

Abstract
A four‐membered oxygen ring (oxetane) can be readily grafted into native peptides and proteins through site‐selective bis‐alkylation of cysteine residues present as disulfides under mild and biocompatible conditions. The selective installation of the oxetane graft enhances stability and activity, as demonstrated for a range of biologically relevant cyclic peptides, including somatostatin, proteins, and antibodies, such as a Fab arm of the antibody Herceptin and a designed antibody DesAb‐Aβ against the human Amyloid‐β peptide. Oxetane grafting of the genetically detoxified diphtheria toxin CRM197 improves significantly the immunogenicity of this protein in mice, which illustrates the general utility of this strategy to modulate the stability and biological activity of therapeutic proteins containing disulfides in their structures.
Keywords: antibodies, disulfides, immunogenic proteins, oxetanes, stapling
The rational modification of the structure of peptides and proteins offers a wide range of opportunities for the modulation of their biological activity.1 Many efforts have been made to develop strategies that induce such conformational changes and modulation. Towards this end, macrocyclization and stapling have emerged as useful tactics to chemically manipulate peptides and proteins, increasing their proteolytic stability, cell permeability, and producing changes in polarity, binding activity, and pharmacokinetic properties.2 During the last years, different approaches have been developed for the covalent tethering of the side chains of natural or non‐canonical amino acids.2 Considering natural residues, cysteine (Cys) has been the residue of choice for stapling through alkylation,3 arylation,4 cycloaddition,4b and disulfide forming reactions both at native5 or engineered6 Cys residues. More recently, nitrogen arylation has also been shown to be a useful strategy for macrocyclization of lysine residues on peptides.7 Otherwise, efficient macrocyclization of linear peptides through the formation of an oxadiazole has also been reported.8 However, a large number of stapling/macrocyclization/re‐bridging strategies consist of the introduction of non‐canonical amino acids and their subsequent ligation by ring‐closing metathesis,9 lactamization,10 or cycloaddition reactions.2c, 4b Common to many of these strategies is either the requirement for complicated orthogonal protection procedures, sequence engineering, the appendage of bulky/constrained linkers between the two residues, or the use of organic solvents. These conditions have limited, for instance, the application of such methods for the stapling of residues on intact, full‐length proteins to impart structural conformational constraints leading to enhanced stability and activity. Thus, there remains a need for simple and robust strategies for stapling native peptides and proteins.
Herein we report a method for site‐selective peptide and protein stapling through the bis‐alkylation of the sulfhydryl side chain of Cys residues resulting from disulfide reduction, using commercially available 3,3‐bis(bromomethyl)oxetane 1 (Figure 1). Oxetanes have become common motifs in drug design due to their ability to modulate parameters including solubility, basicity, lipophilicity, and metabolic stability.11 While there are examples of the modification of small peptides with oxetanes,12 their incorporation and modulation of the structure and activity of complex biomolecules13 remain mostly unexplored.
Figure 1.
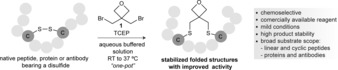
Stabilization of folded structures of peptides and proteins through bis‐alkylation of Cys residues present in the form of a native disulfide using an oxetane graft.
A requirement for a direct method to graft Cys residues present as disulfides on proteins is the compatibility of the reagent with a reducing agent, such as tris(2‐carboxyethyl)phosphine (TCEP). Importantly, we found that a model pentapeptide bearing two Cys residues reacted with 1 in the presence of TCEP to afford the corresponding stapled cyclic peptide in 75 % yield (see the Supporting Information, Figures S2–S5, S34–S39, 48–49 for characterization and discussion of structural features). We then explored the use of 1 to graft two cyclic and biologically relevant peptides: somatostatin 2 and its analogue octreotide 4, which can be used for imaging and treating neuroendocrine tumors.14 Peptides 2 and 4 were reacted simultaneously with TCEP and oxetane 1 in a 1:9 mixture of DMF/H2O at 25 °C for 12 h. After HPLC purification, the grafted cyclic peptides 3 and 5 were obtained in 76 % and 81 % yield, respectively (Figure 2 a). The affinity of these derivatives to the natural somatostatin receptor 2 (SSTR2) was experimentally determined by tryptophan fluorescence spectroscopy (Supporting Information, Figure S1). While octreotide 4 and surrogate peptide 5 showed a similar affinity against this receptor, grafted somatostatin 3 displayed improved binding properties, showing a 4‐fold enhancement in K D value (Figure 2 b). The improvement of binding activity is a considerable advantage of the incorporation of the oxetane graft when compared, for instance, with the recently reported methylene thioacetal that led to a decrease in binding affinity to SSTR2.3b Interestingly, 0.5 μs MD simulations performed on these derivatives in explicit water and using ff14SB amber force‐field15 suggested that octreotide 4 and its stapled derivative 5 presented a similar conformational behavior in solution (Figure 2 d; Supporting Information, Figure S8),16 displaying an equilibrium between antiparallel β‐sheet structures and conformations in which the C‐terminal residues form a 310 helix‐like fold, as reported in DMSO solution. In contrast, stapled somatostatin 3 was more rigid and displayed a more defined conformation in solution than 2 (Figure 2 c; Supporting Information, Figures S6 and S7).17 In fact, 3 showed a closely related β‐sheet arrangement in solution stabilized by a typical hydrogen bond network, which is apparently ideal for a more efficient binding to the receptor. Finally, analysis of the stability of 3 and 5 both in human plasma as well in the presence glutathione (GSH) showed that the oxetane grafted peptides remain intact under these conditions (Supporting Information, Figures S13–S18).
Figure 2.
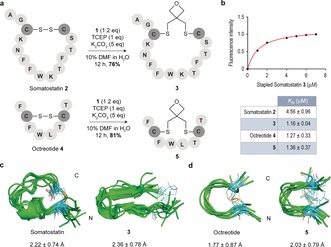
a) Stapling of disulfide‐containing cyclic peptides 2 and 4. b) Binding affinity studies. K D values were determined by tryptophan fluorescence spectroscopy. c),d) Structural ensembles obtained by 0.5 μs MD simulations. The peptide backbone is in green. Carbon atoms of Cys residues as well as of the oxetane moiety are in cyan. The numbers indicate the root‐mean‐square deviation (RMSD) for heavy‐atom superimposition of the backbone with respect to the average structure.
To initially test the potential of using this one‐pot, site‐selective bis‐alkylation oxetane stapling method directly on proteins, we chose thioredoxin (Trx) as a model protein that features a naturally occurring, solvent‐exposed disulfide bond. We could reduce and staple the disulfide bond in a straightforward manner through selective bis‐alkylation with 1 in the presence of TCEP and 10 % DMF in sodium phosphate buffer at pH 8.5. Complete conversion was achieved after 24 h at 37 °C, as confirmed by HPLC‐MS analysis (Supporting Information, Figures S19 and S20). Furthermore, analysis of the CD spectra of the native and stapled Trx‐1 (Supporting Information, Figure S22) indicated that both molecules present very similar conformational preferences in solution. Although this is supported by MD simulations performed on both proteins in explicit water, the calculations indicate a small increase in flexibility for the peptide backbone of stapled Trx‐1 (Supporting Information, Figure S11). This result may be explain attending to the greater S−S distance in Thrx‐1 when compared to the native Trx (4.18 and 2.04 Å, respectively). Finally, we confirmed the suppression of Trx redox activity18 through the selective and covalent disulfide stapling (Supporting Information, Figure S21).
Next, we demonstrated the utility of the oxetane graft to build stapled antibodies. First, the exposed disulfide bond tethering the heavy and light chains of a Fab fragment of Herceptin (Fab‐Her), an antibody currently used to treat Her2+ breast cancer patients,19 was readily stapled using 1 under aqueous buffered conditions in the presence of TCEP at pH 8.5 and at 37 °C (Figures 3 a,b; Supporting Information, Figures S23 and S24). The oxetane stapled Fab‐Her‐1, unlike the disulfide native antibody, was stable under reducing conditions and in human plasma (Supporting Information, Figures S25 and S26). This stability is a key aspect of antibody therapeutics design as thiol‐exchange reactions in plasma lowers efficacy and adds side‐toxicity.20 Importantly, a relatively small but significant increase in binding affinity to the Her2 receptor, as determined by bio‐layer interferometry (BLI) experiments (Figure 3 d; Supporting Information, Figure S27), was observed for Fab‐Her‐1 when compared with the native antibody. Next, we extended our stapling strategy to the antibody DesAb‐Aβ3‐9, which was designed to target the region 3–9 of human Amyloid‐β (Aβ42) peptide, the aggregation of which is a hallmark of Alzheimer's disease.21 This antibody features a challenging, hindered intra‐domain disulfide typical of VH domains. Of note, complete conversion into the oxetane grafted antibody DesAb‐Aβ3‐9‐1 was achieved using our method (Figure 3 c; Supporting Information, Figures S28 and S29). Owing to the fact that the disulfide is deeply buried, an excess of TCEP (40 equiv) and longer reaction times were required (Supporting Information). Unlike the reduced antibody that readily reacts with thiol‐specific Elman's reagent, the stapled DesAb‐Aβ3‐9‐1 did not react suggesting complete consumption of the reduced Cys during stapling (Supporting Information). Finally, analysis of secondary structural content by CD showed no significant differences between the original and stapled antibodies (Figure 3 e). MD simulations performed on a 3D model of DesAb‐Aβ3‐9, previously generated using ABodyBuilder,22 and on the stapled derivative DesAb‐Aβ3‐9‐1, suggest that, although the 3D structure is maintained upon the chemical modification, the oxetane motif provokes a slightly increase in the degree of flexibility (Supporting Information, Figure S31). Collectively, these data demonstrate the suitability of the oxetane motif to staple solvent accessible disulfide bonds on proteins with minimal secondary structure alterations.
Figure 3.
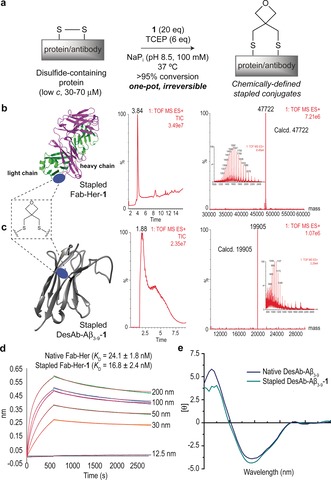
a) Representation of disulfide stapling of native antibody sequences using an oxetane graft. b),c) Total ion chromatogram, combined ion series and deconvoluted mass spectrum reconstructed from the ion series using the MaxEnt algorithm. b) Fab‐Her‐1 (pdb ID Fab‐Her: 1N8Z) and c) DesAb‐Aβ3‐9‐1 (3D model of DesAb‐Aβ3‐9 generated using ABodyBuilder, see main text). d) BLI and fit curves obtained for Fab‐Her‐1, together with the derived K D constants for Fab‐Her and Fab‐Her‐1. e) CD spectra of DesAb‐Aβ3‐9 and DesAb‐Aβ3‐9‐1.
To demonstrate the practical application of our method to therapeutic proteins, we investigated the effects of the selective introduction of the oxetane staple into the genetically detoxified diphtheria toxin CRM197, which features four Cys residues in the form of two disulfides. Recently, it has been shown that antibodies against CRM197 neutralized diphtheria toxin in HIV infected young individuals.23 Furthermore, CRM197 is a clinically used carrier in many glycoconjugate vaccines.24 Previous structural studies showed that only the disulfide C186‐C201 connecting the fragment A (C domain) and B (T/R domain) of CRM197 is selectively reduced in the presence of the highly hindered C461–C471 disulfide upon treatment with dithiothreitol.24b Addition of a slight excess of TCEP to CRM197 under aqueous buffered conditions at pH 8.5 and 37 °C, followed by an excess of 1, led to the introduction of one oxetane graft (Figure 4 a,b and the Supporting Information, Figure S32 for mass spectrometry analysis), presumably at C186–C201 according to our previous findings.25 The impact of the installation of the oxetane moiety into CRM197 on its structure and thermal stability was studied by CD and differential scanning calorimetry (DSC) analysis, respectively, and compared with the native protein (Figure 4 c–e). We found that both the far and near UV CD spectra of CRM197‐1 were nearly identical to those of CRM197, which indicates that the 3D structure is preserved upon the chemical stapling. The DSC curves also corroborate this finding. Although CRM197‐1 exhibited a broader DSC peak when compared to the sharp change in heat capacity, in the range of 40–55 °C for CRM197, both proteins present an identical transition midpoint (T m) of 46 °C.
Figure 4.
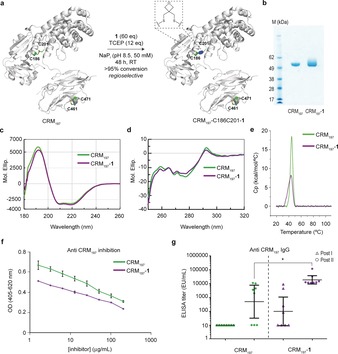
Enhancing the immunogenicity of a protein carrier through disulfide oxetane stapling. a) The functional stapling of CRM197 (pdb ID CRM197: 4AE0) with 1. b) SDS‐page of native and stapled CRM197 ‐1. c) Far UV CD spectrum. d) Near UV CD spectrum. e) DCS analysis. f) Competition of anti‐CRM197 serum binding to the protein with CRM197 and its stapled form as inhibitors. g) Anti‐CRM197 IgG levels of CRM197 and CRM197‐1 after first and second boost immunizations in mice, 2 weeks apart.
To evaluate the biological effects resulting from the introduction of the oxetane staple into CRM197, we first assayed the capacity of competing with the binding of anti‐CRM197 serum to the proteins. We found that the stapled CRM197‐1 induces an inhibition that was slightly lower compared to the unmodified protein (Figure 4 f). In contrast, a much better inhibition of the binding to a commercial anti‐diphtheria toxoid human recombinant monoclonal antibody was observed for CRM197‐1 compared to the unmodified protein (Supporting Information, Figure S33a). To ascertain that protein epitopes were not impaired by the chemical modification of the disulfide bond, groups of 8 BALB/c mice were immunized with both unmodified CRM197 and the stapled CRM197‐1 (Figure 4 g). Remarkably, these in vivo experiments demonstrated that CRM197‐1 induced a statistically significant higher level of anti‐protein antibodies respect to the unmodified protein. The antibodies generated by CRM197‐1 had a threefold higher avidity for the protein antigen compared to the anti‐CRM197 serum (avidity index=0.8±0.4 m for CRM197‐1 vs. 0.3±0.1 m for CRM197), as determined by ELISA using thiocyanate elution (Supporting Information, Figure S33b). These data, together with 200 ns MD simulations performed on both proteins (Supporting Information, Figure S12), suggest that the oxetane bridging of the disulfide bond does not cause relevant structural modifications on the protein but results in improved immunogenic activity in vivo, most likely through chemical stabilization of the antigen against proteases and/or other degradation factors.
In summary, we have presented an efficient method for oxetane stapling of Cys residues present as native disulfides on peptides and proteins under mild and biocompatible aqueous conditions. The four‐membered oxetane ring has an ideal distance to enable direct stapling of native disulfides on several protein scaffolds, including antibodies. This approach is however dependent on solvent accessibility of the disulfide within the protein of interest. Furthermore, and unlike current protocols, this method does not require prior sequence engineering neither purification after the disulfide reduction step. The selective installation of the oxetane motif enables stabilization of folded structures and results in disulfide‐grafted products with enhanced bioactivity that are stable under biological conditions. We demonstrate the value of oxetane graft installation on protein through the regioselective disulfide stapling of the protein carrier CRM197 that showed a significant increase in its immunogenicity in vivo. Because many therapeutic proteins feature Cys residues in the form of disulfide bonds, we anticipate that their direct modulation through oxetane grafting can, in principle, be used as a general strategy to enhance their in vivo stability and to fine‐tune their structure for optimal pharmacokinetics and activity.
Conflict of interest
N.M.S., S.S., O.B., F.Corzana, and G.J.L.B. are listed as inventors on a pending patent application related to the technology described in this work. F.Carboni, D.O., and R.A. are employees of GSK companies.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank FCT Portugal, the EU (Marie Curie IEF to O.B.; Marie Sklodowska‐Curie ITN GlycoVax to G.J.L.B. and R.A.), Cambridge Trust and China Scholarship Council (PhD studentship to S.S.), MINECO (CTQ2015‐67727‐R and Salvador de Madariaga mobility grant to F.Corzana) and the EPSRC for funding. I.C. thanks Universidad de La Rioja for a FPI fellowship. We thank Dr. Vijay Chudasama for providing the Fab‐Her antibody, Dr. Werner Pansegrau (GSK Vaccines) for acquiring CD spectrum and DSC profile, and Dr. Francesco Aprile for providing the construct of the single domain antibody DesAb‐Aβ3‐9. G.J.L.B. is a Royal Society URF and the recipient of a ERC Grant (TagIt).
N. Martínez-Sáez, S. Sun, D. Oldrini, P. Sormanni, O. Boutureira, F. Carboni, I. Compañón, M. J. Deery, M. Vendruscolo, F. Corzana, R. Adamo, G. J. L. Bernardes, Angew. Chem. Int. Ed. 2017, 56, 14963.
References
- 1.
- 1a. Bock J. E., Gavenonis J., Kritzer J. A., ACS Chem. Biol. 2013, 8, 488–499; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Krall N., da Cruz F. P., Boutureira O., Bernardes G. J. L., Nat. Chem. 2016, 8, 103–113. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Góngora-Benítez M., Tulla-Puche J., Albericio F., Chem. Rev. 2014, 114, 901–926; [DOI] [PubMed] [Google Scholar]
- 2b. White C. J., Yudin A. K., Nat. Chem. 2011, 3, 509–524; [DOI] [PubMed] [Google Scholar]
- 2c. Lau Y. H., De Andrade P., Quah S. T., Rossmann M., Laraia L., Sköld N., Sum T. J., Rowling P. J. E., Joseph T. L., Verma C., Hyvönen M., Itzhaki L. S., Venkitaraman A. R., Brown C. J., Lane D. P., Spring D. R., Chem. Sci. 2014, 5, 1804–1809. [Google Scholar]
- 3.
- 3a. Assem N., Ferreira D. J., Wolan D. W., Dawson P. E., Angew. Chem. Int. Ed. 2015, 54, 8665–8668; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8789–8792; [Google Scholar]
- 3b. Kourra C. M. B. K., Cramer N., Chem. Sci. 2016, 7, 7007–7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Spokoyny A. M., Zou Y., Ling J. J., Yu H., Lin Y.-S., Pentelute B. L., J. Am. Chem. Soc. 2013, 135, 5946–5949; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Brown S. P., Smith A. B., J. Am. Chem. Soc. 2015, 137, 4034–4037; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Kalhor-Monfared S., Jafari M. R., Patterson J. T., Kitov P. I., Dwyer J. J., Nuss J. M., Derda R., Chem. Sci. 2016, 7, 3785–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuan S. L., Wang T., Weil T., Chem. Eur. J. 2016, 22, 17112–17129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu T., Wang Y., Luo X., Li J., Reed S. A., Xiao H., Young T. S., Schultz P. G., Proc. Natl. Acad. Sci. USA 2016, 113, 5910–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lautrette G., Touti F., Lee H. G., Dai P., Pentelute B. L., J. Am. Chem. Soc. 2016, 138, 8340–8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frost J. R., Scully C. C. G., Yudin A. K., Nat. Chem. 2016, 8, 1105–1111. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Schafmeister C. E., Po J., Verdine G. L., J. Am. Chem. Soc. 2000, 122, 5891–5892; [Google Scholar]
- 9b. Blackwell H. E., Grubbs R. H., Angew. Chem. Int. Ed. 1998, 37, 3281–3284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 3469–3472. [Google Scholar]
- 10. Shepherd N. E., Hoang H. N., Abbenante G., Fairlie D. P., J. Am. Chem. Soc. 2005, 127, 2974–2983. [DOI] [PubMed] [Google Scholar]
- 11. Burkhard J. A., Wuitschik G., Rogers-Evans M., Müller K., Carreira E. M., Angew. Chem. Int. Ed. 2010, 49, 9052–9067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9236–9251. [Google Scholar]
- 12.
- 12a. McLaughlin M., Yazaki R., Fessard T. C., Carreira E. M., Org. Lett. 2014, 16, 4070–4073; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Powell N. H., Clarkson G. J., Notman R., Raubo P., Martin N. G., Shipman M., Chem. Commun. 2014, 50, 8797–8800. [DOI] [PubMed] [Google Scholar]
- 13. Boutureira O., Martínez-Sáez N., Brindle K. M., Neves A. A., Corzana F., Bernardes G. J. L., Chem. Eur. J. 2017, 23, 6483–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolin E. M., Gastrointest. Cancer Res. 2012, 5, 161–168. [PMC free article] [PubMed] [Google Scholar]
- 15. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., Simmerling C., J. Chem. Theory Comput. 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Melacini G., Zhu Q., Goodman M., Biochemistry 1997, 36, 1233–1241. [DOI] [PubMed] [Google Scholar]
- 17. Anoop A., Ranganathan S., Dhaked B. D., Jha N. N., Pratihar S., Ghosh S., Sahay S., Kumar S., Das S., Kombrabail M., Agarwal K., Jacob R. S., Singru P., Bhaumik P., Padinhateeri R., Kumar A., Maji S. K., J. Biol. Chem. 2014, 289, 16884–16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holmgren A., Annu. Rev. Biochem. 1985, 54, 237–271. [DOI] [PubMed] [Google Scholar]
- 19. Hudis C. A., N. Engl. J. Med. 2007, 357, 39–51. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Chudasama V., Maruani A., Caddick S., Nat. Chem. 2016, 8, 114–119; [DOI] [PubMed] [Google Scholar]
- 20b. Donaghy H., mAbs 2016, 8, 659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Sormanni P., Aprile F. A., Vendruscolo M., Proc. Natl. Acad. Sci. USA 2015, 112, 9902–9907; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Aprile F. A., Sormanni P., Perni M., Arosio P., Linse S., Knowles T. P. J., Dobson C. M., Vendruscolo M., Sci. Adv. 2017, 3, e1700488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leem J., Dunbar J., Georges G., Shi J., Deane C. M., mAbs 2016, 8, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silva G. P., Santos R. S., Pereira-Manfro W. F., Ferreira B., Barreto D. M., Frota A. C. C., Hofer C. B., Milagres L. G., Vaccine 2017, 35, 3803–3807. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Tontini M., Romano M. R., Proietti D., Balducci E., Micoli F., Balocchi C., Santini L., Masignani V., Berti F., Costantino P., Vaccine 2016, 34, 4235–4242; [DOI] [PubMed] [Google Scholar]
- 24b. Malito E., Bursulaya B., Chen C., Surdo P. L., Picchianti M., Balducci E., Biancucci M., Brock A., Berti F., Bottomley M. J., Nissum M., Costantino P., Rappuoli R., Spraggon G., Proc. Natl. Acad. Sci. USA 2012, 109, 5229–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stefanetti G., Hu Q.-Y., Usera A., Robinson Z., Allan M., Singh A., Imase H., Cobb J., Zhai H., Quinn D., Lei M., Saul A., Adamo R., MacLennan C. A., Micoli F., Angew. Chem. Int. Ed. 2015, 54, 13198–13203; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13396–13401. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary