

Summary
Background
Leucopenia is a common side effect in patients treated with thiopurines. Variants in the thiopurine S‐methyltransferase (TPMT) gene are the best‐known risk factor, but only explain up to 25% of leucopenia cases.
Aim
To identify the clinical risk factors for thiopurine‐induced leucopenia in patients without a common TPMT variant, and explore if these patients are at increased risk for infections.
Methods
Post hoc analysis of the Thiopurine response Optimisation by Pharmacogenetic testing in Inflammatory bowel disease Clinics (TOPIC) trial. For this analysis, patients without a variant in TPMT (*2, *3A or*3C) were included. Uni‐ and multivariate Cox‐proportional hazard models were used to identify risk factors for leucopenia and infections. Leucopenia was defined as a white blood cell (WBC) count <3.0 × 109/L and infections were classified according to the Common Terminology Criteria for Adverse Events.
Results
Sixty hundred and ninety‐five patients (90.6%) included in the TOPIC‐trial had no variant in TPMT, of which 45 (6.5%) developed leucopenia. Median time to leucopenia was 56 (29‐112) days. Multivariate analysis showed that use of mercaptopurine compared to azathioprine was associated with leucopenia (hazard ratio [HR] 2.61 [95% CIs, 1.39‐4.88; P < .01]) and a higher baseline WBC count was protective (HR 0.80 [95% CIs, 0.71‐0.89; P < .01]). Risk factors for infections were older age (per 10 year; HR 2.07 [95% CIs, 1.18‐3.63; P = .01]) and concomitant use of biologic drugs (HR 2.15 [95% CIs, 1.14‐4.07; P = .02]).
Conclusions
Low baseline WBC count and mercaptopurine, due to a relatively higher dose, were risk factors for thiopurine‐induced leucopenia in patients without a TPMT variant.
1. INTRODUCTION
Thiopurines are immunosuppressive drugs that have an important role in the treatment of inflammatory bowel diseases (IBD), both as monotherapy and as combination treatment with biologics.1, 2, 3 Yet many patients have to discontinue treatment in an early phase due to ineffectiveness or side effects. Leucopenia as a result of thiopurine use is a frequent reason for dose reductions and/or (temporary) treatment discontinuation in patients with IBD, leading to suboptimal treatment conditions.4, 5 Dose reductions or treatment discontinuation in case of leucopenia is mainly driven by fear for infectious complications.4
The best‐known risk factor for the development of thiopurine‐induced leucopenia is the presence of genetic variants in the thiopurine S‐methyltransferase (TPMT) gene, which leads to a reduced or negligible TPMT activity and subsequently to excessive cytotoxic 6‐thioguanine nucleotide (6‐TGN) metabolite formation.6, 7 In the Caucasian population, 95% of the decreased TPMT enzyme activity follows from three common variants (TPMT*2, TPMT*3A or TPMT*3C).7 Especially patients with a homozygous genetic variant in TPMT (~0.3% of the population), and to a lesser degree patients with a heterozygous variant in TPMT (~10% of the population) are at increased risk for severe, life‐threatening, myelosuppression when treated with the standard recommended dose of azathioprine (AZA) or mercaptopurine (MP).8, 9, 10 Leucopenia can be prevented by genotyping for these variants prior to initiation of thiopurine treatment and subsequently taper the thiopurine dose to 50% of the original dose in those heterozygous for a variant in TPMT and 0%‐10% of the original dose in patients with a homozygous variant.11, 12, 13 However, only up to 25% of the leucopenia cases can be explained by variants in TPMT, which makes that leucopenia is frequently observed in patients without a TPMT variant.1, 5, 14, 15, 16 This suggests that also other genes and environmental or clinical factors may contribute to the risk of leucopenia during thiopurine treatment.17, 18, 19, 20, 21
It is unknown to what degree leucopenia attributes to the risk for infections. Recent studies in the treatment of hepatitis C did not reveal a relation between interferon‐induced leucopenia and infectious complications. 22, 23 If this also applies for thiopurine treatment in IBD, strict dose reductions in case of leucopenia might be redundant.
In the present post hoc analysis of the “Thiopurine response Optimisation by Pharmacogenetic testing in Inflammatory bowel disease Clinics” (TOPIC) trial, we aimed to (1) identify clinical risk factors for the development of TPMT‐independent leucopenia in patients with IBD treated with thiopurines and (2) evaluate whether TPMT‐independent leucopenia is a risk factor for the occurrence of infections in these patients.
2. METHODS
2.1. Patients
We designed a case‐control study with patient data from the TOPIC trial, which evaluated the efficacy of pre‐treatment TPMT genotyping on the occurrence of myelosuppression during thiopurine treatment.14 In brief, in the TOPIC trial thiopurine‐naive patients with IBD with an indication for thiopurine treatment (indicated by the treating physician) were randomised 1:1 for the intervention group with prior to treatment genotyping of the three most common genetic variants (*2, *3A and *3C) in TPMT vs the standard of care group with thiopurine dosing according to current standard of care; AZA (2.0‐2.5 mg/kg) and MP (1.0‐1.5 mg/kg). In patients allocated to the intervention arm, a 50% dose reduction was recommended in case of a heterozygous TPMT variant and in case of a homozygous TPMT variant 10% of the standard dose or an alternative treatment was advised. Patients were followed for 20 ± 6 weeks with laboratory measurements including white blood cell (WBC) count at week 0, 1, 2, 4, 6, 8, 20 and when indicated by the treating physician. Neutrophil count was not routinely measured in the TOPIC trial. Physicians were free in their choice for AZA or MP and were advised to start full dose. After the follow‐up period, TPMT‐genotype was assessed in patients randomised to the standard of care arm and in addition TPMT activity was assessed in all patients. Main exclusion criteria of the TOPIC trial were previous use of thiopurines, co‐treatment with allopurinol, baseline WBC count <3.0 × 109/L, baseline liver test abnormalities (alanine transaminase, aspartate transaminase, alkaline phosphatase and/or gamma glutamate transpeptidase ≥ two times the normal upper limit), known TPMT enzyme activity or TPMT‐genotype. 6‐Methylmercaptopurine ribonucleotides (6‐MMPR) and 6‐TGN levels were assessed at week eight in the first 301 patients included in the trial. For a detailed description of the study design, patient selection and patient data, we refer to Coenen et al14 The TOPIC trial was approved by the institutional ethics committee and all patients provided written informed consent (clinicaltrials.gov, NCT00521950).
The primary aim of the current study was to indentify risk factors for leucopenia in patients without one of the tested genetic variants in TPMT. All randomised patients who started with a thiopurine and tested negative for a variant in TPMT were included in an intention‐to‐treat analysis (Figure 1). Patients allocated to the genotyping arm with a TPMT variant (treated with 50% of the regular dose), as well as patients assigned to the standard of care arm with a TPMT variant (treated with 100% of the regular dose) were excluded. The Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 was used to classify the severity of leucopenia.24 According to these criteria, mild leucopenia (grade 1) is defined as a WBC count between the lower limit of normal and ≥3.0 × 109/L, moderate leucopenia (grade 2) was defined as a WBC count between ≥2.0 × 109/L and <3.0 × 109/L and severe leucopenia (grade 3) if the WBC count was <2.0 × 109/L. For the current analysis, only cases of moderate and severe leucopenia were included. Leucopenia which developed 2 weeks or later after discontinuation of the thiopurine was not considered to be associated with thiopurine use.
Figure 1.
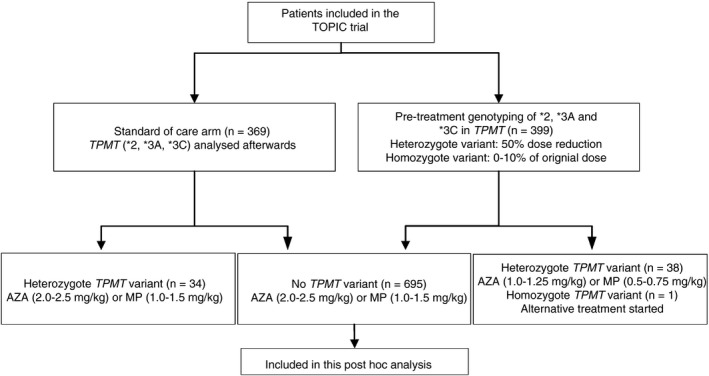
Flowchart of patient selection for this post hoc analysis. All patients who participated in the TOPIC trial, without a genetic variant in TPMT, were included. AZA, azathioprine; MP, mercaptopurine; TPMT; thiopurine S‐methyltransferase
The second goal of this study was to explore if patients with TPMT‐independent leucopenia during thiopurine treatment were at increased risk for infections. For this analysis, infections were classified according to the CTCAE.24 The source of infection was reported and severity was subdivided in grade 1‐5, in which grade 1 means no indication for infection treatment, grade 2: oral intervention (antibiotics, antifungal or anti‐viral) treatment indicated, grade 3: intravenous treatment (antibiotics, antifungal or anti‐viral) or operative intervention indicated, grade 4: life‐threatening consequences and grade 5 indicates death from the infection. Only clinical relevant infections, grade 2 or higher, were included in the analysis, ie, infections for which oral or intravenous antibiotic, antifungal or anti‐viral treatment was indicated. Infections up to 2 weeks after thiopurine treatment discontinuation were included. Infections were classified as ‘leucopenia‐associated infection’ when the infection occurred while the patient was leucopenic (WBC count <3.0 × 109/L) or within 2 weeks after the leucopenia was resolved.
Covariates included in the analysis were baseline (week 0) WBC count and week one decrease in WBC count, defined as the decrease in WBC level after 7 (±1) days of thiopurine use. Furthermore, use of concomitant drugs, ie, use of 5‐aminosalicylic acid (5‐ASA), steroids, biologics and ACE‐inhibitors at baseline or initiated during the use of the initial thiopurine was reported. Steroids with limited systemic bioavailability, such as budesonide were not included. Information about co‐morbidities (diabetes mellitus, chronic pulmonary disease and rheumatoid diseases) was reported. Patients were classified as active smoker or nonsmoker, determined at baseline. Disease severity at baseline was classified according to the Harvey‐Bradshaw Index in patients with Crohn's disease (CD) and partial Mayo score for ulcerative colitis (UC). AZA dose in mg/kg was divided by 2.08 for comparison with MP dose.12, 25
2.2. TPMT enzyme activity and TPMT genotyping
TPMT enzyme activity was measured in red blood cells (RBCs) using a high‐performance liquid chromatography method described previously.14 Genotyping of variants *2, *3A and *3C in TPMT was described previously.14 Possible cases of TPMT‐induced leucopenia caused by rare variants, other than *2, *3A and *3C in TPMT, might have been missed. For this reason, we sequenced all exons including splice sites in the TPMT gene in patients with a low TPMT activity (<60 mg 6‐methylguanine/mmolHb.h) or who developed leucopenia (WBC <3.0 × 109/L). Polymerase chain reaction was performed on 10 ng DNA using AmpliTaq Gold 360 mastermix (Life Techologies, Bleiswijk, The Netherlands). Primers are described elsewhere.14 Annealing temperature was 56°C for all exons. PCR products were purified using Multiscreen filter plates (Millipore Carrigtwohill, Cork, Ireland). Purified products were used for Sanger sequence analysis (bidirectional) with dye‐termination chemistry (BigDye Terminator, version 3) on a 3730 DNA analyser (Applied Biosystems, Inc., Foster City, CA, USA.
2.3. 6‐MMPR and 6‐TGN metabolite measurements
By protocol, 6‐TGN and 6‐MMPR levels at week eight were assessed at a routine check‐up in the first 301 patients included in the TOPIC trial. 6‐TGN and 6‐MMPR levels were determined in RBCs by high‐performance liquid chromatography according to the Lennard method.14 Median (IQR) thiopurine metabolite concentrations are presented in pmol/8x108RBCs.
2.4. Statistical analysis
All analyses were performed using spss version 20.0.0.1 (SPSS Inc., Chicago, IL, USA). First, leucopenia rate was calculated for patients without one of the tested TPMT variants. Characteristics of patients with and without TPMT‐independent leucopenia were compared by Chi‐squared test (categorical data) or independent t test (continuous data). Nonparametric tests were used when data were not distributed normally. In a similar fashion, characteristics of patients with and without an infection during the use of the initial thiopurine were analysed. In patients with steady‐state metabolite levels at week 8, the 6‐MMPR, the 6‐TGN levels and the 6‐MMPR/6‐TGN ratio were compared by Mann‐Whitney U test in patients with and without TPMT‐independent leucopenia.
Univariate Cox‐proportional hazard models were used to identify clinical factors associated with the development of leucopenia in patients without one of the tested TPMT variants. The time to leucopenia and in patients without leucopenia the number of days of thiopurine use served as reference. Variables with a P < .2 were included in a multivariate Cox‐proportional hazard model with backward elimination to explore which factors were independently associated with the development of TPMT‐independent leucopenia during thiopurine treatment. In the same way, uni‐ and multivariate Cox‐proportional analysis was performed to identify factors associated with the occurrence of infections during thiopurine treatment. The variable leucopenia was retained in this model, since we wanted to explore whether these patients were at increased risk for infections. A P < .05 was considered as statistically significant.
3. RESULTS
3.1. Patients
Of the 796 patients with IBD randomised in the TOPIC trial, 29 patients were excluded because they did not start with a thiopurine or were lost to follow‐up. Of the remaining 767 patients who actually started with thiopurine treatment, 72 (9.4%) carried a genetic variant in TPMT and were excluded, which leaves 695 patients with IBD for this analysis (Figure 1). Of the 695 included patients, 247 patients (35.5%) were treated with MP (mean dose ± SD 1.20 ± 0.15 mg/kg) and 448 patients (64.5%) were treated with AZA (mean dose ± SD 2.19 ± 0.20 mg/kg). To compare the AZA dose with the MP dose, a conversion rate of 2.08 was applied, resulting in a mean converted dose of 1.05 ± 0.09 mg/kg. T test showed that the difference was significant (P < .001).
3.2. Leucopenia
Of the 695 patients without a variant in TPMT, 45 (6.5%) developed leucopenia in the first 5 months of treatment; 41 (5.9%) patients developed moderate leucopenia and four (0.6%) patients developed severe leucopenia. See Table S1 for detailed information of the 45 patients with TPMT‐independent leucopenia. Median time (IQR) to onset of leucopenia was 56 (29‐112) days. In 11 patients (24.4%) with TPMT‐independent leucopenia the thiopurine treatment was discontinued, while in 20 patients (44.5%) the thiopurine dose was reduced, in 12 patients (26.7%) leucopenia recovered without an intervention and in two patients (4.4%) the intervention was unknown. TPMT enzyme activity mean (±SD) was not different between patients with and without leucopenia (95.8 ± 21.8 vs 94.4 ± 18.8 mg/mmol Hb.h, P = .65; Figure 2).
Figure 2.
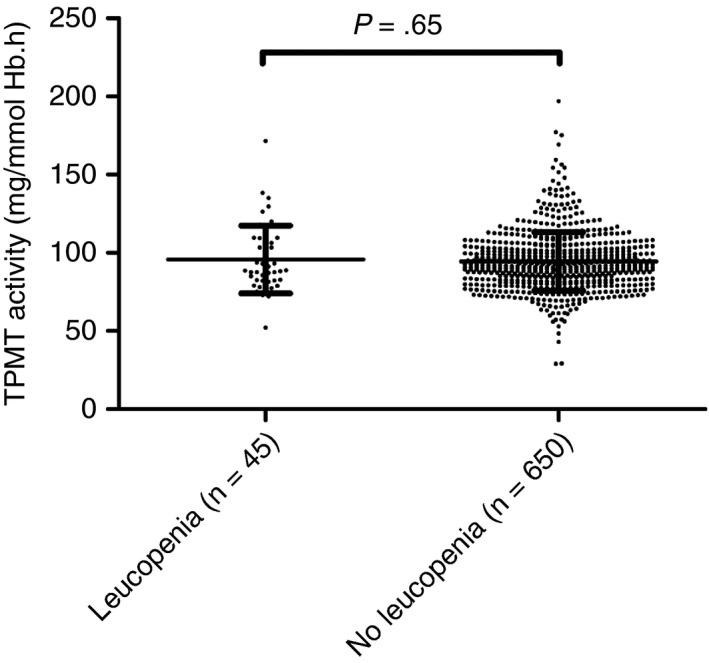
Thiopurine S‐methyltransferase (TPMT) activity in patients with and without TPMT‐independent thiopurine‐induced leucopenia. Leucopenia was defined as white blood cell count of <3.0 × 109/L
Table 1 shows the characteristics of patients with and without TPMT‐independent leucopenia during thiopurine treatment. Table 2 provides the results of the uni‐ and multivariate Cox‐proportional hazard model and shows that the choice for MP compared to AZA, Hazard ratio (HR) 2.61 (95% CIs 1.39‐4.88; P < .01) was an independent risk factor for TPMT‐independent thiopurine‐induced leucopenia. Furthermore, a higher baseline WBC count protected the patient from TPMT‐independent thiopurine‐induced leucopenia, HR 0.80 (95% CIs 0.71‐0.89; P < .01): for every increase of WBC at baseline with 1.0 × 109/L the risk of leucopenia decreased with 0.80.
Table 1.
Characteristics of the patients with and without TPMT‐independent thiopurine‐induced leucopenia
| Characteristics | Leucopenia | P‐value | |
|---|---|---|---|
| Yes (n = 45) | No (n = 650) | ||
| Male sex, n (%) | 21 (47) | 289 (45) | .77 |
| Age, y, median (IQR) | 43 (28‐51) | 40 (26‐53) | .52 |
| BMI, mean (SD) | 24.9 (5.6) | 24.8 (4.8) | .89 |
| Current smoker, n (%) | 4 (9) | 156 (24) | .02 |
| Drug, AZA, n (%) | 18 (40) | 430 (66) | <.01 |
| Thiopurine dose (mg/kg), median (IQR)a | 1.15 (1.03‐1.31) | 1.09 (1.02‐1.17) | .03 |
| TPMT activity, mg/mmol Hb.h, mean (SD) | 95.8 (21.7) | 94.4 (18.8) | .65 |
| Disease, CD, n (%)a | 24 (53) | 399 (61) | .33 |
| Wk 0 WBC count, 109/L, mean(SD) | 8.4 (3.5) | 10.6 (3.6) | <.01 |
| Wk 1 decrease WBC count, 109/L, mean (SD)c | 1.2 (2.9) | 0.8 (2.8) | .42 |
| Concomitant drugs, n (%) | |||
| 5‐Aminosalicylic acid | 25 (56) | 320 (49) | .41 |
| Systemic steroids | 27 (60) | 371 (57) | .74 |
| Biologics | 9 (20) | 66 (10) | .04 |
| ACE‐inhibitor | 2 (4) | 20 (3) | .61 |
| Co‐morbidity, n (%)b | |||
| Diabetes mellitus | 3 (7) | 22 (3) | .25 |
| Rheumatic disease | 2 (5) | 36 (6) | .75 |
| Asthma/COPD | 3 (8) | 66 (12) | .45 |
| Disease severity | |||
| CD, HBI, mean (SD)e | 3.7 (3.6) | 3.6 (3.1) | .83 |
| UC, partial Mayo, mean (SD)c | 3.7 (1.5) | 3.8 (1.7) | .83 |
Leucopenia was defined as a white blood cell count (WBC) of <3.0 × 109/L.
AZA, azathioprine; BMI, body mass index; CD, Crohn's disease; HBI; Harvey‐Bradshaw Index; TPMT, thiopurine S‐methyltransferase; UC, ulcerative colitis; WBC, white blood cell.
AZA dose in mg/kg was divided by 2.08 to compare with MP dosage.
Seven patients were diagnosed with indeterminate colitis.
Known for 492 patients.
Known for 602 patients.
Known for 325 patients with CD.
Known for 225 patients with UC.
Table 2.
Uni‐ and multivariate Cox‐proportional hazards regression analysis to explore factors associated with development of thiopurine‐induced leucopenia in the first five months of thiopurine treatment in patients without a TPMT variant
| Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|
| Hazard ratio (95% CIs) | P‐value | Hazard ratio (95% CIs) | P‐value | |
| Drug type, MP vs AZA | 2.45 (1.35‐4.48) | .003 | 2.61 (1.39‐4.88) | .003 |
| Week 0 WBC count, 109/L | 0.78 (0.70‐0.88) | <.001 | 0.80 (0.71‐0.89) | <.001 |
| Concomitant biologic drugs | 1.67 (0.80‐3.49) | .17 | ||
| Thiopurine dosage (mg/kg) | 5.59 (0.68‐46.1) | .11 | ||
| Active smoking | 0.37 (0.13‐1.03) | .06 | ||
AZA dose in mg/kg was divided by 2.08 to compare with MP dosage.
AZA, azathioprine; MP, mercaptopurine; WBC, white blood cell.
In 17 patients who developed leucopenia after 8 weeks, steady‐state 6‐MMPR and 6‐TGN levels were assessed. Neither 6‐TGN levels, nor 6‐MMPR levels, nor the 6‐MMPR/6‐TGN ratio were different from patients without leucopenia in whom steady‐state week eight levels were assessed; 267 (205‐364) pmol/8 × 108 RBCs vs 242 (186‐341) pmol/8 × 108 RBCs, P = .39 and 3596 (1304‐12 824) pmol/8×108 RBCs vs 3600 (1326‐7415) pmol/8 × 108 RBCs, P = .63. The 6‐MMPR/6‐TGN median (IQR) ratio was 13 (6‐35) in patients with leucopenia and 14 (6‐32) in patients without leucopenia, P = .80.
3.3. Infections
Sixty‐five (9.4%) of the 695 patients developed an infection while being treated with a thiopurine during the study period of 5 months. Fifty‐nine (8.5%) infections were classified as grade 2 or higher according to the CTCAE, of these, 32 patients had a grade 2 infection, 25 patients a grade 3 infection and 2 patients were classified as grade 4 infection. Two patients died during the TOPIC trial, both were excluded from this analysis, one patient had a heterozygous TPMT variant and the other patient developed an infection while he was treated with steroids, infliximab and methotrexate, more than 2 weeks after thiopurine treatment was discontinued.14 Median (IQR) time after the start of thiopurine treatment to an infection grade 2 or higher was 54 (18‐88) days. Of the 45 patients with TPMT‐independent leucopenia five patients (11.1%), of whom three had a severe leucopenia (WBC count <2.0 × 109/L), experienced a “leucopenia‐associated infection”, while the infection rate in patients without leucopenia was 54 in 650 patients (8.3%) (P = .41). Details about infection localisations are depicted in Table 3. Characteristics of patients with and without an infection are presented in Table 4. In the multivariate Cox‐proportional hazard model only age (per 10 years), HR 2.07 (95% CIs, 1.18‐3.63; P = .01) and concomitant use of biologic drugs, HR 2.15 (95% CIs, 1.14‐4.07; P = .02), were associated with the occurrence of infections (Table 5).
Table 3.
Infection localisation according to the common terminology criteria for adverse events (version 4.0). Only patients with infections grade 2 or higher were included
| Localisation | Frequency |
|---|---|
| Lung infection | 16 |
| Urinary tract infection | 8 |
| Skin infection | 7 |
| Abdominal infection | 5 |
| Infectious enterocolitis | 4 |
| Upper respiratory infection | 2 |
| Meningitis | 1 |
| Unknown | 16 |
| Overall | 59 |
Table 4.
Characteristics of the patients with and without an infection during thiopurine treatment
| Characteristics | Infection during thiopurine treatment | P‐value | |
|---|---|---|---|
| Yes (n = 59) | No (n = 636) | ||
| Male sex, n (%) | 21 (36) | 289 (45) | .15 |
| Age,y, median (IQR) | 42 (33‐60) | 40 (26‐53) | .11 |
| BMI, mean (SD) | 24.9 (6.1) | 24.7 (4.7) | .82 |
| Current smoker, n (%) | 18 (31) | 142 (22) | .16 |
| Drug, AZA, n (%) | 42 (71) | 406 (64) | .26 |
| Thiopurine dose in mg/kg, median (IQR)c | 1.08 (1.02‐1.16) | 1.10 (1.02‐1.19) | .50 |
| TPMT activity, mg/mmol Hb.h, mean (SD) | 96.2 (19.6) | 94.4 (18.9) | .47 |
| Disease, CD, n (%)a | 38 (64) | 385 (61) | .63 |
| Wk 0 WBC count, 109/L, mean (SD) | 9.9 (3.5) | 10.5 (3.6) | .21 |
| Wk 1 decrease WBC count, 109/L, mean (SD)c | 0.4 (3.0) | 0.9 (2.8) | .29 |
| Leucopenia | 5 (9)c | 37 (6) | .41 |
| Concomitant drugs, n (%) | |||
| 5‐Aminosalicylic acid | 28 (48) | 317 (50) | .47 |
| Systemic steroids | 33 (56) | 632 (58) | .79 |
| Biologics | 12 (20) | 63 (10) | .01 |
| ACE‐inhibitor | 2 (4) | 20 (3) | .61 |
| Co‐morbidity, n (%)b | |||
| Diabetes mellitus | 1 (2) | 24 (4) | .41 |
| Rheumatic disease | 8 (16) | 61 (11) | .32 |
| Asthma/COPD | 5 (10) | 33 (6) | .28 |
| Disease severity | |||
| CD, HBI, mean (SD)e | 3.7 (2.8) | 3.6 (3.1) | .80 |
| UC, partial Mayo, mean (SD)f | 3.9 (1.7) | 3.8 (1.7) | .82 |
Leucopenia was defined as a WBC count of <3.0 × 109/L. Only infections grade 2 or higher according to the Common Terminology Criteria for Adverse Events were included.
AZA, azathioprine; BMI, body mass index; CD, Crohn's disease; HBI; Harvey‐Bradshaw Index; TPMT, thiopurine S‐methyltransferase; UC, ulcerative colitis; WBC, white blood cell.
AZA dose in mg/kg was divided by 2.08 to compare with MP dosage.
Seven patients were diagnosed with indeterminate colitis.
Known for 492 patients.
Known for 602 patients.
Known for 325 patients with CD.
Known for 225 patients with UC.
As follows from Table 3, 45 patients experienced leucopenia, however in 3 patients with leucopenia during thiopurine treatment who also had an infection, the infection was not considered to be related to leucopenia, either because leucopenia was already resolved for more than 2 weeks (2 patients), or the infection was already known before the leucopenia occurred (1 patient). For the analysis, these cases were counted as an infection without leucopenia.
Table 5.
Uni‐ and multivariate Cox‐proportional hazards regression analysis to explore factors associated with infections during thiopurine treatment
| Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|
| Hazard ratio (95% CIs) | P‐value | Hazard ratio (95% CIs) | P‐value | |
| Sex, male | 0.67 (0.40‐1.15) | .15 | ||
| Age, per 10‐year | 1.84 (1.06‐3.21) | .03 | 2.07 (1.18‐3.63) | .01 |
| Active smoker | 1.58 (0.90‐2.76) | .11 | ||
| Week 0 WBC count, 109/L | 0.95 (0.88‐1.02) | .17 | ||
| Leucopenia | 1.66 (0.75‐3.65) | .21 | ||
| Concomitant use of biologics | 2.03 (1.08‐3.83) | .03 | 2.15 (1.14‐4.07) | .02 |
Infections were classified according to the Common Terminology Criteria for Adverse Events 4.0 (version 4.0). Only infections grade 2 or higher were included.
WBC, white blood cell.
4. DISCUSSION
In this study, we showed that in patients with IBD without a variant in TPMT, baseline WBC count and the choice of MP compared to AZA, were independent risk factors for thiopurine‐induced leucopenia during the first five months of treatment. However, this did not result in a higher risk for infections. Infections during thiopurine treatment were more frequently seen in elderly and in patients with concomitant use of biologic drugs.
The overall incidence of thiopurine‐induced leucopenia is estimated to be 7%.4 Importantly, in 75% of the patients this is not correlated with a genetic variant in TPMT. As shown in this study, leucopenia still occurs in 6.5% of the patients without a genetic variant in TPMT, often leading to dose reductions or treatment discontinuation.5 Thiopurine‐induced leucopenia is at least partially dose‐dependent.26 In a previous analysis of all patients included in the TOPIC trial, we already showed that patients treated with MP were relatively higher dosed than patients treated with AZA and that this resulted in amongst others a higher rate of leucopenia.25 This difference annulled in a secondary analysis with adjustment for dose and metabolite levels, therefore an interdrug difference between AZA and MP seems unlikely. Also, other studies showed a relatively higher dose and leucopenia rate in patients on MP compared to AZA.20, 21, 27 The difference might be attributed to the absence of MP tablets with a content of 25 mg, which allow a more personalised dosing. Another explanation might be derived from the conversion rate of 2.08, which is used to calculate the MP dose from the AZA dose. This conversion rate includes assumptions such as a 100% bioavailability.28 The higher rate of dose‐dependent side effects might be prevented with a slight reduction of the MP starting dose.25
In our study, 6‐TGN and 6‐MMPR metabolite levels were not increased in patients with leucopenia. It must be stressed that most patients with leucopenia were subjected to dose reductions or treatment discontinuation before week eight. Therefore, steady‐state week eight 6‐TGN levels were available only in a limited number of cases, which most likely explains why we found no difference. Besides 6‐TGN levels, also excessively elevated 6‐MMPR levels have been associated with leucopenia.21 The increased risk for leucopenia with a lower baseline WBC count has also been reported in the setting of ganciclovir‐ and chemotherapy‐induced leucopenia.29, 30 In contrast to a previous study, we did not find a correlation between concomitant 5‐ASA use and myelosuppression.31 It has been suggested that 5‐ASA might increase 6‐TGN levels by a weak inhibition of the TPMT enzyme activity and therefore may increase the risk of myelosuppression.32 Previous studies using data of the TOPIC trial already showed that 6‐TGN levels were not significantly higher in patients on 5‐ASA, which explains why we found no increased leucopenia rate in this group.33, 34 It is known that smoking leads to an increase of WBC count of 0.5–1.0 × 109/L as a result of a low‐grade systemic inflammation.35 Indeed, in our study univariate analysis showed that smokers had less often leucopenia, however, this did not reach significance in the multivariate analysis.
Pharmacogenetic testing of TPMT is a cost‐effective strategy to prevent severe, life‐threatening, myelosuppression, in those who are TPMT‐deficient.36, 37 This strategy is strongly recommended by several guidelines.9, 11, 38, 39 TPMT‐related and TPMT‐independent leucopenia may be considered as two separate entities. Most studies reporting on thiopurine‐induced leucopenia provide no information regarding the TPMT status, which hampers extrapolation to daily clinical practice.40, 41, 42 For this reason, we specifically focused on the patients without a variant in TPMT. Thiopurine‐induced leucopenia in patients without genetic variants in TPMT occurred at any moment during the first 5 months of therapy, with a slight peak in the first 2 months. Therefore, the current routine laboratory tests should remain mandatory to identify patients with myelosuppression in an early stage.1 In more than 90% of the cases leucocyte levels did not drop below 2.0 × 109/L. Yet thiopurine dosage was reduced or treatment was discontinued in two‐thirds of these patients, probably due to fear for infectious complications. Overall, most cases of TPMT‐independent leucopenia are mild and therefore did not require immediate treatment discontinuation or dose reduction. However in the setting of an unknown TPMT‐genotype, all cases of leucopenia in the first months of thiopurine treatment should be considered as potentially TPMT‐induced, because otherwise life‐threatening myelosuppression might occur in the patients whom actually have a genetic variant in TPMT.1 Previous studies estimated a cumulative infection rate of 6.5% among patients with thiopurine‐induced leucopenia.4 In the present study, we showed that infection rates are comparable for patients with and without TPMT‐independent leucopenia. Both viral and bacterial infections were seen, however in most of the cases no specific pathogen was isolated. The risk for bacterial infections increases with neutrophil levels below 0.5 × 109/L. 43 These levels are often observed in patients with a homozygous variant in TPMT when treated with standard thiopurine doses due to the very high 6‐TGN levels.1, 44 In our study, neutrophil levels at time of leucopenia were available in ~50% of the patients and a neutrophil count below 0.5 × 109/L was observed in only one patient, which suggests that severe neutropenia is not frequently observed in patients without a TPMT variant. This, in combination with the fact that in most cases leucopenia was only moderate (WBC count ≥2.0 × 109/L), might explain why we did not find a higher infection rate in patients with a thiopurine‐induced leucopenia. It must be mentioned that in 69% of patients with leucopenia the thiopurine dose was reduced or treatment was discontinued. It cannot be excluded that this prevented the aggravation of the leucopenia and/or the development of infectious complications, which is an important limitation of our study. Yet, our findings corroborate studies exploring the risk of infections in hepatitis C treatment. In these studies, interferon‐induced leucopenia was not a risk factor for infectious complications.22, 23 The lack of association between leucopenia and infections might have great implications for clinical practice. Some studies showed a relation between mild leucopenia (defined as WBC count ≥3.0 and <5.0 × 109/L) and treatment efficacy.45, 46 This increased treatment efficacy in case of leucopenia was independent from 6‐TGN levels.45 Therefore, if our observations are correct, a more preserved strategy in case of moderate leucopenia would not increase infection risk, prevents a patient from under‐dosing, and possibly increases treatment efficacy. Future studies are needed to evaluate whether such a strategy is safe and does increase the infection rate or lead to more patients with severe leucopenia.
Age and concomitant use of biologics were the only factors associated with the occurrence of infections. Increasing age is a known risk factor for infections, which is thought to result from an impaired immune response in the elderly.47, 48 This has also been reported in patients with IBD.49, 50 In contrast to many clinical trials, there was no upper age limit with inclusion in the TOPIC trial. As a consequence, 105 (15.1%) of the patients included in current study were 60 years or older, of whom 24 patients were 70 years or older. This makes it a representative sample for the real life setting in the era of an ageing IBD population.51 The higher risk for infections with concomitant use of biologic drugs is not unexpected, since use of biologics is an independent risk factor for (opportunistic) infections.49, 52 This observation is not limited to IBD, but is also observed in other immune‐mediated diseases.53, 54 In contrast to previous studies, we found no relation with concomitant steroid use.49 A likely explanation is that steroid use was not protocolised and as a consequence the group of patients on oral steroids was very heterozygous and included patients on low maintenance dose, short‐term use during thiopurine initiation, but also patients on a high dose for a longer period. Furthermore, patients currently taking no steroids still have an increased infection risk, when treated with steroids in the past 2 years.55 We found no association between diabetes mellitus or smoking and infection risk, probably because of the relatively small study size and short follow‐up period.56, 57
This study comes with some limitations. First, current analyses were not included in the original study protocol. Second, patients in the TOPIC trial were only tested for the three common variants in TPMT (*2, *3A and *3C), which cover more than 90% of the prevalence of genetic variants in TPMT in Caucasians. Therefore, it is possible that we missed some patients with other TPMT variants.58 This was circumvented by sequencing the coding region of TPMT in all patients with a low TPMT activity or those who developed leucopenia; which did not reveal other variants (unpublished data). Furthermore, we assumed that when the treating physician started anti‐infectious therapy there was a true suspicion of an infection. This comes with some restrictions, since exacerbations of IBD or flu‐like side effects (which might include elevation of C‐reactive protein) can mimic (gastro‐intestinal) infections leading to the prescription of antibiotics. Finally, (viral) infections may induce leucopenia.19 However, if this is the case it would have resulted in an overestimation of ‘leucopenia‐associated infections’ and further bolster our findings that (moderate) leucopenia itself is not a risk factor for infectious complications.
In conclusion, in the present study we showed that the choice for MP compared to AZA (as a result of a relatively higher dose) and baseline WBC count are risk factors for the development of thiopurine‐induced leucopenia in patients without a genetic variant in TPMT; however, this was not associated with an increased risk for infections. Multivariate analysis showed that older patients and patients with concomitant use of biologics had a higher risk for infections during thiopurine treatment and should therefore be treated with more caution. Our data suggest that moderate TPMT‐independent leucopenia during thiopurine treatment might be accepted in the light of close monitoring of WBC count and metabolite levels.
ACKNOWLEDGEMENT
We thank the patients for participation in the study. We thank Rene H.M. te Morsche and Wilbert H.M. Peters from the Department of Gastroenterology, Radboud University Medical Center, Nijmegen, The Netherlands, for the measurement of TPMT activity. Furthermore, we thank Mariëlle Maas, Miet Fiddelaers, Milevis Reitsma, Leonie Peters and Jean Cilissen from the department of Clinical Pharmacy and Toxicology, Zuyderland Medical Center, Sittard‐Geleen, The Netherlands for technical assistance with metabolite measurement and Debbie Heinen, Marjolein M.J. van Donkelaar, Freshteh Golestani, Marlies E. de Vos, J.G. Angelien M. Heister, Doménique M.W. Nijsten, Mascha M.V.A.P. Schijvenaars, and Martine E.C. Cranen from the department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands for their support in data‐management. We thank Dr. Sita H. Vermeulen, department for Health Evidence, Radboud Institute for Health Sciences, Radboud University Medical Center, Nijmegen, The Netherlands, and Professor Barbara Franke, department of Human Genetics, Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Center, Nijmegen, The Netherlands, for their contribution to the design of the TOPIC trial. At last, we thank Professor Joost P.H. Drenth from the Department of Gastroenterology, Radboud University Medical Center, Nijmegen, The Netherlands, for intellectual contribution to the content of the manuscript.
The TOPIC recruitment team was responsible for patient recruitment and collection of clinical data. Compensation was given to the members of the recruitment team for additional biochemical measurements and examinations that had to be performed for the TOPIC study. TOPIC recruitment team members are: Department of Gastroenterology, Academisch Ziekenhuis Maastricht, Maastricht, The Netherlands—AAM Masclee, MD; PhD, M Pierik, MD, PhD; W Mares, MD; W Hameeteman, MD, PhD; Department of Gastroenterology, Rijnstate Ziekenhuis Arnhem, Arnhem, The Netherlands—PJ Wahab, MD; PhD, H Seinen, MD, PhD; Department of Gastroenterology, Amphia Ziekenhuis, Breda, The Netherlands—MCM Rijk, MD, PhD; IM Harkema, MD; Department of Gastroenterology, Atrium Medisch Centrum, Heerlen, The Netherlands—M de Bièvre, MD; L Oostenbrug, MD, PhD; CM Bakker, MD, PhD; M Aquarius, MD; C van Deursen, MD, PhD; AB van Nunen, MD, PhD; JG Goedhard, MD, PhD; M Hamacher, MD; Department of Gastroenterology, Bernhoven Hospital, Oss, The Netherlands—IAM Gisbertz, MD, PhD; BJ Brenninkmeijer, MD, PhD; Department of Gastroenterology, Canisius Wilhelmina Ziekenhuis, Nijmegen, The Netherlands—ACITL Tan, MD, PhD; MN Aparicio‐Pagés, MD, PhD, EM Witteman, MD, PhD; Department of Gastroenterology, Diakonessenhuis, Utrecht, The Netherlands—SAC van Tuyl, MD; R Breumelhof, MD, PhD; Department of Gastroenterology, Catharina Ziekenhuis, Eindhoven, The Netherlands—A Stronkhorst, MD, PhD; LPL Gilissen, MD, PhD; EJ Schoon, MD, PhD; Department of Gastroenterology, Elkerliek Ziekenhuis, Helmond, The Netherlands—JWM Tjhie‐Wensing, MD; A Temmerman, MD; HagaZiekenhuis, ‘s‐Gravenhage, The Netherlands—JJ Nicolaï, MD, PhD; Department of Gastroenterology, Gelderse Vallei Hospital, Ede, The Netherlands—JD van Bergeijk, MD, PhD; DJ Bac, MD, PhD; BJM Witteman, MD, PhD; N Mahmmod, MD; JJ Uil, MD, PhD; H Akol, MD, PhD; Department of Gastroenterology, Ikazia Hospital, Rotterdam, The Netherlands—RJTh Ouwendijk, MD, PhD; Department of Gastroenterology, Jeroen Bosch Hospital, ‘s‐Hertogenbosch, The Netherlands—IP van Munster, MD, PhD; M Pennings, MD; AMP De Schryver, MD, PhD; ThJM van Ditzhuijsen, MD, PhD; RCH Scheffer, MD, PhD; TEH Römkens, MD; DL Schipper, MD, PhD; Department of Gastroenterology, Laurentius Hospital, Roermond, The Netherlands—PJ Bus, MD; Department of Gastroenterology, Máxima Medisch Centrum, Eindhoven‐Veldhoven, The Netherlands—JWA Straathof, MD, PhD; ML Verhulst, MD, PhD; PJ Boekema, MD, PhD; JTh Kamphuis, MD; HJ van Wijk, MD, PhD; JMJL Salemans, MD, PhD; Department of Gastroenterology, Meander MC, Amersfoort, The Netherlands—JR Vermeijden, MD; Department of Gastroenterology, MC Haaglanden, Den Haag, The Netherlands—SDJ van der Werf, MD, PhD; RJ Verburg MD, PhD; Department of Gastroenterology, Medisch Centrum Leeuwarden, Leeuwarden, The Netherlands—P Spoelstra, MD, PhD; JML de Vree, MD, PhD; K van der Linde, MD, PhD; HJA Jebbink, MD, PhD; M. Jansen; H. Holwerda; Department of Gastroenterology, Medisch Spectrum Twente, Enschede, The Netherlands—N van Bentem, MD; JJ Kolkman, MD, PhD; MGVM Russel, MD, PhD; GH van Olffen, MD; MJ Kerbert‐Dreteler, MD; M Bargeman, MD, PhD; JM Götz, MD, PhD; R Schröder, MD; Department of Gastroenterology, Onze Lieve Vrouwe Gasthuis, Amsterdam, The Netherlands—JM Jansen, MD; Department of Gastroenterology, Orbis Medisch Centrum, Sittard‐Geleen, The Netherlands—LP Bos, MD, PhD; LGJB Engels, MD, PhD; MJL Romberg‐Camps, MD; ETP Keulen, MD, PhD; Department of Gastroenterology, Radboud university medical center, Nijmegen, The Netherlands ‐ AAJ van Esch, MD; JPH Drenth, MD, PhD; MCA van Kouwen, MD, PhD; GJA Wanten, MD, PhD; TJ Bisseling, MD, PhD; TEH Römkens, MD; MWJ van Vugt; Department of Gastroenterology, Slingeland Hospital, Doetinchem, The Netherlands—PC van de Meeberg, MD, PhD; SJ van den Hazel, MD, PhD; Department of Gastroenterology, St Elisabeth Ziekenhuis, Tilburg, The Netherlands—WNHM Stuifbergen, MD, PhD; MJAL Grubben, MD, PhD; U de Wit, MD, PhD; GAH Dodemont, MD, PhD; RF Eichhorn, MD; Department of Gastroenterology, Tergooiziekenhuizen, Blaricum‐Hilversum, The Netherlands—JMH van den Brande, MD, PhD; AHJ Naber, MD, PhD; EJ van Soest, MD, PhD; PJ Kingma, MD, PhD; Department of Gastroenterology, TweeSteden Ziekenhuis, Tilburg, The Netherlands ‐ NC Talstra, MD; KF Bruin, MD, PhD; FHJ Wolfhagen, MD, PhD; Department of Gastroenterology, University Medical Centre Leiden, Leiden, The Netherlands—DW Hommes, MD, PhD; PPJ van der Veek, MD, PhD; JCA Hardwick, MD, PhD; RJ Stuyt, MD, PhD; HH Fidder, MD; Department of Gastroenterology, University Medical Centre Utrecht, Utrecht, The Netherlands—B Oldenburg, MD, PhD; Department of Gastroenterology, Ziekenhuisgroep Twente, Hengelo, The Netherlands—TG Tan, MD.
Declaration of personal interests: None.
AUTHORSHIP
Guarantor of the article: M.M.T.J. Broekman.
Author contributions: All authors listed were involved in the study design and interpretation of data for the work. All authors had access to the data and have approved the final version of the manuscript for publication. MB, MC responsibility for the integrity of the data and the accuracy of the data analysis. CM, MC, DJ collected data. Metabolite measurements were performed under guidance of DW and PH. Data analysis was performed by MB, MC, DJ, GW, DW with input from the other investigators. The manuscript was written by MB, MC, DJ, GW and all other authors edited the manuscript and gave final approval for publication. MB, MC had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Supporting information
Broekman MMTJ, Coenen MJH, Wanten GJA, et al. Risk factors for thiopurine‐induced myelosuppression and infections in inflammatory bowel disease patients with a normal TPMT genotype. Aliment Pharmacol Ther. 2017;46:953–963. https://doi.org/10.1111/apt.14323
Funding information
The TOPIC trial was funded by The Netherlands Organisation for Health Research and Development grant number 94507606 and the participating institutes. No separate funding was received for this study.
The Handling Editor for this article was Professor Ailsa Hart, and it was accepted for publication after full peer‐review.
REFERENCES
- 1. Colombel JF, Ferrari N, Debuysere H, et al. Genotypic analysis of thiopurine S‐methyltransferase in patients with Crohn's disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118:1025‐1030. [DOI] [PubMed] [Google Scholar]
- 2. Strik AS, van den Brink GR, Ponsioen C, Mathot R, Lowenberg M, D'Haens GR. Suppression of anti‐drug antibodies to infliximab or adalimumab with the addition of an immunomodulator in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2017;45:1128‐1134. [DOI] [PubMed] [Google Scholar]
- 3. Kirchgesner J, Lemaitre M, Rudnichi A, et al. Therapeutic management of inflammatory bowel disease in real‐life practice in the current era of anti‐TNF agents: analysis of the French administrative health databases 2009‐2014. Aliment Pharmacol Ther. 2017;45:37‐49. [DOI] [PubMed] [Google Scholar]
- 4. Gisbert JP, Gomollon F. Thiopurine‐induced myelotoxicity in patients with inflammatory bowel disease: a review. The American journal of gastroenterology. 2008;103:1783‐1800. [DOI] [PubMed] [Google Scholar]
- 5. Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2006;24:331‐342. [DOI] [PubMed] [Google Scholar]
- 6. Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651‐662. [PMC free article] [PubMed] [Google Scholar]
- 7. Moon W, Loftus EV Jr. Review article: recent advances in pharmacogenetics and pharmacokinetics for safe and effective thiopurine therapy in inflammatory bowel disease. Aliment Pharmacol Ther. 2016;43:863‐883. [DOI] [PubMed] [Google Scholar]
- 8. Lennard L. Implementation of TPMT testing. Br J Clin Pharmacol. 2014;77:704‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther. 2013;93:324‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Higgs JE, Payne K, Roberts C, Newman WG. Are patients with intermediate TPMT activity at increased risk of myelosuppression when taking thiopurine medications? Pharmacogenomics. 2010;11:177‐188. [DOI] [PubMed] [Google Scholar]
- 11. DiPiero J, Teng K, Hicks JK. Should thiopurine methyltransferase (TPMT) activity be determined before prescribing azathioprine, mercaptopurine, or thioguanine? Clevel Clin J Med. 2015;82:409‐413. [DOI] [PubMed] [Google Scholar]
- 12. Sandborn WJ. Rational dosing of azathioprine and 6‐mercaptopurine. Gut. 2001;48:591‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Moorsel SA, Bevers N, Meurs M, van Rossum LK, Hooymans PM, Wong DR. Azathioprine therapy in a pediatric TPMT‐deficient patient‐still an option. Ther Drug Monit. 2017;39:1‐4. [DOI] [PubMed] [Google Scholar]
- 14. Coenen MJ, deJong DJ , van Marrewijk CJ, et al. Identification of patients with variants in TPMT and dose reduction reduces hematologic events during thiopurine treatment of inflammatory bowel disease. Gastroenterology. 2015;149:907‐917 e7. [DOI] [PubMed] [Google Scholar]
- 15. Derijks LJ, Gilissen LP, Engels LG, et al. Pharmacokinetics of 6‐mercaptopurine in patients with inflammatory bowel disease: implications for therapy. Ther Drug Monit. 2004;26:311‐318. [DOI] [PubMed] [Google Scholar]
- 16. Goldberg R, Irving PM. Toxicity and response to thiopurines in patients with inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2015;9:891‐900. [DOI] [PubMed] [Google Scholar]
- 17. McGovern D, Kugathasan S, Cho JH. Genetics of inflammatory bowel diseases. Gastroenterology. 2015;149:1163‐1176.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine‐induced leukopenia. Nat Genet. 2014;46:1017‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Asseldonk DP, Kanis BM, de Boer NK, van Bodegraven AA. Leukopenia due to parvovirus B19 in a Crohn's disease patient using azathioprine. Digestion. 2009;79:211‐214. [DOI] [PubMed] [Google Scholar]
- 20. Chaparro M, Ordas I, Cabre E, et al. Safety of thiopurine therapy in inflammatory bowel disease: long‐term follow‐up study of 3931 patients. Inflamm Bowel Dis. 2013;19:1404‐1410. [DOI] [PubMed] [Google Scholar]
- 21. Meijer B, Kreijne JE, van Moorsel SAW, et al. 6‐methylmercaptopurine‐induced leukocytopenia during thiopurine therapy in inflammatory bowel disease patients. J Gastroenterol Hepatol. 2017a;32:1183‐1190. [DOI] [PubMed] [Google Scholar]
- 22. Maan R, van der Meer AJ, Hansen BE, et al. Risk of infections during interferon‐based treatment in patients with chronic hepatitis C virus infection and advanced hepatic fibrosis. J Gastroenterol Hepatol. 2015;30:1057‐1064. [DOI] [PubMed] [Google Scholar]
- 23. Berden FA, van Zwietering IM, Maan R, de Knegt RJ, Kievit W, Drenth JP. high risk of infection during triple therapy with first‐generation protease inhibitors: a nationwide cohort study. J Gastrointest Liver Dis. 2016;25:197‐204. [DOI] [PubMed] [Google Scholar]
- 24. Common Terminology Criteria for Adverse Events (CTCAE) versio 4.0. Available from: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf [PubMed]
- 25. Broekman MM, Coenen MJH, van Marrewijk CJ, et al. More dose‐dependent side effects with mercaptopurine over azathioprine in IBD treatment due to relatively higher dosing. Inflamm Bowel Dis. 2017; https://doi.org/10.1097/MIB.0000000000001163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dubinsky MC, Lamothe S, Yang HY, et al. Pharmacogenomics and metabolite measurement for 6‐mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705‐713. [DOI] [PubMed] [Google Scholar]
- 27. Meijer B, Seinen ML, van Egmond R, et al. Optimizing thiopurine therapy in inflammatory bowel disease among 2 real‐life intercept cohorts: effect of allopurinol comedication? Inflamm Bowel Dis. 2017; https://doi.org/10.1097/MIB.0000000000001168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther. 2006;24:715‐729. [DOI] [PubMed] [Google Scholar]
- 29. Kwon WA, Oh TH, Lee JW, Park SC. Predictive factors for neutropenia after docetaxel‐based systemic chemotherapy in Korean patients with castration‐ resistant prostate cancer. Asian Pac J Cancer Prev. 2014;15:3443‐3446. [DOI] [PubMed] [Google Scholar]
- 30. Venton G, Crocchiolo R, Furst S, et al. Risk factors of Ganciclovir‐related neutropenia after allogeneic stem cell transplantation: a retrospective monocentre study on 547 patients. Clin Microbiol Infect. 2014;20:160‐166. [DOI] [PubMed] [Google Scholar]
- 31. Fangbin Z, Xiang G, Minhu C, et al. Should thiopurine methyltransferase genotypes and phenotypes be measured before thiopurine therapy in patients with inflammatory bowel disease? Ther Drug Monit. 2012;34:695‐701. [DOI] [PubMed] [Google Scholar]
- 32. de Graaf P, de Boer NK, Wong DR, et al. Influence of 5‐aminosalicylic acid on 6‐thioguanosine phosphate metabolite levels: a prospective study in patients under steady thiopurine therapy. Br J Pharmacol. 2010;160:1083‐1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wong DR, Coenen MJ, Vermeulen SH, et al. Early assessment of thiopurine metabolites identifies patients at risk of thiopurine‐induced leukopenia in inflammatory bowel disease. J Crohn's Colitis. 2017;11:175‐184. [DOI] [PubMed] [Google Scholar]
- 34. Broekman MM, Wong DR, Wanten GJ, et al. The glutathione transferase Mu null genotype leads to lower 6‐MMPR levels in patients treated with azathioprine but not with mercaptopurine. Pharmacogenomics J. 2017; https://doi.org/10.1038/tpj.2016.87 [DOI] [PubMed] [Google Scholar]
- 35. Lavi S, Prasad A, Yang EH, et al. Smoking is associated with epicardial coronary endothelial dysfunction and elevated white blood cell count in patients with chest pain and early coronary artery disease. Circulation. 2007;115:2621‐2627. [DOI] [PubMed] [Google Scholar]
- 36. Thompson AJ, Newman WG, Elliott RA, Roberts SA, Tricker K, Payne K. The cost‐effectiveness of a pharmacogenetic test: a trial‐based evaluation of TPMT genotyping for azathioprine. Value Health. 2014;17:22‐33. [DOI] [PubMed] [Google Scholar]
- 37. Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost‐effectiveness analysis of alternative disease management strategies in patients with Crohn's disease treated with azathioprine or 6‐mercaptopurine. Am J Gastroenterol. 2005;100:2239‐2247. [DOI] [PubMed] [Google Scholar]
- 38. Lichtenstein GR, Abreu MT, Cohen R, Tremaine W; American Gastroenterological Association Institute technical review on corticosteroids, immunomodulators, and infliximab in inflammatory bowel disease. Gastroenterology. 2006;130:940‐987. [DOI] [PubMed] [Google Scholar]
- 39. Mowat C, Cole A, Windsor A, et al. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2011;60:571‐607. [DOI] [PubMed] [Google Scholar]
- 40. Lewis JD, Abramson O, Pascua M, et al. Timing of myelosuppression during thiopurine therapy for inflammatory bowel disease: implications for monitoring recommendations. Clin Gastroenterol Hepatol. 2009;7:1195‐1201; quiz 41‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Warman JI, Korelitz BI, Fleisher MR, Janardhanam R. Cumulative experience with short‐ and long‐term toxicity to 6‐mercaptopurine in the treatment of Crohn's disease and ulcerative colitis. J Clin Gastroenterol. 2003;37:220‐225. [DOI] [PubMed] [Google Scholar]
- 42. Connell WR, Kamm MA, Ritchie JK, Lennard‐Jones JE. Bone marrow toxicity caused by azathioprine in inflammatory bowel disease: 27 years of experience. Gut. 1993;34:1081‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bodey GP, Buckley M, Sathe YS, Freireich EJ. Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med. 1966;64:328‐340. [DOI] [PubMed] [Google Scholar]
- 44. Anstey A, Lennard L, Mayou SC, Kirby JD. Pancytopenia related to azathioprine–an enzyme deficiency caused by a common genetic polymorphism: a review. J R Soc Med. 1992;85:752‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nguyen TV, Vu DH, Nguyen TM, Lachaux A, Boulieu R. Exploring associations of 6‐thioguanine nucleotide levels and other predictive factors with therapeutic response to azathioprine in pediatric patients with IBD using multilevel analysis. Inflamm Bowel Dis. 2013;19:2404‐2410. [DOI] [PubMed] [Google Scholar]
- 46. Park MS, Kim DH, Kim DH, et al. Leukopenia predicts remission in patients with inflammatory bowel disease and Behcet's disease on thiopurine maintenance. Dig Dis Sci. 2015;60:195‐204. [DOI] [PubMed] [Google Scholar]
- 47. van Langevelde P, Joop K, van Loon J, et al. Endotoxin, cytokines, and procalcitonin in febrile patients admitted to the hospital: identification of subjects at high risk of mortality. Clin Infect Dis. 2000;31:1343‐1348. [DOI] [PubMed] [Google Scholar]
- 48. Miller RA. The aging immune system: primer and prospectus. Science. 1996;273:70‐74. [DOI] [PubMed] [Google Scholar]
- 49. Lichtenstein GR, Feagan BG, Cohen RD, et al. Serious infection and mortality in patients with Crohn's disease: more than 5 years of follow‐up in the TREAT registry. Am J Gastroenterol. 2012;107:1409‐1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toruner M, Loftus EV Jr, Harmsen WS, et al. Risk factors for opportunistic infections in patients with inflammatory bowel disease. Gastroenterology. 2008;134:929‐936. [DOI] [PubMed] [Google Scholar]
- 51. Gisbert JP, Chaparro M. Systematic review with meta‐analysis: inflammatory bowel disease in the elderly. Aliment Pharmacol Ther. 2014;39:459‐477. [DOI] [PubMed] [Google Scholar]
- 52. Nyboe Andersen N, Pasternak B, Friis‐Moller N, Andersson M, Jess T. Association between tumour necrosis factor‐alpha inhibitors and risk of serious infections in people with inflammatory bowel disease: nationwide Danish cohort study. BMJ. 2015;350:h2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kalb RE, Fiorentino DF, Lebwohl MG, et al. Risk of serious infection with biologic and systemic treatment of psoriasis: results from the Psoriasis Longitudinal Assessment and Registry (PSOLAR). JAMA Dermatol. 2015;151:961‐969. [DOI] [PubMed] [Google Scholar]
- 54. Singh JA, Cameron C, Noorbaloochi S, et al. Risk of serious infection in biological treatment of patients with rheumatoid arthritis: a systematic review and meta‐analysis. Lancet. 2015;386:258‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dixon WG, Abrahamowicz M, Beauchamp ME, et al. Immediate and delayed impact of oral glucocorticoid therapy on risk of serious infection in older patients with rheumatoid arthritis: a nested case‐control analysis. Ann Rheum Dis. 2012;71:1128‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ananthakrishnan AN, Cagan A, Cai T, et al. Diabetes and the risk of infections with immunomodulator therapy in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015;41:1141‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bagaitkar J, Demuth DR, Scott DA. Tobacco use increases susceptibility to bacterial infection. Tob Induc Dis. 2008;4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dewit O, Moreels T, Baert F, et al. Limitations of extensive TPMT genotyping in the management of azathioprine‐induced myelosuppression in IBD patients. Clin Biochem. 2011;44:1062‐1066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials