occurrence of neural tube defects in Men1 null mice: Relevance to genetic modifiers. Journal of Endocrinology, 203(1), 133–142. [DOI] [PubMed] [Google Scholar]
- Li, X. , Gonzalez, M. E. , Toy, K. , Filzen, T. , Merajver, S. D. , & Kleer, C. G. (2009). Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. American Journal of Pathology, 175(3), 1246–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, D. J. , Hahn, M. A. , Howell, V. M. , & Gill, A. J. (2007). Molecular diagnosis of primary hyperparathyroidism in familial cancer syndromes. Expert Opinion on Medical Diagnostics, 1(3), 377–392. [DOI] [PubMed] [Google Scholar]
- Marx, S. J. (2000). Hyperparathyroid and hypoparathyroid disorders. New England Journal of Medicine, 343(25), 1863–1875. [DOI] [PubMed] [Google Scholar]
- Masi, G. , Barzon, L. , Iacobone, M. , Viel, G. , Porzionato, A. , Macchi, V. , … Palu, G. (2008). Clinical, genetic, and histopathologic investigation of CDC73‐related familial hyperparathyroidism. Endocrine‐Related Cancer, 15(4), 1115–1126. [DOI] [PubMed] [Google Scholar]
- Masi, G. , Iacobone, M. , Sinigaglia, A. , Mantelli, B. , Pennelli, G. , Castagliuolo, I. , … Barzon, L. (2014). Characterization of a new CDC73 missense mutation that impairs parafibromin expression and nucleolar localization. PLoS One, 9(5), e97994 https://doi.org/10.1371/journal.pone.0097994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew, C. G. , Chin, K. S. , Easton, D. F. , Thorpe, K. , Carter, C. , Liou, G. I. , … Ponder, B. A. J. (1987). A linked genetic marker for multiple endocrine neoplasia type 2A on chromosome 10. Nature, 328(6130), 527–528. [DOI] [PubMed] [Google Scholar]
- Mathews, J. W. , Winchester, R. , Alsaygh, N. , Bartlett, A. M. , & Luttrell, L. (2016). Hyperparathyroidism‐jaw tumor syndrome: An overlooked cause of severe hypercalcemia. American Journal of the Medical Sciences, 352(3), 302–305. [DOI] [PubMed] [Google Scholar]
- Mehlen, P. , & Thibert, C. (2004). Dependence receptors: Between life and death. Cellular and Molecular Life Sciences, 61(15), 1854–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta, A. , Patel, D. , Rosenberg, A. , Boufraqech, M. , Ellis, R. J. , Nilubol, N. , … Kebebew, E. (2014). Hyperparathyroidism‐jaw tumor syndrome: Results of operative management. Surgery, 156(6), 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele, M. , Rolighed, L. , Jespersen, M. , Rejnmark, L. , & Christiansen, P. (2016). Recurrence of hyperparathyroid hypercalcemia in a patient with the HRPT‐2 mutation and a previous parathyroid carcinoma in hyperparathyroidism‐jaw tumor syndrome. International Journal of Endocrinology and Metabolism, 14(2), e35424 https://doi.org/10.5812/Ijem.35424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer‐Rochow, G. Y. , Alvarado, R. , Sywak, M. S. , Sidhu, S. B. , Delbridge, L. W. , & Gill, A. J. (2007). Letter 2: Intraoperative diagnosis and treatment of parathyroid cancer and atypical parathyroid adenoma (Br J Surg 2007; 94: 566–570). British Journal of Surgery, 94(8), 1043; author reply 1043–1044. [DOI] [PubMed] [Google Scholar]
- Miedlich, S. , Lohmann, T. , Schneyer, U. , Lamesch, P. , & Paschke, R. (2001). Familial isolated primary hyperparathyroidism—A multiple endocrine neoplasia type 1 variant? European Journal of Endocrinology, 145(2), 155–160. [DOI] [PubMed] [Google Scholar]
- Minikel, E. V. , Vallabh, S. M. , Lek, M. , Estrada, K. , Samocha, K. E. , Sathirapongsasuti, J. F. , … MacArthur, D. G. (2016). Quantifying prion disease penetrance using large population control cohorts. Science Translational Medicine, 8(322), 322ra329 https://doi.org/10.1126/scitranslmed.aad5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizusawa, N. , Uchino, S. , Iwata, T. , Tsuyuguchi, M. , Suzuki, Y. , Mizukoshi, T. , … Yoshimoto, K. (2006). Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism‐jaw tumour syndrome. Clinical Endocrinology (Oxford), 65(1), 9–16. [DOI] [PubMed] [Google Scholar]
- Moon, S. D. , Park, J. H. , Kim, E. M. , Kim, J. H. , Han, J. H. , Yoo, S. J. , … Cha, B. Y. (2005). A Novel IVS2‐1G>A mutation causes aberrant splicing of the HRPT2 gene in a family with hyperparathyroidism‐jaw tumor syndrome. Journal of Clinical Endocrinology and Metabolism, 90(2), 878–883. [DOI] [PubMed] [Google Scholar]
- Mosimann, C. , Hausmann, G. , & Basler, K. (2006). Parafibromin/Hyrax activates Wnt/Wg target gene transcription by direct association with β‐catenin/Armadillo. Cell, 125(2), 327–341. [DOI] [PubMed] [Google Scholar]
- Mulligan, L. M. , Kwok, J. B. , Healey, C. S. , Elsdon, M. J. , Eng, C. , Gardner, E. , … Ponder, B. A. J. (1993). Germ‐line mutations of the RET proto‐oncogene in multiple endocrine neoplasia type 2A. Nature, 363(6428), 458–460. [DOI] [PubMed] [Google Scholar]
- Nagamura, Y. , Yamazaki, M. , Shimazu, S. , Sano, K. , Tsukada, T. , & Sakurai, A. (2012). A novel splice site mutation of the MEN1 gene identified in a patient with primary hyperparathyroidism. Endocrine Journal, 59(6), 523–530. [DOI] [PubMed] [Google Scholar]
- Newey, P. J. , Bowl, M. R. , Cranston, T. , & Thakker, R. V. (2010). Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism‐jaw tumor syndrome (HPT‐JT) and parathyroid tumors. Human Mutation, 31(3), 295–307. [DOI] [PubMed] [Google Scholar]
- Newey, P. J. , & Thakker, R. V. (2011). Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice. Endocrine Practice, 17(Suppl 3), 8–17. [DOI] [PubMed] [Google Scholar]
- Newey, P. J. , Nesbit, M. A. , Rimmer, A. J. , Attar, M. , Head, R. T. , Christie, P. T. , … Thakker, R. V. (2012). Whole‐exome sequencing studies of nonhereditary (sporadic) parathyroid adenomas. Journal of Clinical Endocrinology and Metabolism, 97(10), E1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, J. W. , Lama, D. , Lukman, S. , Lane, D. P. , Verma, C. S. , & Sim, A. Y. (2015). R248Q mutation ‐ beyond p53‐DNA binding. Proteins, 83(12), 2240–2250. [DOI] [PubMed] [Google Scholar]
- Ohye, H. , Sato, M. , Matsubara, S. , Miyauchi, A. , Imachi, H. , Murao, K. , & Takahara, J. (1998). Germline mutation of the multiple endocrine neoplasia type 1 (MEN1) gene in a family with primary hyperparathyroidism. Endocrine Journal, 45(6), 719–723. [DOI] [PubMed] [Google Scholar]
- Paez, J. G. , Janne, P. A. , Lee, J. C. , Tracy, S. , Greulich, H. , Gabriel, S. , … Meyerson, M. (2004). EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science, 304(5676), 1497–1500. [DOI] [PubMed] [Google Scholar]
- Panicker, L. M. , Zhang, J. H. , Dagur, P. K. , Gastinger, M. J. , & Simonds, W. F. (2010). Defective nucleolar localization and dominant interfering properties of a parafibromin L95P missense mutant causing the hyperparathyroidism‐jaw tumor syndrome. Endocrine‐Related Cancer, 17(2), 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannett, A. A. , Kennedy, A. M. , Turner, J. J. , Forbes, S. A. , Cavaco, B. M. , Bassett, J. H. , … Thakker, R. V. (2003). Multiple endocrine neoplasia type 1 (MEN1) germline mutations in familial isolated primary hyperparathyroidism. Clinical Endocrinology (Oxford), 58(5), 639–646. [DOI] [PubMed] [Google Scholar]
- Parfitt, J. , Harris, M. , Wright, J. M. , & Kalamchi, S. (2015). Tumor suppressor gene mutation in a patient with a history of hyperparathyroidism‐jaw tumor syndrome and healed generalized osteitis fibrosa cystica: A case report and genetic pathophysiology review. Journal of Oral and Maxillofacial Surgery, 73(1), 194 e191‐199. [DOI] [PubMed] [Google Scholar]
- Pazienza, V. , la Torre, A. , Baorda, F. , Alfarano, M. , Chetta, M. , Muscarella, L. A. , … Guarnieri, V. (2013). Identification and functional characterization of three NoLS (nucleolar localisation signals) mutations of the CDC73 gene. PLoS One, 8(12), e82292 https://doi.org/10.1371/journal.pone.0082292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce, S. H. , Trump, D. , Wooding, C. , Sheppard, M. N. , Clayton, R. N. , & Thakker, R. V. (1996). Loss of heterozygosity studies at the retinoblastoma and breast cancer susceptibility (BRCA2) loci in pituitary, parathyroid, pancreatic and carcinoid tumours. Clinical Endocrinology (Oxford), 45(2), 195–200. [DOI] [PubMed] [Google Scholar]
- Petitjean, A. , Mathe, E. , Kato, S. , Ishioka, C. , Tavtigian, S. V. , Hainaut, P. , & Olivier, M. (2007). Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Human Mutation, 28(6), 622–629. [DOI] [PubMed] [Google Scholar]
- Pichardo‐Lowden, A. R. , Manni, A. , Saunders, B. D. , & Baker, M. J. (2011). Familial hyperparathyroidism due to a germline mutation of the CDC73 gene: Implications for management and age‐appropriate testing of relatives at risk. Endocrine Practice, 17(4), 602–609. [DOI] [PubMed] [Google Scholar]
- Pimenta, F. J. , Gontijo Silveira, L. F. , Tavares, G. C. , Silva, A. C. , Perdigao, P. F. , Castro, W. H. , … Gomez, R. S. (2006). HRPT2 gene alterations in ossifying fibroma of the jaws. Oral Oncology, 42(7), 735–739. [DOI] [PubMed] [Google Scholar]
- Plaza‐Menacho, I. , Mologni, L. , & McDonald, N. Q. (2014). Mechanisms of RET signaling in cancer: Current and future implications for targeted therapy. Cellular Signalling, 26(8), 1743–1752. [DOI] [PubMed] [Google Scholar]
- Poncin, J. , Abs, R. , Velkeniers, B. , Bonduelle, M. , Abramowicz, M. , Legros, J. J. , … Beckers, A. (1999). Mutation analysis of the MEN1 gene in Belgian patients with multiple endocrine neoplasia type 1 and related diseases. Human Mutation, 13(1), 54–60. [DOI] [PubMed] [Google Scholar]
- Pontikides, N. , Karras, S. , Kaprara, A. , Anagnostis, P. , Mintziori, G. , Goulis, D. G. , … Krassas, G. (2014). Genetic basis of familial isolated hyperparathyroidism: A case series and a narrative review of the literature. Journal of Bone and Mineral Metabolism, 32(4), 351–366. [DOI] [PubMed] [Google Scholar]
- Posada‐Gonzalez, M. , Gomez‐Ramirez, J. , Luque‐Ramirez, M. , Guijarro, M. , Martin‐Perez, E. , Rodriguez‐Sanchez, A. , … Larranaga, E. (2014). Nonfunctional metastatic parathyroid carcinoma in the setting of multiple endocrine neoplasia Type 2A syndrome. Surgery Research and Practice, 2014, 731481 https://doi.org/10.1155/2014/731481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quervain, F. (1909). Parastruma maligna aberrata. Deutsche Zeitschrift für Chirurgie, 100(1), 334–353. [Google Scholar]
- Rahbari, R. , Holloway, A. K. , He, M. , Khanafshar, E. , Clark, O. H. , & Kebebew, E. (2011). Identification of differentially expressed microRNA in parathyroid tumors. Annals of Surgical Oncology, 18(4), 1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raue, F. , Haag, C. , & Frank‐Raue, K. (2007). Hyperparathyreoidismus‐Kiefertumor‐Syndrom. Eine hereditäre Form des primären Hyperparathyreoidismus mit Nebenschilddrüsenkarzinom. Deutsche Medizinische Wochenschrift, 132(27), 1459–1462. [DOI] [PubMed] [Google Scholar]
- Raue, F. , & Frank‐Raue, K. (2012). Genotype‐phenotype correlation in multiple endocrine neoplasia type 2. Clinics (Sao Paulo), 67(Suppl 1), 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea, S. , Eisenhaber, F. , O'Carroll, D. , Strahl, B. D. , Sun, Z. W. , Schmid, M. , … Jenuwein, T. (2000). Regulation of chromatin structure by site‐specific histone H3 methyltransferases. Nature, 406(6796), 593–599. [DOI] [PubMed] [Google Scholar]
- Rekik, N. , Ben Naceur, B. , Mnif, M. , Mnif, F. , Mnif, H. , Boudawara, T. , & Abid, M. (2010). Hyperparathyroidism‐jaw tumor syndrome: A case report. Annals of Endocrinology (Paris), 71(2), 121–126. [DOI] [PubMed] [Google Scholar]
- Rozenblatt‐Rosen, O. , Hughes, C. M. , Nannepaga, S. J. , Shanmugam, K. S. , Copeland, T. D. , Guszczynski, T. , … Meyerson, M. (2005). The parafibromin tumor suppressor protein is part of a human Paf1 complex. Molecular and Cellular Biology, 25(2), 612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin, M. R. , Bilezikian, J. P. , Birken, S. , & Silverberg, S. J. (2008). Human chorionic gonadotropin measurements in parathyroid carcinoma. European and Journal of Endocrinology, 159(4), 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruda, J. M. , Hollenbeak, C. S. , & Stack, B. C., Jr. (2005). A systematic review of the diagnosis and treatment of primary hyperparathyroidism from 1995 to 2003. Otolaryngology Head and Neck Surgery, 132(3), 359–372. [DOI] [PubMed] [Google Scholar]
- Salameh, A. , Lee, A. K. , Cardo‐Vila, M. , Nunes, D. N. , Efstathiou, E. , Staquicini, F. I. , … Arap, W. (2015). PRUNE2 is a human prostate cancer suppressor regulated by the intronic long noncoding RNA PCA3. Proceedings of the National Academy of Sciences of the United States of America, 112(27), 8403–8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanpaolo, E. , Miroballo, M. , Corbetta, S. , Verdelli, C. , Baorda, F. , Balsamo, T. , … Guarnieri, V. (2016). EZH2 and ZFX oncogenes in malignant behaviour of parathyroid neoplasms. Endocrine, 54(1), 55–59. [DOI] [PubMed] [Google Scholar]
- Sarquis, M. S. , Silveira, L. G. , Pimenta, F. J. , Dias, E. P. , Teh, B. T. , Friedman, E. , … De Marco, L. (2008). Familial hyperparathyroidism: Surgical outcome after 30 years of follow‐up in three families with germline HRPT2 mutations. Surgery, 143(5), 630–640. [DOI] [PubMed] [Google Scholar]
- Sato, M. , Matsubara, S. , Miyauchi, A. , Ohye, H. , Imachi, H. , Murao, K. , & Takahara, J. (1998). Identification of five novel germline mutations of the MEN1 gene in Japanese multiple endocrine neoplasia type 1 (MEN1) families. Journal of Medical Genetics, 35(11), 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, M. , Miyauchi, A. , Namihira, H. , Bhuiyan, M. M. , Imachi, H. , Murao, K. , & Takahara, J. (2000). A newly recognized germline mutation of MEN1 gene identified in a patient with parathyroid adenoma and carcinoma. Endocrine, 12(3), 223–226. [DOI] [PubMed] [Google Scholar]
- Schaapveld, M. , Jorna, F. H. , Aben, K. K. , Haak, H. R. , Plukker, J. T. , & Links, T. P. (2011). Incidence and prognosis of parathyroid gland carcinoma: A population‐based study in the Netherlands estimating the preoperative diagnosis. American Journal of Surgery, 202(5), 590–597. [DOI] [PubMed] [Google Scholar]
- Schantz, A. , & Castleman, B. (1973). Parathyroid carcinoma. A study of 70 cases. Cancer, 31(3), 600–605. [DOI] [PubMed] [Google Scholar]
- Schmidt, B. P. , Bradrick, J. P. , & Gabali, A. (2009). Hyperparathyroidism‐jaw tumor syndrome: A case report. Journal of Oral and Maxillofacial Surgery, 67(2), 423–427. [DOI] [PubMed] [Google Scholar]
- Semba, S. , Kusumi, R. , Moriya, T. , & Sasano, H. (2000). Nuclear accumulation of β‐catenin in human endocrine tumors: Association with Ki‐67 (MIB‐1) proliferative activity. Endocrine Pathology, 11(3), 243–250. [DOI] [PubMed] [Google Scholar]
- Serrano‐Gonzalez, M. , Shay, S. , Austin, J. , Maceri, D. R. , & Pitukcheewanont, P. (2016). A germline mutation of HRPT2/CDC73 (70 G>T) in an adolescent female with parathyroid carcinoma: First case report and a review of the literature. Journal of Pediatric Endocrinology and Metabolism, 29(9), 1005–1012. [DOI] [PubMed] [Google Scholar]
- Shane, E. , & Bilezikian, J. P. (1982). Parathyroid carcinoma: A review of 62 patients. Endocrine Reviews, 3(2), 218–226. [DOI] [PubMed] [Google Scholar]
- Shattuck, T. M. , Kim, T. S. , Costa, J. , Yandell, D. W. , Imanishi, Y. , Palanisamy, N. , … Arnold, A. (2003a). Mutational analyses of RB and BRCA2 as candidate tumour suppressor genes in parathyroid carcinoma. Clinical Endocrinology (Oxford), 59(2), 180–189. [DOI] [PubMed] [Google Scholar]
- Shattuck, T. M. , Valimaki, S. , Obara, T. , Gaz, R. D. , Clark, O. H. , Shoback, D. , … Arnold, A. (2003b). Somatic and germ‐line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. New England Journal of Medicine, 349(18), 1722–1729. [DOI] [PubMed] [Google Scholar]
- Shih, R. Y. , Fackler, S. , Maturo, S. , True, M. W. , Brennan, J. , & Wells, D. (2009). Parathyroid carcinoma in multiple endocrine neoplasia type 1 with a classic germline mutation. Endocrine Practice, 15(6), 567–572. [DOI] [PubMed] [Google Scholar]
- Shimizu, S. , Tsukada, T. , Futami, H. , Ui, K. , Kameya, T. , Kawanaka, M. , … Yamaguchi, K. (1997). Germline mutations of the MEN1 gene in Japanese kindred with multiple endocrine neoplasia type 1. Japanese Journal of Cancer Research, 88(11), 1029–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira, L. G. , Dias, E. P. , Marinho, B. C. , Gomez, R. S. , De Marco, L. , & Sarquis, M. S. (2008). HRPT2‐related familial isolated hyperparathyroidism: Could molecular studies direct the surgical approach? Arquivos Brasileiros de Endocrinologia & Metabologia, 52(8), 1211–1220. [DOI] [PubMed] [Google Scholar]
- Silverberg, S. J. , Shane, E. , Jacobs, T. P. , Siris, E. S. , Gartenberg, F. , Seldin, D. , … Bilezikian, J. P. (1990). Nephrolithiasis and bone involvement in primary hyperparathyroidism. American Journal of Medicine, 89(3), 327–334. [DOI] [PubMed] [Google Scholar]
- Simonds, W. F. , James‐Newton, L. A. , Agarwal, S. K. , Yang, B. , Skarulis, M. C. , Hendy, G. N. , & Marx, S. J. (2002). Familial isolated hyperparathyroidism: Clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore), 81(1), 1–26. [DOI] [PubMed] [Google Scholar]
- Simonds, W. F. , Robbins, C. M. , Agarwal, S. K. , Hendy, G. N. , Carpten, J. D. , & Marx, S. J. (2004). Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism‐jaw tumor syndrome. Journal of Clinical Endocrinology and Metabolism, 89(1), 96–102. [DOI] [PubMed] [Google Scholar]
- Simpson, N. E. , Kidd, K. K. , Goodfellow, P. J. , McDermid, H. , Myers, S. , Kidd, J. R. , … White, B. N. (1987). Assignment of multiple endocrine neoplasia type 2A to chromosome 10 by linkage. Nature, 328(6130), 528–530. [DOI] [PubMed] [Google Scholar]
- Singh Ospina, N. , Sebo, T. J. , Thompson, G. B. , Clarke, B. L. , & Young, W. F., Jr. (2016). Prevalence of parathyroid carcinoma in 348 patients with multiple endocrine neoplasia type 1 ‐ case report and review of the literature. Clinical Endocrinology (Oxford), 84, 244–249. [DOI] [PubMed] [Google Scholar]
- Siu, W. K. , Law, C. Y. , Lam, C. W. , Mak, C. M. , Wong, G. W. , Ho, A. Y. , … Chan, A. Y. (2011). Novel nonsense CDC73 mutations in Chinese patients with parathyroid tumors. Familial Cancer, 10(4), 695–699. [DOI] [PubMed] [Google Scholar]
- Sriphrapradang, C. , Sornmayura, P. , Chanplakorn, N. , Trachoo, O. , Sae‐Chew, P. , & Aroonroch, R. (2014). Fine‐needle aspiration cytology of parathyroid carcinoma mimic hurthle cell thyroid neoplasm. Case Reports in Endocrinology, 2014, 680876 https://doi.org/10.1155/2014/680876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starker, L. F. , Svedlund, J. , Udelsman, R. , Dralle, H. , Akerstrom, G. , Westin, G. , … Carling, T. (2011). The DNA methylome of benign and malignant parathyroid tumors. Genes, Chromosomes and Cancer, 50(9), 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulaiman, L. , Juhlin, C. C. , Nilsson, I. L. , Fotouhi, O. , Larsson, C. , & Hashemi, J. (2013). Global and gene‐specific promoter methylation analysis in primary hyperparathyroidism. Epigenetics, 8(6), 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedlund, J. , Auren, M. , Sundstrom, M. , Dralle, H. , Akerstrom, G. , Bjorklund, P. , & Westin, G. (2010). Aberrant WNT/β‐catenin signaling in parathyroid carcinoma. Molecular Cancer, 9, 294 https://doi.org/10.1186/1476-4598-9-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedlund, J. , Koskinen Edblom, S. , Marquez, V. E. , Akerstrom, G. , Bjorklund, P. , & Westin, G. (2012). Hypermethylated in cancer 1 (HIC1), a tumor suppressor gene epigenetically deregulated in hyperparathyroid tumors by histone H3 lysine modification. Journal of Clinical Endocrinology and Metabolism, 97(7), E1307–1315. [DOI] [PubMed] [Google Scholar]
- Svedlund, J. , Barazeghi, E. , Stalberg, P. , Hellman, P. , Akerstrom, G. , Bjorklund, P. , & Westin, G. (2014). The histone methyltransferase EZH2, an oncogene common to benign and malignant parathyroid tumors. Endocrine‐Related Cancer, 21(2), 231–239. [DOI] [PubMed] [Google Scholar]
- Szijan, I. , Orlow, I. , Dalamon, V. , Vergani, P. , Danilowicz, K. , Mezzadri, N. , … Bruno, O. D. (2000). Alterations in the retinoblastoma pathway of cell cycle control in parathyroid tumors. Oncology Reports, 7(2), 421–425. [DOI] [PubMed] [Google Scholar]
- Takami, H. , Shirahama, S. , Ikeda, Y. , Sasaki, Y. , Wada, N. , Niimi, M. , & Kameyama, K. (2000). Familial hyperparathyroidism. Biomedicine and Pharmacotherapy, 54(Suppl 1), 21–24. [DOI] [PubMed] [Google Scholar]
- Takeuchi, T. , Yoto, Y. , Tsugawa, T. , Kamasaki, H. , Kondo, A. , Ogino, J. , … Tsutsumi, H. (2015). An adolescent case of familial hyperparathyroidism with a germline frameshift mutation of the CDC73 gene. Clinical Pediatric Endocrinology, 24(4), 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, G. , Miyoshi, H. , Ogata, S. Y. , Sasou, S. , Kudoh, S. , Kikuchi, J. , … Motoyama, T. (2009). Parathyroid carcinoma with anaplastic feature: Association of a p53 gene mutation with anaplastic transformation. Pathology International, 59(2), 107–110. [DOI] [PubMed] [Google Scholar]
- Tan, M. H. , Morrison, C. , Wang, P. , Yang, X. , Haven, C. J. , Zhang, C. , … Teh, B. T. (2004). Loss of parafibromin immunoreactivity is a distinguishing feature of parathyroid carcinoma. Clinical Cancer Research, 10(19), 6629–6637. [DOI] [PubMed] [Google Scholar]
- Teh, B. T. , Esapa, C. T. , Houlston, R. , Grandell, U. , Farnebo, F. , Nordenskjold, M. , … Harris, P. E. (1998). A family with isolated hyperparathyroidism segregating a missense MEN1 mutation and showing loss of the wild‐type alleles in the parathyroid tumors. American Journal of Human Genetics, 63(5), 1544–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh, B. T. , Howell, V. M. , Haven, C. J. , Kahnoski, K. , Khoo, S. K. , Petillo, D. , … Morreau, H. (2004). Human gene mutations. Gene symbol: HRPT2. Disease: Hyperparathyroidism jaw‐tumor syndrome. Human Genetics, 114(2), 221–224. [PubMed] [Google Scholar]
- Thakker, R. V. , Bouloux, P. , Wooding, C. , Chotai, K. , Broad, P. M. , Spurr, N. K. , … O'Riordan, J. L. (1989). Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. New England Journal of Medicine, 321(4), 218–224. [DOI] [PubMed] [Google Scholar]
- Thakker, R. V. (1993). The molecular genetics of the multiple endocrine neoplasia syndromes. Clinical Endocrinology (Oxford), 38(1), 1–14. [DOI] [PubMed] [Google Scholar]
- Thakker, R. V. (1998). Multiple endocrine neoplasia ‐ syndromes of the twentieth century. Journal of Clinical Endocrinology and Metabolism, 83(8), 2617–2620. [DOI] [PubMed] [Google Scholar]
- Thakker, R. V. (2004). Diseases associated with the extracellular calcium‐sensing receptor. Cell Calcium, 35(3), 275–282. [DOI] [PubMed] [Google Scholar]
- Thakker, R. V. (2010). Multiple endocrine neoplasia type 1 (MEN1). Best Practice & Research: Clinical Endocrinology and Metabolism, 24(3), 355–370. [DOI] [PubMed] [Google Scholar]
- Thakker, R. V. , Newey, P. J. , Walls, G. V. , Bilezikian, J. , Dralle, H. , Ebeling, P. R. , … Brandi, M. L. (2012). Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). Journal of Clinical Endocrinology and Metabolism, 97(9), 2990–3011. [DOI] [PubMed] [Google Scholar]
- Thakker, R. V. (2014). Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Molecular and Cellular Endocrinology, 386(1‐2), 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker, R. V. (2016). Genetics of parathyroid tumours. Journal of Internal Medicine, 280(6), 574–583. [DOI] [PubMed] [Google Scholar]
- Tham, E. , Grandell, U. , Lindgren, E. , Toss, G. , Skogseid, B. , & Nordenskjold, M. (2007). Clinical testing for mutations in the MEN1 gene in Sweden: A report on 200 unrelated cases. Journal of Clinical Endocrinology and Metabolism, 92(9), 3389–3395. [DOI] [PubMed] [Google Scholar]
- The Universal Mutation Database . (2017). The UMD‐MEN1 mutations database Retrieved from https://www.umd.be/MEN1/ [accessed 14/08/2017).
- Trump, D. , Farren, B. , Wooding, C. , Pang, J. T. , Besser, G. M. , Buchanan, K. D. , … Thakker, R. V. (1996). Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM, 89(9), 653–669. [DOI] [PubMed] [Google Scholar]
- Vaira, V. , Elli, F. , Forno, I. , Guarnieri, V. , Verdelli, C. , Ferrero, S. , … Corbetta, S. (2012). The microRNA cluster C19MC is deregulated in parathyroid tumours. Journal of Molecular Endocrinology, 49(2), 115–124. [DOI] [PubMed] [Google Scholar]
- Vasef, M. A. , Brynes, R. K. , Sturm, M. , Bromley, C. , & Robinson, R. A. (1999). Expression of cyclin D1 in parathyroid carcinomas, adenomas, and hyperplasias: A paraffin immunohistochemical study. Modern Pathology, 12(4), 412–416. [PubMed] [Google Scholar]
- Veiguela, B. , Isidro, M. L. , Jorge, S. , & Ruano, B. (2010). Una causa rara de hipercalcemia: Carcinoma sincrónico de dos paratiroides en el contexto del síndrome de hiperparatiroidismo familiar‐tumor mandibular. Endocrinologia y Nutricion, 57(8), 391–393. [DOI] [PubMed] [Google Scholar]
- Villablanca, A. , Wassif, W. S. , Smith, T. , Hoog, A. , Vierimaa, O. , Kassem, M. , … Larsson, C. (2002). Involvement of the MEN1 gene locus in familial isolated hyperparathyroidism. European Journal of Endocrinology, 147(3), 313–322. [DOI] [PubMed] [Google Scholar]
- Villablanca, A. , Calender, A. , Forsberg, L. , Hoog, A. , Cheng, J. D. , Petillo, D. , … Larsson, C. (2004). Germline and de novo mutations in the HRPT2 tumour suppressor gene in familial isolated hyperparathyroidism (FIHP). Journal of Medical Genetics, 41(3), e32 https://doi.org/10.1136/jmg.2003.012369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vire, E. , Brenner, C. , Deplus, R. , Blanchon, L. , Fraga, M. , Didelot, C. , … Fuks, F. (2006). The polycomb group protein EZH2 directly controls DNA methylation. Nature, 439(7078), 871–874. [DOI] [PubMed] [Google Scholar]
- Walsh, R. , Thomson, K. L. , Ware, J. S. , Funke, B. H. , Woodley, J. , McGuire, K. J. , … Watkins, H. (2017). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genetics in Medicine, 19(2), 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. A. , & Gaz, R. D. (1985). Natural history of parathyroid carcinoma. Diagnosis, treatment, and results. American Journal of Surgery, 149(4), 522–527. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Han, D. , Chen, W. , Zhang, S. , Wang, Z. , Li, K. , … Yang, A. (2015). Non‐functional parathyroid carcinoma: A case report and review of the literature. Cancer Biology and Therapy, 16(11), 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, O. , Wang, C. , Nie, M. , Cui, Q. , Guan, H. , Jiang, Y. , … Xing, X. (2012). Novel HRPT2/CDC73 gene mutations and loss of expression of parafibromin in Chinese patients with clinically sporadic parathyroid carcinomas. PLoS One, 7(9), e45567 https://doi.org/10.1371/journal.pone.0045567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P. , Bowl, M. R. , Bender, S. , Peng, J. , Farber, L. , Chen, J. , … Teh, B. T. (2008). Parafibromin, a component of the human PAF complex, regulates growth factors and is required for embryonic development and survival in adult mice. Molecular and Cellular Biology, 28(9), 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner, J. , Epstein, M. , Sweet, A. , Singh, D. , Burgess, J. , Stranks, S. , … Cardinal, J. (2004). Genetic testing in familial isolated hyperparathyroidism: Unexpected results and their implications. Journal of Medical Genetics, 41(3), 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner, J. V. , Nyholt, D. R. , Busfield, F. , Epstein, M. , Burgess, J. , Stranks, S. , … Cardinal, J. W. (2006). Familial isolated hyperparathyroidism is linked to a 1.7 Mb region on chromosome 2p13.3‐14. Journal of Medical Genetics, 43(3), e12 https://doi.org/10.1136/jmg.2005.035766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wautot, V. , Vercherat, C. , Lespinasse, J. , Chambe, B. , Lenoir, G. M. , Zhang, C. X. , … Calender, A. (2002). Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: Search for correlation between phenotype and the functional domains of the MEN1 protein. Human Mutation, 20(1), 35–47. [DOI] [PubMed] [Google Scholar]
- Wells, S. A., Jr. , Asa, S. L. , Dralle, H. , Elisei, R. , Evans, D. B. , & Gagel, R. F. (2015). American Thyroid Association Guidelines Task Force on Medullary Thyroid C Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid, 25(6), 567–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, C. , Cavaco, B. M. , Jauch, A. , Dixon, P. H. , Forbes, S. , Harding, B. , … Thakker, R. V. (1999). Mapping the gene causing hereditary primary hyperparathyroidism in a Portuguese kindred to chromosome 1q22‐q31. Journal of Bone and Mineral Research, 14(2), 230–239. [DOI] [PubMed] [Google Scholar]
- Willmore‐Payne, C. , Holden, J. A. , Tripp, S. , & Layfield, L. J. (2005). Human malignant melanoma: Detection of BRAF‐ and c‐kit‐activating mutations by high‐resolution amplicon melting analysis. Human Pathology, 36(5), 486–493. [DOI] [PubMed] [Google Scholar]
- Witteveen, J. E. , Hamdy, N. A. , Dekkers, O. M. , Kievit, J. , van Wezel, T. , Teh, B. T. , … Morreau, H. (2011). Downregulation of CASR expression and global loss of parafibromin staining are strong negative determinants of prognosis in parathyroid carcinoma. Modern Pathology, 24(5), 688–697. [DOI] [PubMed] [Google Scholar]
- Woodard, G. E. , Lin, L. , Zhang, J. H. , Agarwal, S. K. , Marx, S. J. , & Simonds, W. F. (2005). Parafibromin, product of the hyperparathyroidism‐jaw tumor syndrome gene HRPT2, regulates cyclin D1/PRAD1 expression. Oncogene, 24(7), 1272–1276. [DOI] [PubMed] [Google Scholar]
- Wu, C. W. , Huang, C. I. , Tsai, S. T. , Chiang, H. , Lui, W. Y. , & P'Eng F, K. (1992). Parathyroid carcinoma in a patient with non‐secretory pituitary tumor: A variant of multiple endocrine neoplasia type‐I? European Journal of Surgical Oncology, 18(5), 517–520. [PubMed] [Google Scholar]
- Wynne, A. G. , van Heerden, J. , Carney, J. A. , & Fitzpatrick, L. A. (1992). Parathyroid carcinoma: Clinical and pathologic features in 43 patients. Medicine (Baltimore), 71(4), 197–205. [PubMed] [Google Scholar]
- Yamashita, Y. , Akiyama, T. , Mizusawa, N. , Yoshimoto, K. , & Goto, M. (2007). A case of hyperparathyroidism‐jaw tumour syndrome found in the treatment of an ossifying fibroma in the maxillary bone. International Journal of Oral and Maxillofacial Surgery, 36(4), 365–369. [DOI] [PubMed] [Google Scholar]
- Yang, Y. J. , Song, T. Y. , Park, J. , Lee, J. , Lim, J. , Jang, H. , … Cho, E. J. (2013). Menin mediates epigenetic regulation via histone H3 lysine 9 methylation. Cell Death and Disease, 4, e583 https://doi.org/10.1038/cddis.2013.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yart, A. , Gstaiger, M. , Wirbelauer, C. , Pecnik, M. , Anastasiou, D. , Hess, D. , & Krek, W. (2005). The HRPT2 tumor suppressor gene product parafibromin associates with human PAF1 and RNA polymerase II. Molecular and Cellular Biology, 25(12), 5052–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip, L. , Seethala, R. R. , Nikiforova, M. N. , Nikiforov, Y. E. , Ogilvie, J. B. , Carty, S. E. , & Yim, J. H. (2008). Loss of heterozygosity of selected tumor suppressor genes in parathyroid carcinoma. Surgery, 144(6), 949–955. [DOI] [PubMed] [Google Scholar]
- Yu, W. , McPherson, J. R. , Stevenson, M. , van Eijk, R. , Heng, H. L. , Newey, P. , … Morreau, H. (2015). Whole‐exome sequencing studies of parathyroid carcinomas reveal novel PRUNE2 mutations, distinctive mutational spectra related to APOBEC‐catalyzed DNA mutagenesis and mutational enrichment in kinases associated with cell migration and invasion. Journal of Clinical Endocrinology and Metabolism, 100(2), E360–364. [DOI] [PubMed] [Google Scholar]
- Zhang, C. , Kong, D. , Tan, M. H. , Pappas, D. L., Jr. , Wang, P. F. , Chen, J. , … Teh, B. T. (2006). Parafibromin inhibits cancer cell growth and causes G1 phase arrest. Biochemical and Biophysical Research Communications, 350(1), 17–24. [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Li, Q. , Zhang, L. , Fu, R. , Wang, Y. , Chen, S. , … Cui, Y. (2012). Identification of a germline mutation in the HRPT2 gene in a Chinese family with parathyroid carcinomas. Intractable and Rare Diseases Research, 1(1), 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Sun, L. H. , Liu, D. M. , He, X. Y. , Tao, B. , Ning, G. , … Zhao, H. Y. (2014). Copy number variation in CCND1 gene is implicated in the pathogenesis of sporadic parathyroid carcinoma. World Journal of Surgery, 38(7), 1730–1737. [DOI] [PubMed] [Google Scholar]