

Abstract
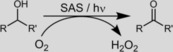
The aerobic organocatalytic oxidation of alcohols was achieved by using water‐soluble sodium anthraquinone sulfonate. Under visible‐light activation, this catalyst mediated the aerobic oxidation of alcohols to aldehydes and ketones. The photo‐oxyfunctionalization of alkanes was also possible under these conditions.
Keywords: alcohols, organocatalysis, oxidation, oxyfunctionalization, photocatalysis
The search for selective and benign oxidation protocols represents a very active research area in organic chemistry. Catalytic methods utilizing molecular oxygen or hydrogen peroxide are preferred owing to the availability of the terminal oxidants,1 their atom efficiency, and the unproblematic side products formed. Today, transition‐metal‐catalyzed methods2 prevail, but organocatalytic approaches are developing rapidly.3 A particularly interesting field is organo‐photocatalysis, which exploits the unique reactivities of photoexcited organic dyes.3a, 4 Benzophenones and anthraquinones are among these promising photoredox catalysts. Upon light initiation, anthraquinone catalysts allow C−H bond or alcohol oxidation, oxidative esterification, and cleavage of cyclic acetals.5
As early as the 1960s, water‐soluble sodium anthraquinone sulfonate (SAS) has been reported as a photocatalyst for the oxidation of alcohols.6 However, whereas its mode of action is today fairly understood, synthetic applications are scarce. Interestingly, SAS has so far only been evaluated as a catalyst for the oxidation of alcohols. The oxyfunctionalization of C−H bonds has only been scarcely considered with SAS.
Inspired by recent contributions from Wolf3d and König,3e–3 who reported the use of flavin photocatalysts for the light‐driven organocatalytic oxyfunctionalization of alkyl benzenes, we decided to evaluate the usefulness of SAS as a photocatalyst for the oxygenation of alkyl benzenes (Scheme 1).
Scheme 1.
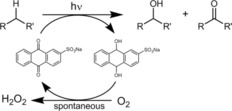
Schematic representation of the photocatalytic, SAS‐mediated oxidation of C−H bonds.
A first experiment with homogeneously dissolved toluene gave clean conversion of the starting material into benzaldehyde (81 % yield) with minor amounts of benzyl alcohol (5.6 %) and benzoic acid (2.5 %) formed (Figure S1, Supporting Information). The apparent gap in mass balance can most likely be attributed to the volatility of the starting material and the product under the reaction conditions. Notably, in the absence of either the photocatalyst (i.e., SAS) and/or light, no conversion of the starting material was observed. Performing the reaction under (near) anaerobic conditions resulted in a significantly reduced rate of product formation (Figure S3).
Encouraged by these results we advanced to higher substrate loadings by adding toluene as the organic phase to the reaction mixture (Figure 1). The oxidation of toluene proceeded almost linearly for more than 2 days, and more than 170 mm of benzaldehyde was accumulated in the organic phase. The concentration of benzyl alcohol steadily rose to approximately 7 mm within the first 8 h and then remained constant at this concentration throughout the experiment, which suggested that the second oxidation of benzyl alcohol to benzaldehyde was significantly faster than the first step (toluene to benzyl alcohol).
Figure 1.
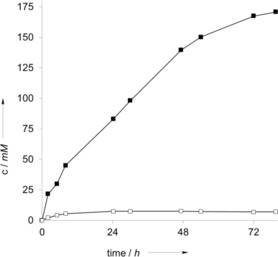
Representative time course for the photocatalytic oxidation of toluene in a biphasic system. Conditions: toluene/water [c(SAS)aq=1 mm]=3:7, ambient atmosphere, T=30 °C, λ>400 nm. ▪: benzaldehyde, □: benzyl alcohol. Values correspond to the concentration in the organic phase.
Indeed, repeating the same experiment but using benzyl alcohol as the organic phase resulted in a dramatic increase in the rate relative to the rate of oxidation of toluene (Figure 2). Whereas in the first case an average product formation rate of approximately 5.2 mm h−1 was observed, this value was roughly one order of magnitude higher in the case of benzyl alcohol as the starting material (49.5 mm h−1). In this experiment, approximately 24 % conversion of the benzyl alcohol was achieved within 80 h. The only byproduct detected was benzoic acid (60 mm in the aqueous phase corresponding to ≈5 % of the total product). Qualitatively, this trend was also observed with all other starting materials evaluated here (Tables 1 and 2). This “overoxidation activity” was pH dependent, and higher rates were found at more alkaline pH values. We attribute this to the formation of gem‐diols, which have abstractable H atoms and the pH‐dependent hydration equilibrium of benzaldehyde.7 In any case, under buffered conditions (pH 7) the overoxidation was practically negligible (Figure S5). Using benzaldehyde as the starting material gave only minor conversion under otherwise identical conditions.
Figure 2.
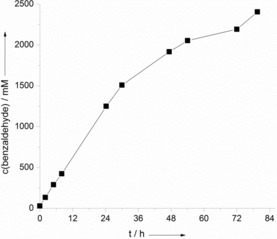
Time course for the photocatalytic oxidation of benzyl alcohol in a biphasic system. Conditions: benzyl alcohol/water [c(SAS)aq=1 mm]=3:7, ambient atmosphere, T=30 °C, λ>400 nm. Values correspond to the concentration in the organic phase.
Table 1.
Photocatalytic oxidation of some C−H bonds.[a]
| Entry | Substrate | Product | Product [mm] | Selectivity [%][b] | TON | TOF [h−1] |
|---|---|---|---|---|---|---|
| 1 |
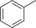
|
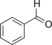
|
86.6 | 89.4 | 37.1 | 1.5 |
| 2 |
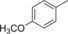
|
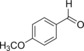
|
58.8 | 80 | 25.2 | 1.0 |
| 3 |
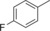
|
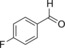
|
8.6 | 69.5 | 3.7 | 0.2 |
| 4 |
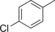
|
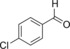
|
21.4 | 76 | 9.2 | 0.4 |
| 5 |
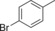
|
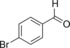
|
14.5 | n.d. | 6.2 | 0.3 |
| 6 |
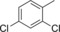
|
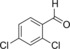
|
17 | n.d. | 7.3 | 0.3 |
| 7 |
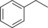
|
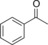
|
34.3 | 80.7 | 14.7 | 0.6 |
| 8[c] |
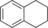
|
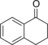
|
195 | 96 | 41.8 | 0.9 |
| 9 |
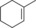
|
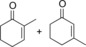
|
29.3+38.8 | n.d. | 12.6+16.6 | 0.5+0.7 |
| 10 |
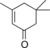
|
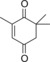
|
94.3 | n.d. | 40.4 | 1.7 |
| 11 |
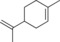
|
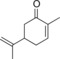
|
14.4 | n.d. | 6.2 | 0.3 |
| 12 |
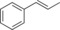
|
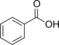
|
19.2 | n.d. | 8.2 | 0.34 |
| 13[d] |
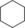
|
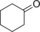
|
7.5/0.6[e] | n.d. | 3.2/0.4[e] | 0.13/0.02[e] |
| 14[d] |
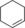
|
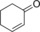
|
11.9/4.4[e] | n.d. | 5.1/3.1[e] | 0.21/0.1[e] |
[a] Conditions: alkane/water [c(SAS)aq=1 mm]=3:7, O2 atmosphere, T=30 °C, λ>400 nm, reaction time: 24 h; values correspond to the concentration in the organic phase; n.d.=not determined; TON=molproduct×molSAS −1; TOF=TON×24 h−1. [b] Selectivity was calculated on the basis of the sum of the aldehyde/ketone products and intermediates alcohols. [c] c(SAS)aq=2 mm, t=48 h. [d] Minor amounts of side products were detected by GC (Figures S16 and S17). [e] Product concentration in water phase.
Table 2.
Photocatalytic oxidation of some alcohols.[a]
| Entry | Substrate | Product | Product [mm] | TON | TOF [h−1] |
|---|---|---|---|---|---|
| 1 |
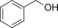
|
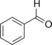
|
989 | 424 | 17.7 |
| 2 |
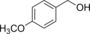
|
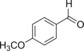
|
87.5 | 37.5 | 1.6 |
| 3 |
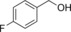
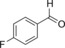
|
|
330 | 142 | 5.9 |
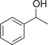
| 4 |
|
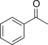
|
235 | 101 | 4.2 |
| 5 |
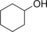
|
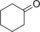
|
179/15.7[c] | 76.8/11.0[c] | 3.2/0.5[c] |
| 6 |
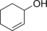
|
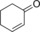
|
359/63.6[c] | 154/44.5[c] | 6.4/1.9[c] |
| 7 |
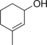
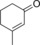
|
|
440 | 188 | 7.8 |
| 8[b] |
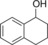
|
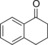
|
307 | 65.7 | 2.7 |
| 9 |
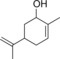
|
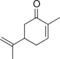
|
412 | 177 | 7.4 |
| 10 |
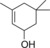
|
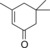
|
514 | 220 | 9.2 |
| 11 |

|
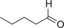
|
27.5 | 11.8 | 0.5 |
| 12 |
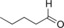
|
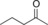
|
69.9 | 30.0 | 1.3 |
| 13 |
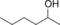
|
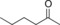
|
44.3 | 19.0 | 0.8 |
[a] Conditions: alcohol/water [c(SAS)aq=1 mm]=3:7, O2 atmosphere, T=30 °C, λ>400 nm, reaction time: 24 h; values correspond to the concentration in the organic phase; TON=molproduct×molSAS −1; TOF=TON×24 h−1. [b] c(SAS)aq=2 mm was used, t=48 h. [c] Product concentration in water phase.
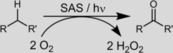
Next, we set out to characterize the novel photocatalytic reaction system further. As shown in Scheme 1, H2O2 was expected as a byproduct of the aerobic oxidation reaction. Indeed, we observed the accumulation of H2O2 in the course of the reaction (Figure S6); however, it was not formed in a stoichiometric amount. Most likely this can be attributed to competing H2O2‐oxidation activity of photoexcited SAS (Figure S7). To exclude a possible contribution from a H2O2 oxidation reaction, some control experiments were performed. Performing the reaction in the presence of H2O2 (100 mm) in the dark gave no conversion. Furthermore, performing the photochemical oxidation reaction either in the presence of catalase (to dismutate any H2O2 formed) or in the presence of an excess amount of H2O2 under conditions otherwise identical to those in the experiments outlined above had no significant influence on the time course of the reaction (Figure S8). Therefore, we concluded that H2O2 was not involved in the actual oxidation reaction.
The reaction rate correlated with the concentration of the photocatalyst applied (Figure 3). This correlation, however, was not linear with the photocatalyst concentration. The turnover frequency (TOF) of SAS (as determined within the first 3 h of reaction) decreased from 26.4 to 13.9 and 10.8 h−1 by using 0.25, 0.5, and 1 mm of SAS, respectively. Possibly, this may be attributed to decreasing transparency of the reaction medium, which leads to reduced photoexcitation of SAS. The rate of the reaction directly correlated with the light intensity applied (Figure S9), which supports this assumption. However, at present we also cannot fully rule out that the diffusion of O2 into the reaction system may become overall rate limiting at high overall conversion rates. Previously, we observed similar effects in the case of photoenzymatic oxidation reactions.8
Figure 3.
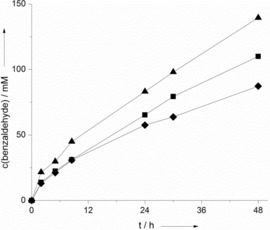
Time course for benzaldehyde formation at different SAS concentrations. The general conditions are described in Figure 1. c(SAS)aq=0.25 mm (♦), 0.5 mm (▪), and 1 mm(▴).
We also investigated the stability of SAS under the reaction conditions (transformation of toluene into benzaldehyde). As shown in Figure 4, SAS could be reused in consecutive batch reactions but continuously lost its catalytic activity (as indicated by the decreasing rate of toluene oxidation). After approximately 72 h, SAS exhibited 50 % of its initial activity (overall a turnover number, TON, of 71 was estimated for SAS). This loss in catalytic performance was also accompanied by significant structural modifications, as judged by 1H NMR spectroscopy (Figure S15). Probably, aromatic hydroxylation of SAS led to (photocatalytically) inactive species and, eventually, resulted in structural decomposition of the catalyst.9 This is partially supported by the occurrence of hydroxyl (.OH) radicals in the course of the photocatalytic reactions (Figure S16). It is, however, worth mentioning here that significantly higher TON values are possible, for example, with benzyl alcohol as the starting material (Table 2).
Figure 4.
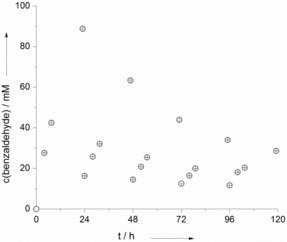
SAS recycling experiments. Conditions: toluene/water [c(SAS)aq=1 mm]=3:7, ambient atmosphere, T=30 °C, λ>400 nm. The organic phase was replaced every 24 h by fresh toluene and the reaction was continued.
To explore the scope of the photocatalytic oxidation reaction further, a range of activated and nonactivated alkanes and alcohols were evaluated (Tables 1 and 2). Again, the oxidation reactions of the alcohols were significantly faster than those of the alkanes and yielded up to 10 times more product than the corresponding alkanes. Also, activated C−H bonds (e.g., benzylic or allylic ones) yielded higher product concentrations.
Notably, the conversions of the experiments shown in Table 2 are generally below 20 %, which can be attributed to the low catalyst loading [substrate/catalyst (S/C) =<4000 mol mol−1]. Comparative experiments at more favorable S/C ratios (<M;<50:1 mol mol−1) gave full conversion within 2 days (Figure S25).
Wells and co‐workers6a,6b suggested H‐atom abstraction to be the overall rate‐limiting step followed by a series of deprotonation and secondary electron‐transfer steps. Accordingly, an increasing substitution pattern as well as electron‐donating substituents should increase the overall reaction rate. The results shown in Tables 1 and 2, however, do not allow for such a conclusion. Most probably, this can be assigned to the biphasic character of the reaction mixtures used. As a consequence, the aqueous concentrations of the different starting materials may differ significantly owing to their different partitioning coefficients. Hence, the different in situ concentrations of the reagents in the aqueous (SAS‐containing) phase may influence the oxidation rate to an extent similar to that of their oxidizability and thereby lead to the somewhat confusing correlation. In addition, the oxidation of aliphatic alcohols, as expected, is much slower than that of aromatic and allylic alcohols (Table 2, entries 11–13).
For anthraquinone, a H‐atom‐abstraction mechanism from the starting material to the photoexcited (triplet) catalyst has been established.4a,4b This radical mechanism most likely also applies to the SAS‐catalyzed reaction reported here (Scheme 2). Nevertheless, further studies clarifying the catalytic mechanism in more detail are currently underway in our laboratory.
Scheme 2.
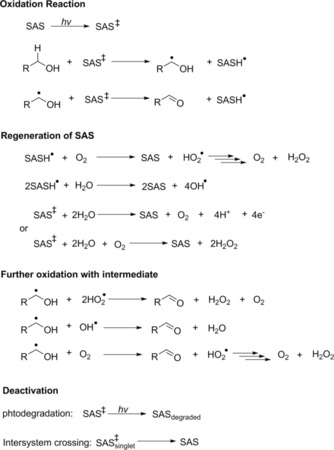
Plausible elementary steps involved in photochemical alcohol oxidation by using SAS.4a,4b, 6a,6b
Overall, we demonstrated that simple and commercially available sodium anthraquinone sulfonate could be used as a photocatalyst for the selective oxidation/oxyfunctionalization of a range of (non)activated C−H bonds in alkanes and alkanols. In particular, the selective oxidation of alcohols appears to be a promising method for photocatalytic, metal‐free oxidation by using molecular oxygen as the oxidant. Preliminary experiments in our laboratory also indicated that this reaction could be extended to amines and other functionalities. Future studies will concentrate on exploring the functional‐group preference of the present photocatalyst as well as further optimization of this reaction based on more in‐depth understanding of the catalytic mechanism.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by the European Research Council (ERC Consolidator Grant No. 648026) is gratefully acknowledged. The authors thank Prof. Fred Hagen (Delft University of Technology) for EPR measurements, Dr. S. Schmidt (Delft University of Technology) for GC analysis, and M. van Schie (Delft University of Technology) for characterization of the light source.
W. Zhang, J. Gacs, I. W. C. E. Arends, F. Hollmann, ChemCatChem 2017, 9, 3821.
Contributor Information
Dr. Wuyuan Zhang, Email: W.Zhang-1@tudelft.nl
Dr. Frank Hollmann, Email: F.Hollmann@tudelft.nl.
References
- 1. Sheldon R. A., Arends I. W. C. E., Hanefeld U., Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007. [Google Scholar]
- 2.
- 2a. ten Brink G. J., Arends I., Sheldon R. A., Science 2000, 287, 1636–1639; [DOI] [PubMed] [Google Scholar]
- 2b. Enache D. I., Edwards J. K., Landon P., Solsona-Espriu B., Carley A. F., Herzing A. A., Watanabe M., Kiely C. J., Knight D. W., Hutchings G. J., Science 2006, 311, 362–365; [DOI] [PubMed] [Google Scholar]
- 2c. Stahl S. S., Angew. Chem. Int. Ed. 2004, 43, 3400–3420; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3480–3501; [Google Scholar]
- 2d. Roduner E., Kaim W., Sarkar B., Urlacher V. B., Pleiss J., Gläser R., Einicke W.-D., Sprenger G. A., Beifuß U., Klemm E., Liebner C., Hieronymus H., Hsu S.-F., Plietker B., Laschat S., ChemCatChem 2013, 5, 82–112; [Google Scholar]
- 2e. Abad A., Concepcion P., Corma A., Garcia H., Angew. Chem. Int. Ed. 2005, 44, 4066–4069; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 4134–4137. [Google Scholar]
- 3.
- 3a. Fukuzumi S., Ohkubo K., Chem. Sci. 2013, 4, 561–574; [Google Scholar]
- 3b. Pandey G., Pal S., Laha R., Angew. Chem. Int. Ed. 2013, 52, 5146–5149; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5250–5253; [Google Scholar]
- 3c. Pandey G., Laha R., Angew. Chem. Int. Ed. 2015, 54, 14875–14879; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15088–15092; [Google Scholar]
- 3d. Mühldorf B., Wolf R., Angew. Chem. Int. Ed. 2016, 55, 427–430; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 437–441; [Google Scholar]
- 3e. Lechner R., Kummel S., König B., Photochem. Photobiol. Sci. 2010, 9, 1367–1377; [DOI] [PubMed] [Google Scholar]
- 3f. Schmaderer H., Hilgers P., Lechner R., König B., Adv. Synth. Catal. 2009, 351, 163–174; [Google Scholar]
- 3g. Neumann M., Füldner S., König B., Zeitler K., Angew. Chem. Int. Ed. 2011, 50, 951–954; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 981–985. [Google Scholar]
- 4.
- 4a. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]
- 4b. Ravelli D., Fagnoni M., Albini A., Chem. Soc. Rev. 2013, 42, 97–113; [DOI] [PubMed] [Google Scholar]
- 4c. Liu X., Lin L., Ye X., Tan C.-H., Jiang Z., Asian J. Org. Chem. 2017, 6, 422–425; [Google Scholar]
- 4d. Ohkubo K., Hirose K., Fukuzumi S., RSC Adv. 2016, 6, 41011–41014. [Google Scholar]
- 5.
- 5a. Nagasawa Y., Tanba K., Tada N., Yamaguchi E., Itoh A., Synlett 2015, 26, 412–415; [Google Scholar]
- 5b. Yamaguchi T., Kudo Y., Hirashima S.-I., Yamaguchi E., Tada N., Miura T., Itoh A., Tetrahedron Lett. 2015, 56, 1973–1975; [Google Scholar]
- 5c. Fujiya A., Nobuta T., Yamaguchi E., Tada N., Miura T., Itoh A., RSC Adv. 2015, 5, 39539–39543; [Google Scholar]
- 5d. Shimada Y., Hattori K., Tada N., Miura T., Itoh A., Synthesis 2013, 45, 2684–2688; [Google Scholar]
- 5e. Tada N., Ikebata Y., Nobuta T., Hirashima S.-i., Miura T., Itoh A., Photochem. Photobiol. Sci. 2012, 11, 616–619; [DOI] [PubMed] [Google Scholar]
- 5f. Tada N., Hattori K., Nobuta T., Miura T., Itoh A., Green Chem. 2011, 13, 1669–1671. [Google Scholar]
- 6.
- 6a. Wells C. F., Trans. Faraday Soc. 1961, 57, 1719–1731; [Google Scholar]
- 6b. Wells C. F., Trans. Faraday Soc. 1961, 57, 1703–1718; [Google Scholar]
- 6c. Clark K. P., Stonehil H. I., J. Chem. Soc. Faraday Trans. 1 1972, 68, 577–590; [Google Scholar]
- 6d. Clark K. P., Stonehil H. I., J. Chem. Soc. Faraday Trans. 1 1972, 68, 1676–1686. [Google Scholar]
- 7. Greenzaid P., J. Org. Chem. 1973, 38, 3164–3167. [Google Scholar]
- 8. Churakova E., Arends I. W. C. E., Hollmann F., ChemCatChem 2013, 5, 565–568. [Google Scholar]
- 9.
- 9a. Kosydar R., Drelinkiewicz A., Ganhy J. P., Catal. Lett. 2010, 139, 105–113; [Google Scholar]
- 9b. Theurich J., Bahnemann D. W., Vogel R., Ehamed F. E., Alhakimi G., Rajab I., Res. Chem. Intermed. 1997, 23, 247–274. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary