

Abstract
Antimonene, a novel group 15 two‐dimensional material, is functionalized with a tailormade perylene bisimide through strong van der Waals interactions. The functionalization process leads to a significant quenching of the perylene fluorescence, and surpasses that observed for either graphene or black phosphorus, thus allowing straightforward characterization of the flakes by scanning Raman microscopy. Furthermore, scanning photoelectron microscopy studies and theoretical calculations reveal a remarkable charge‐transfer behavior, being twice that of black phosphorus. Moreover, the excellent stability under environmental conditions of pristine antimonene has been tackled, thus pointing towards the spontaneous formation of a sub‐nanometric oxide passivation layer. DFT calculations revealed that the noncovalent functionalization of antimonene results in a charge‐transfer band gap of 1.1 eV.
Keywords: antimonene, electron microscopy, charge transfer, phosphorus, surface chemistry
Two‐dimensional (2D) materials have attracted enormous attention during the last few years because of their outstanding properties and applications.1, 2, 3 Beyond gapless graphene, other elemental 2D materials have been successfully synthesized, with black phosphorus (BP) being the only semiconducting material reported so far.4, 5, 6, 7 This result together with its excellent charge‐carrier mobility and current on/off ratios renders BP a perfect candidate for nanoelectronics and nanophotonics. However, its (in)stability represents a major drawback for the development of the real applications.8, 9, 10, 11 In contrast, its relative in the periodic table, antimonene, that is, a single layer of antimony, exhibits a band gap of about 1.8–2.4 eV and an outstanding stability under ambient conditions. Antimonene has been recently isolated for the very first time both by mechanical exfoliation12 and liquid‐phase exfoliation13 as reported by our groups. A number of theoretical calculations predict extraordinary physical properties like high carrier mobility,14 thermal conductivity,15 and strain‐induced band transition,16 among others.
Therefore, antimonene appears to be a promising platform for high‐performance sensors,17 double‐gate MOSFETs,18 spintronics,19, 20 optoelectronic applications,21, 22 energy storage and conversion,23 and biomedicine.24 Recently, van der Waals and molecular beam epitaxy (MBE) have been applied for the synthesis of antimonene on different surfaces, thereby exhibiting good stabilities and high electrical conductivities of up to 104 S m−1 in about 30 nm thick flakes.25, 26 Despite synthetic efforts, the chemistry and therefore the molecular doping of antimonene remains completely unexplored. In relation to that, we have recently reported on the noncovalent functionalization of BP with electron‐poor and polarizable polycyclic aromatic molecules, thus observing a remarkable charge‐transfer behavior, and improving the resistance of the flakes against oxygen degradation.27, 28 In this context, the present work nicely illustrates, for the first time, the noncovalent functionalization of antimonene with a perylene bisimide (PDI; Figure 1 a), thereby showing a more pronounced charge‐transfer behavior than BP. Moreover, we have performed scanning X‐ray photoelectron microscopy experiments combined with DFT calculations to shed light on the charge‐transfer process.
Figure 1.
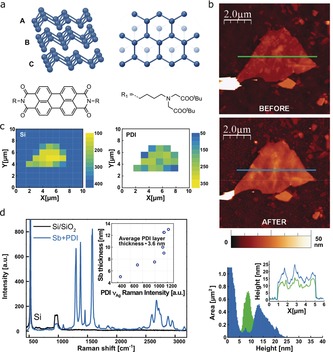
a) Structure of β‐antimonene (top panel) and the perylene bisimide (PDI) molecule (bottom panel). b) AFM topographic images showing an antimonene flake of about 10 nm of thickness. Top: flake as deposited. Middle: same flake after the functionalization with PDI molecules. Bottom: height histograms of the flake before (green) and after functionalization (blue), showing an average thickness increase of 4.1 nm. The average PDI coverage in all the studied flakes was 3.6 nm. The inset shows representative profiles corresponding to the lines in the images. c) Scanning Raman microscopy (SRM) of the same flake. Left: silicon intensity Raman map showing a decrease in the 521 cm−1 signal which clearly reveals the morphology of the flake. Right: Raman intensity mapping shows the exclusive self‐assembly of the PDI on the antimonene flakes and not on the Si/SiO2 substrate. d) Mean Raman spectra (excitation at 532 nm) of the flake showing the PDI bands as a consequence of the quenching of its fluorescence. The correlation between the PDI νAg Raman intensities and the flake thicknesses is highlighted in the inset.
Here, to investigate the noncovalent functionalization of antimonene, we chose micromechanically exfoliated flakes supported on SiO2/Si and gold surfaces.12 Specifically, we carried out the functionalization process by drop‐casting a THF solution (1E−5 m) of a tailormade EDTA‐PDI (EDTA=ethylenediaminetetraacetic acid) derivative on the exfoliated flakes, followed by a thorough washing process after the functionalization (see the Supporting Information for details). This family of compounds exhibits high performance in the functionalization of several nanomaterials such as carbon nanotubes, graphene, molybdenum disulfide (MoS2), and BP.27, 29, 30, 31 The Raman characterization of very thin antimonene flakes, obtained by microexfoliation, is challenging because of the very low non‐resonant Raman intensities, as recently reported by our group.12, 13 Therefore, fast detection of the exfoliated flakes is precluded by Raman spectroscopy, so we have recently optimized optical microscopy conditions to facilitate the identification of few‐layer antimonene flakes.32 The use of scanning probe microscopy techniques, however, is still imperative for an accurate determination of their thicknesses. Prior to the functionalization process, we characterized several flakes by atomic‐force microscopy (AFM), showing thicknesses in the 2.7–15 nm range (see Figures S1–S3 in the Supporting Information). Interestingly, the statistical AFM characterization of all samples after the functionalization process revealed an average increase in the thicknesses of the flakes of about 3.6 nm because of the self‐assembly of the PDI molecules on their surfaces (Figure 1 b; see Figure S4). This result is in excellent accordance to PDI‐protected BP flakes, covalent aryl diazonium functionalization of graphene and BP, and recent theoretical calculations on BP‐protected with perylene‐3,4,9,10‐tetracarboxylic dianhydride (PTCDA).27, 33, 34 It is worth noting that the protection of the flakes with layers of greater than 2 nm self‐assembled PDI molecules results in an effective passivation of the outer layers from the ambient O2 and H2O.34 Moreover, there is no apparent correlation between flake thickness and organic covering (see Figure S4). Optical microscopy analysis of functionalized samples revealed an appreciable increase of the optical contrast under white‐light illumination, and increases with the thickness of the PDI organic layer (see Figures S5–S8). When scanning Raman microscopy (SRM) measurements of the same flakes after the deposition of the PDI molecules were performed, a significant quenching of the fluorescence was observed, thus allowing the characterization of the PDI spectrum with an outstanding quality. Even more appealing was the exclusive self‐assembly of the PDI molecules on the antimonene surface (Figure 1; see Figures S9–S11), thereby leaving the SiO2 substrate free of significant traces of this molecule. This quenching of the fluorescence allowed us to easily image the flakes in seconds by SRM without needing to resolve the elusive Sb peaks.35 Remarkably, the quenching of the fluorescence is stronger than that observed for BP or even monolayer graphene, thus suggesting a more effective interaction by charge transfer (see below).27, 36 There is a correlation between the PDI Raman intensities, that is, the quenching of the fluorescence, and the thicknesses of the flakes when the organic coverage is below 4.5 nm (about 10 monolayers), as depicted in the inset of Figure 1 d. Interestingly, for thicker organic coverage, a slight increase of the background fluorescence, accompanied by a decrease in the PDI intensities, was observed. These results indicate that if the number of PDI layers exceeds a certain value, the fluorescence of the outer PDI layers contribute to the Raman spectrum. To shed light on the fluorescence quenching we also studied the effect of the excitation wavelength (namely: 785, 633 and 532 nm) on the Raman spectra of Sb‐PDI. For this experiment we used a flake of about 50 nm in which both the signatures of antimonene and the PDI moieties can be clearly observed, thus confirming the quenching of the fluorescence for all the wavelengths (see Figure S12).
Whereas for BP a slight change in its electronic properties is expected with a negligible influence on the band gap after functionalization,27, 34 in the case of antimonene this behavior is still an open question. In addition, antimonene exhibits excellent stability against environmental degradation, but a detailed analysis by electron energy loss spectroscopy (EELS) revealed that the flakes present a small amount of oxygen contribution.13 The same effect was observed by energy dispersive spectroscopy (EDS) for flakes grown by van der Waals epitaxy.25 To shine light on the possible presence of oxygen on the antimonene flakes and gain knowledge on the functionalization process, a forefront technique, namely XPS photoelectron microscopy using synchrotron radiation on pristine and functionalized antimonene flakes was performed (Figure 2). This technique allowed us to obtain both spatial and chemical resolution of the surface of the flakes with outstanding precision. In particular, the XPS data on the different flakes, deposited on gold surfaces, exhibited similar results, thus showing the presence of an oxidation layer with a binding energy (BE; 530.9 eV) larger than the value expected for Sb2O3 (530 eV) and slightly higher than the value expected for Sb2O5 (530.8 eV). Following the same trend, the O2 peak presented a higher BE value (532.61 eV) compared to most of the metallic oxides (531–528 eV), thus pointing to a superficial oxidized component present only on the outer surface and not presenting a conventional oxidation pattern. It is worth mentioning that these flakes were prepared several weeks before the XPS experiments and stored under atmospheric conditions. Accordingly, the oxidation took place in the presence of oxygen, thus creating a static passivation layer. Indeed, recent theoretical predictions claim the feasibility of superficial oxidation of antimonene flakes creating the so‐called antimonene oxides.37 These results clearly point towards a sub‐nanometric oxide passivation layer as the origin of the environmental stability shown by antimonene. Moreover, this oxidation could have an influence in observed fluorescence quenching.
Figure 2.
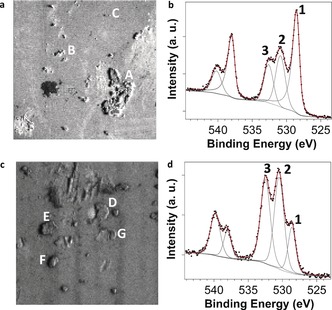
a) Sb 3d image for the reference sample. Letters refer to antimonene flakes specifically studied. b) Sb 3d peak for the reference sample, corresponding to point B in a). Numbers identify the different components: 1 (Sb 3d5/2), 2 (oxidized Sb 3d5/2), and 3 (O 1s). Peaks at higher BEs are the Sb 3d3/2 components. Black dots are experimental points and the red line is the results of fit (see the Supporting Information). Grey lines correspond to the different components used and the background. c) The Sb 3d image for the functionalized sample. Letters refer to antimonene flakes specifically studied. d) Sb 3d peak for the functionalized sample, corresponding to point E in c). The peak identification and other details are as in b). The size of images in a) and c) are 40×40 μm.
Figure 2 shows the Sb 3d image of the functionalized sample prepared on gold substrates, and its corresponding overall spectra, which is representative for all other recorded spectra. The overall line shape is similar to that observed within the reference sample. Again, the spectrum exhibits a metallic‐like Sb 3d contribution, which is now less intense than the fraction of oxidized Sb (see Table S1 for BE values). Besides the differences in intensities, an interesting aspect is the change in separation between the metallic and the oxidized components (see Table S1 and Figure S13).
The energy separation for the reference sample is 2.26 eV, while the difference for the functionalized sample is 1.87 eV, thus there is a core‐level shift of about 0.4 eV, and it is induced by the functionalization. The sign of the shift indicates that these Sb species are oxidized with respect to metallic Sb, but the charge transfer is less pronounced than in the oxide. These results indicate that the functionalizing molecule acts as an electron acceptor, which fits perfectly with the electron‐withdrawing behavior exhibited by PDI molecules on other related 2D materials.27, 30
To complement the experimental results, the supramolecular interaction of our PDI on antimonene was systematically investigated by DFT calculations. Concretely, PDI, taking into account the π core and the first methyl group of the peripheral branches for reducing the computational effort, was modeled (Figure 3; see the Supporting Information). We also computed one peripheral branch with half of the PDI core adsorbed on antimonene, and thus further confirmed the parallel disposition of the PDI core with respect to the antimonene surface (see Figure S14). The adsorption energy of the PDI core with single‐layer antimonene is found to be −1.27 eV, which indicates a strong noncovalent interaction (adsorption energies per molecule are defined as: E a=E (PDI‐Sb)−E (PDI)−E (Sb), where E (PDI‐Sb), E (PDI), and E (Sb) stand for the total energy of the PDI‐modified antimonene, the isolated PDI, and the isolated antimonene, respectively). The analysis of the energy contributions reveals that the binding is only due to van der Waals interactions, which is illustrated by the fact that the contribution from the van der Waals correction to the binding energy is −1.36 eV, that is, without this correction PDI would be repelled farther from the antimonene monolayer. In the case of double‐layer antimonene, the adsorption energy increases to −1.72 eV, which matches well with the observed Raman behavior (Figure 1). The distance between PDI and antimonene is 3.4 Å (3.6 Å in the case of the double‐layer), which is very similar to that exhibited by the same PDI on top of monolayer BP (3.3 Å; see the Supporting Information for further details), despite the atomic radius of Sb being significantly larger than that of phosphorus. Interestingly, these short distances, as well as the electron‐withdrawing character of PDIs, lead to a charge transfer from the 2D material to the PDI. For antimonene, this charge transfer is approximately twice as large as for the BP‐PDI system, and primarily based on the contribution of the Sb atoms in the vicinity of the oxygen atoms of PDI (Figure 3 b). Concerning the band gap of antimonene, values of 1.8 eV for the monolayer, 0.4 eV for the double layer, and no band gap for the bulk were predicted. These values are different from those computed by Zhang et al.38 who, however, obtained far too small layer distances for the bulk compared to the experiment. The calculated bulk parameters in this contribution agree with experiment within 1.5 % (see the Supporting Information). With respect to the functionalization process, inspection of the band structures and density of states (DOSs) shows that the highest occupied band of the PDI‐functionalized antimonene sheets is formed by antimonene orbitals, whereas the lowest unoccupied band is built of PDI orbitals (see Figure S15). The HOMO–LUMO excitation of PDI‐functionalized antimonene sheets is thus of charge transfer character. The antimonene/BP band structures themselves show only minor changes upon adsorption of PDI (see the Supporting Information for details).30 However, because of the “PDI band” forming the lowest unoccupied band, the calculated band gap of the PDI‐antimonene system is 1.05 eV, that is, the adsorption‐induced band gap change is −0.75 eV, and in good accordance with the XPS experiments.
Figure 3.
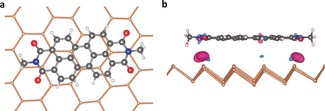
a) Top and b) side view of the optimized geometries of PDI core on single‐layer Sb. The electron density difference between the PDI‐antimonene and the isolated molecules and antimonene is also shown in the side view (for an isovalue of ±0.0005|e| Å−3, blue/red indicates higher/lower electron density, respectively).
To further demonstrate the compatibility of this supramolecular approach with the preparation of devices, we investigated whether it is possible to completely desorb the PDI adlayer from the antimonene surface or not (Figure 4). For this purpose we studied the stability of FL‐Sb layers with temperature. Firstly, we performed a temperature‐dependent Raman analysis of an antimonene flake of about 50 nm (see Figure S16), thus showing that the antimonene flakes are stable below 400 °C (both on SiO2/Si and gold substrates), and a complete degradation takes place at about 450 °C (a temperature remarkably smaller than the bulk melting point of ca. 630 °C). Moreover, to complement our results we performed a temperature‐dependent Raman analysis of the samples previously characterized by XPS microscopy (consisting of antimonene flakes deposited on gold substrates). The desorption of the PDI molecules is reflected in a concomitant decrease of the intensity of the characteristic PDI peaks, as well as a progressive increase of the fluorescence background, until its complete disappearance above about 300 °C (see Figure S17). Furthermore, we performed a controlled study of desorption of the PDI by means of AFM, thus measuring the flakes, previously characterized by optical, Raman spectroscopy and AFM on SiO2/Si substrates, after a controlled thermal treatment under inert atmosphere at 300, 350, and 400 °C (see experimental information and Figures S18–21 for details). We observed that at 300 °C the PDI molecules are partially removed, and by further increasing the temperature to 400 °C we were able to completely eliminate the adlayer from the surface of the antimonene, thus giving access to the pristine original surface. This result represents a big advantage compared to BP and paves the way for the thermal annealing of antimonene flakes in the preparation of devices.
Figure 4.
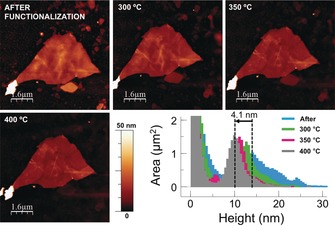
Thermal desorption of the PDI adlayer from the antimonene surface characterized by means of AFM topography images measured after heating at different temperatures, and the corresponding histograms showing the decrease in the thickness of the PDI organic layer (4.1 nm). After heating at 400 °C the original value of the pristine flake was recovered, thus confirming the complete thermal removal of the PDI.
Finally, to explore the validity of this noncovalent functionalization, we performed the functionalization of antimonene dispersions prepared by liquid‐phase exfoliation (LPE). For this purpose, we prepared an propan‐2‐ol antimonene dispersion (0.011 mg mL−1) exhibiting an average thickness of about 8 nm and lateral dimensions below 5 μm2, and added a 10−5 m solution of the PDI under magnetic stirring (see the Supporting Information for experimental procedure and Figures S22–S25 for details). Interestingly, we observed a pronounced quenching of the fluorescence of about 80 %, which is remarkably higher than that observed for BP (ca. 66 %).27 Additionally, to gain unambiguous evidence of the charge‐transfer process we used tetracyanoquinodimethane (TCNQ), a benchmark electron‐withdrawing molecule which has been theoretically predicted to exert a p‐type doping with a pronounced charge transfer of 0.62|e|.39 Thus, we prepared THF antimonene suspensions in an argon‐filled glovebox, added a 10−5 m TCNQ solution, and characterized the UV‐vis spectra under inert conditions before and after the functionalization process (see Figure S26). Remarkably, the almost complete formation of TCNQ2− was observed after 2 days of reaction. It is worthy of a remark here that the generation of TCNQ2− usually requires exhaustive 2 e− reduction (bulk electrolysis) and the rigorous exclusion of O2. This electron transfer from Sb to the TCNQ molecule is similar to that previously observed for BP‐TCNQ hybrids.27
In conclusion, we have presented the noncovalent functionalization of few‐layer antimonene prepared both by micromechanical and liquid‐phase exfoliation with a tailormade PDI and TCNQ for the very first time. Scanning Raman microscopy and statistical Raman spectroscopy revealed the preferential supramolecular functionalization of the antimonene flakes deposited on Si/SiO2 substrates with a significant quenching of the PDI fluorescence. Scanning photoelectron microscopy and computational studies demonstrated the nature of the strong noncovalent interaction between PDI and antimonene as well as the significant charge transfer from the antimonene to the PDI, and it resulted in a charge transfer about twice as large as that observed for BP. In addition, we have characterized the nature of the spontaneous oxide passivation layer of pristine antimonene under environmental conditions resulting in its excellent stability. Moreover, DFT calculations indicate that the noncovalent functionalization of antimonene gives rise to a charge‐transfer band gap of approximately 1.05 eV and is thus a promising route for tailoring the electronic properties of this novel group 15 2D material.
Dedicated to the memory of Pedro Abellán Fenoll
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the ESCA microscopy beamline team at Elettra for technical assistance with the scanning X‐ray photoelectron microscopy measurements. The research leading to these results has received partial funding from the European Union Seventh Framework Programme under grant agreement no. 604391 Graphene Flagship. We thank the Deutsche Forschungsgemeinschaft (DFG‐SFB 953 “Synthetic Carbon Allotropes”, Projects A1 and C2), the Interdisciplinary Center for Molecular Materials (ICMM), and the Graduate School Molecular Science (GSMS) for financial support. We thank the MINECO (Spain) for financial support through the “María de Maeztu” Programme for Units of Excellence in R&D (MDM‐2014‐0377) and the projects: MAT2016‐77608‐C3‐1P and 3P, as well as MAT2014‐52477‐C5‐5 and MAT2015‐66888‐C3‐3‐R. Co‐funding from UE is also acknowledged. G.A. thanks the EU for a Marie Curie Fellowship (FP7/2013‐IEF‐627386), and the FAU for the Emerging Talents Initiative (ETI) grant #WS16‐17_Nat_04.
G. Abellán, P. Ares, S. Wild, E. Nuin, C. Neiss, D. R.-S. Miguel, P. Segovia, C. Gibaja, E. G. Michel, A. Görling, F. Hauke, J. Gómez-Herrero, A. Hirsch, F. Zamora, Angew. Chem. Int. Ed. 2017, 56, 14389.
Contributor Information
Dr. Gonzalo Abellán, Email: gonzalo.abellan@fau.de.
Dr. Félix Zamora, Email: felix.zamora@uam.es
References
- 1. Ferrari A. C., Bonaccorso F., Fal'ko V., Novoselov K. S., Roche S., Bøggild P., Borini S., Koppens F. H. L., Palermo V., Pugno N., et al., Nanoscale 2015, 7, 4598–4810. [DOI] [PubMed] [Google Scholar]
- 2. Bonaccorso F., Colombo L., Yu G., Stoller M., Tozzini V., Ferrari A. C., Ruoff R. S., Pellegrini V., Science 2015, 347, 1246501. [DOI] [PubMed] [Google Scholar]
- 3. Geim A. K., Grigorieva I. V., Nature 2013, 499, 419–425. [DOI] [PubMed] [Google Scholar]
- 4. Liu H., Neal A. T., Zhu Z., Luo Z., Xu X., Tománek D., Ye P. D., ACS Nano 2014, 8, 4033–4041. [DOI] [PubMed] [Google Scholar]
- 5. Ling X., Wang H., Huang S., Xia F., Dresselhaus M. S., Proc. Natl. Acad. Sci. USA 2015, 112, 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castellanos-Gomez A., J. Phys. Chem. Lett. 2015, 6, 4280–4291. [DOI] [PubMed] [Google Scholar]
- 7. Ryder C. R., Wood J. D., Wells S. A., Hersam M. C., ACS Nano 2016, 10, 3900–3917. [DOI] [PubMed] [Google Scholar]
- 8. Wood J. D., Wells S. A., Jariwala D., Chen K.-S., Cho E., Sangwan V. K., Liu X., Lauhon L. J., Marks T. J., Hersam M. C., Nano Lett. 2014, 14, 6964–6970. [DOI] [PubMed] [Google Scholar]
- 9. Island J. O., Steele G. A., van der Zant H. S. J., Castellanos-Gomez A., 2D Mater. 2015, 2, 011002. [Google Scholar]
- 10. Favron A., Gaufrès E., Fossard F., Phaneuf-L'Heureux A.-L., Tang N. Y.-W., Lévesque P. L., Loiseau A., Leonelli R., Francoeur S., Martel R., Nat. Mater. 2015, 14, 826–832. [DOI] [PubMed] [Google Scholar]
- 11. Hanlon D., Backes C., Doherty E., Cucinotta C. S., Berner N. C., Boland C., Lee K., Harvey A., Lynch P., Gholamvand Z., et al., Nat. Commun. 2015, 6, 8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ares P., Aguilar-Galindo F., Rodríguez-San-Miguel D., Aldave D. A., Díaz-Tendero S., Alcamí M., Martín F., Gómez-Herrero J., Zamora F., Adv. Mater. 2016, 28, 6332–6336. [DOI] [PubMed] [Google Scholar]
- 13. Gibaja C., Rodriguez-San-Miguel D., Ares P., Gómez-Herrero J., Varela M., Gillen R., Maultzsch J., Hauke F., Hirsch A., Abellán G., et al., Angew. Chem. Int. Ed. 2016, 55, 14345–14349; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14557–14561. [Google Scholar]
- 14. Zhang S., Xie M., Li F., Yan Z., Li Y., Kan E., Liu W., Chen Z., Zeng H., Angew. Chem. Int. Ed. 2016, 55, 1666–1669; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1698–1701. [Google Scholar]
- 15. Wang G., Pandey R., Karna S. P., ACS Appl. Mater. Interfaces 2015, 7, 11490–11496. [DOI] [PubMed] [Google Scholar]
- 16. Zhao M., Zhang X., Li L., Sci. Rep. 2015, 5, 16108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meng R. S., Cai M., Jiang J. K., Liang Q. H., Sun X., Yang Q., Tan C. J., Chen X. P., IEEE Electron Device Lett. 2017, 38, 134–137. [Google Scholar]
- 18. Pizzi G., Gibertini M., Dib E., Marzari N., Iannaccone G., Fiori G., Nat. Commun. 2016, 7, 12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang L. F., Song Y., Mi W. B., Wang X. C., Appl. Phys. Lett. 2016, 109, 022103. [Google Scholar]
- 20. Yang L., Song Y., Mi W., Wang X., RSC Adv. 2016, 6, 66140–66146. [Google Scholar]
- 21. Singh D., Gupta S. K., Sonvane Y., Lukačević I., J. Mater. Chem. C 2016, 4, 6386–6390. [Google Scholar]
- 22. Lu L., Tang X., Cao R., Wu L., Li Z., Jing G., Dong B., Lu S., Li Y., Xiang Y., et al., Adv. Opt. Mater. 2017, 5, 1700301. [Google Scholar]
- 23. Gu J., Du Z., Zhang C., Ma J., Li B., Yang S., Adv. Energy Mater. 2017, 7, 1700447. [Google Scholar]
- 24. Tao W., Ji X., Xu X., Islam M. A., Li Z., Chen S., Saw P. E., Zhang H., Bharwani Z., Guo Z., et al., Angew. Chem. Int. Ed. 2017, 56, 11896–11900; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12058–12062. [Google Scholar]
- 25. Ji J., Song X., Liu J., Yan Z., Huo C., Zhang S., Su M., Liao L., Wang W., Ni Z., et al., Nat. Commun. 2016, 7, 13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu X., Shao Y., Liu H., Feng Z., Wang Y.-L., Sun J.-T., Liu C., Wang J.-O., Liu Z.-L., Zhu S.-Y., et al., Adv. Mater. 2017, 29, 1605407. [DOI] [PubMed] [Google Scholar]
- 27. Abellán G., Lloret V., Mundloch U., Marcia M., Neiss C., Görling A., Varela M., Hauke F., Hirsch A., Angew. Chem. Int. Ed. 2016, 55, 14557–14562; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14777–14782. [Google Scholar]
- 28. Marcia M., Hirsch A., Hauke F., FlatChem 2017, 9, 89–103. [Google Scholar]
- 29. Backes C., Schmidt C. D., Rosenlehner K., Hauke F., Coleman J. N., Hirsch A., Adv. Mater. 2010, 22, 788–802. [DOI] [PubMed] [Google Scholar]
- 30. Kozhemyakina N. V., Englert J. M., Yang G., Spiecker E., Schmidt C. D., Hauke F., Hirsch A., Adv. Mater. 2010, 22, 5483–5487. [DOI] [PubMed] [Google Scholar]
- 31. Wirtz C., Hallam T., Cullen C. P., Berner N. C., O'Brien M., Marcia M., Hirsch A., Duesberg G. S., Chem. Commun. 2015, 51, 16553–16556. [DOI] [PubMed] [Google Scholar]
- 32. Ares P., Zamora F., Gomez-Herrero J., ACS Photonics 2017, 4, 600–605. [Google Scholar]
- 33. Ryder C. R., Wood J. D., Wells S. A., Yang Y., Jariwala D., Marks T. J., Schatz G. C., Hersam M. C., Nat. Chem. 2016, 8, 597–602. [DOI] [PubMed] [Google Scholar]
- 34. Zhao Y., Zhou Q., Li Q., Yao X., Wang J., Adv. Mater. 2017, 29, 1603990 [DOI] [PubMed] [Google Scholar]
- 35. Englert J. M., Vecera P., Knirsch K. C., Schäfer R. A., Hauke F., Hirsch A., ACS Nano 2013, 7, 5472–5482. [DOI] [PubMed] [Google Scholar]
- 36. Kozlov S. M., Viñes F., Görling A., Adv. Mater. 2011, 23, 2638–2643. [DOI] [PubMed] [Google Scholar]
- 37. Zhang S., Zhou W., Ma Y., Ji J., Cai B., Yang S. A., Zhu Z., Chen Z., Zeng H., Nano Lett. 2017, 17, 3434–3440. [DOI] [PubMed] [Google Scholar]
- 38. Zhang S., Yan Z., Li Y., Chen Z., Zeng H., Angew. Chem. Int. Ed. 2015, 54, 3112–3115; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3155–3158. [Google Scholar]
- 39. Xie M., Zhang S., Cai B., Zou Y., Zeng H., RSC Adv. 2016, 6, 14620–14625. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary