

Abstract
Objective
Spinocerebellar ataxia 38 (SCA38) is caused by mutations in the ELOVL5 gene, which encodes an elongase involved in the synthesis of polyunsaturated fatty acids, including docosahexaenoic acid (DHA). As a consequence, DHA is significantly reduced in the serum of SCA38 subjects. In the present study, we evaluated the safety of DHA supplementation, its efficacy for clinical symptoms, and changes of brain functional imaging in SCA38 patients.
Methods
We enrolled 10 SCA38 patients, and carried out a double‐blind randomized placebo‐controlled study for 16 weeks, followed by an open‐label study with overall 40‐week DHA treatment. At baseline and at follow‐up visit, patients underwent standardized clinical assessment, brain 18‐fluorodeoxyglucose positron emission tomography, electroneurography, and ELOVL5 expression analysis.
Results
After 16 weeks, we showed a significant pre–post clinical improvement in the DHA group versus placebo, using the Scale for the Assessment and Rating of Ataxia (SARA; mean difference [MD] = +2.70, 95% confidence interval [CI] = +0.13 to + 5.27, p = 0.042). At 40‐week treatment, clinical improvement was found significant by both SARA (MD = +2.2, 95% CI = +0.93 to + 3.46, p = 0.008) and International Cooperative Ataxia Rating Scale (MD = +3.8, 95% CI = +1.39 to + 6.41, p = 0.02) scores; clinical data were corroborated by significant improvement of cerebellar hypometabolism (statistical parametric mapping analyses, false discovery rate corrected). We also showed a decreased expression of ELOVL5 in patients’ blood at 40 weeks as compared to baseline. No side effect was recorded.
Interpretation
DHA supplementation is a safe and effective treatment for SCA38, showing an improvement of clinical symptoms and cerebellar hypometabolism. Ann Neurol 2017;82:615–621
Spinocerebellar ataxias (SCAs) are a group of autosomal dominant neurological disorders with a prevalence of 5.5 in 100,000.1 SCAs are phenotypically characterized by gait and limb ataxia, incoordination of eye movements, and speech disturbances. Cerebellar hypometabolism is well documented and considered a main diagnostic marker.2, 3 More than 40 SCA subtypes have been reported, and 34 genes have been identified so far. Three main categories are defined on the basis of the mutation type,4 namely those due to CAG‐coding polyglutamine repeat expansion, noncoding repeat expansions, and conventional mutations (http://neuromuscular.wustl.edu/ataxia/domatax.html).
We recently identified SCA38 (Mendelian Inheritance in Man 611805) as caused by mutations in the ELOVL5 gene.5 The disease onset is in the fourth decade of life, characterized by slowly progressive gait ataxia and associated in most of the cases with pes cavus and hyposmia. The disease progresses with limb ataxia, dysarthria, dysphagia, ophthalmoparesis, and, in the later stages, sensory loss. Brain imaging documented cerebellar hypometabolism with sparing of cerebral cortex.6
ELOVL5 encodes an elongase enzyme involved in the synthesis of very long‐chain fatty acids with a high and specific expression in Purkinje cells.5 Its main products are the 22‐carbon docosahexaenoic acid (DHA) and eicosapentaenoic acid of the omega‐3 polyunsaturated fatty acid class. ELOVL5 mutations likely cause both an altered function of the enzyme and a possible gain of function. As a consequence, SCA38 patients have a reduction of serum DHA, and increased ELOVL5 gene expression and protein levels induced by transcriptional feedback loop regulation.5
In the present work, we performed a clinical trial on 10 SCA38 patients, and we demonstrated that oral DHA supplementation is a safe and effective treatment, exerting clinical efficacy and influencing cerebellar metabolism.
Patients and Methods
Subjects
Ten subjects affected by SCA38 were evaluated at the Center for Ageing Brain and Neurodegenerative Disorders, Department of Clinical and Experimental Sciences, University of Brescia, Italy. Genetic test confirmed the c.689G>T (p.Gly230Val) variant.5 Patients had already been included in a previous work on clinical features of SCA38.6 Written informed consent was obtained from all patients.
In the Table, patients’ demographic and clinical features are reported. Mean age was 48.7 ± 10.8 years, and the mean age at onset was 38.4 ± 6.8 years; 6 patients were females, 4 males.
The study was approved by the ethics committee of Brescia Hospital, Italy (NP1821) and conformed to the Declaration of Helsinki principles.
Study Drug
The study drug was a algal oil derived‐DHA (Sofedus, Milan, Italy) administered as sachets dosed at 600mg/day. Algal DHA contains approximately 75% of DHA by weight and does not contain eicosapentaenoic acid. The DHA dose was established considering a meta‐analysis on several reported trials.7 The intake of 600mg/day of DHA was the highest dose employed in the majority of the studies without side effects. DHA and placebo sachets were indistinguishable and produced by the same company.
Study Design
The study design is shown in Figure 1. We performed a 2‐phase trial: (1) a randomized double‐blind placebo‐controlled phase of 16 weeks and (2) an open‐label phase of 40‐week DHA supplementation in each patient.
Figure 1.
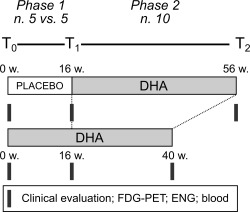
Study design. We conducted a 2‐phase trial consisting of a randomized double‐blind placebo‐controlled phase of 16 weeks, and an open‐label phase of 40‐week docosahexaenoic acid (DHA) supplementation. White bar = placebo treatment; gray bars = DHA treatment. Black blocks indicate the time points of clinical assessment (brain 18‐fluorodeoxyglucose positron emission tomography [FDG‐PET], electromyography/electroneurography [ENG], and blood sampling [blood]). w = weeks.
Patients were initially randomly assigned (1:1) to treatment with 600mg/day DHA (n = 5) or placebo (n = 5) for 16 weeks (see T0 and T1 in Fig 1).
Following the double‐blind phase, all patients (n = 10) underwent the 600mg/day DHA treatment for a total of 40 weeks (see Fig 1).
At enrollment (T0), 16‐week follow‐up (T1), and after 40‐week DHA supplementation (T2), each patient underwent standardized clinical assessment (ie, Scale for the Assessment and Rating of Ataxia [SARA]8 and the International Cooperative Ataxia Rating Scale [ICARS]9, brain 18‐fluorodeoxyglucose [FDG] positron emission tomography [PET] scan, electromyography [EMG]/electroneurography [ENG], and blood sampling for biological analyses).
Inclusion Criteria
Inclusion criteria were: (1) symptomatic p.Gly230Val mutation carriers, (2) age > 18 years old, and (3) ambulant (SARA score at baseline < 23).
Exclusion Criteria
Exclusion criteria were: (1) reported poor compliance with drug regimen, (2) uncontrolled diabetes (exclusion criterion to perform FDG‐PET scan), (3) serum creatinine levels > 2.0mg/dl, (3) alcohol abuse (equivalent to > 12g/day) over the 30 days prior to screening, and (4) evidence of drug abuse within 6 months prior to screening.
Blindness
To ensure blindness in the clinical assessment scoring, at each time point (T0, T1, and T2) neurological examination was video‐recorded and analyzed blind by A.A., who was unaware of both time point and treatment (DHA or placebo), as the videos were presented randomly. Brain FDG‐PET analyses were carried out by Statistical Parametric Mapping (SPM), which is fully automated, unbiased, and operator‐independent software. Biological analyses were conducted by E.D.G., N.M., and D.C. without knowledge of time point and treatment intervention.
Outcome Measures
As primary efficacy measure, we evaluated the significant mean change from enrollment/baseline to endpoint on the clinical scales (SARA and ICARS). Secondary efficacy measures included change from baseline to endpoint on brain FDG‐PET imaging and on DHA and ELOVL5 levels in blood.
Safety Assessments
Safety assessments were conducted at screening and at each visit. Adverse events were elicited by questioning the patient throughout the study and through direct observation by the clinical team.
Clinical Assessment, Instrumental Evaluation, and Molecular Analyses
At each time point, videotapes of the SARA (range = 0–40)8 and the ICARS (range = 0–100)9 were employed to evaluate cerebellar deficits. Intra‐assay variability of SARA and ICARS scores was evaluated between 2 independent neurologists (A.A. and M.M., SARA and ICARS alpha‐Cronbach = 0.983 and 0.995, respectively).
Brain PET image‐processing procedures were carried out using MATLAB (http://it.mathworks.com/products/matlab/; MathWorks, Sherborn, MA) and SPM (http://www.fil.ion.ucl.ac.uk/spm/software/spm12/) software. Details on image preprocessing are given elsewhere.10 Cerebellar metabolism changes were evaluated by nonparametric permutation test (10,000‐permutation Statistical NonParametric Mapping; T0 vs T1 and baseline vs T2), and the threshold was set at p < 0.05, false discovery rate (FDR) cluster level corrected.11
EMG and ENG were performed according to standard procedures.
Blood sampling was performed at fast, between 8 and 9 am. Each patient had undergone a poor‐DHA diet, as recommended by an expert dietician, for 2 weeks before each blood sampling, to avoid possible confounders on blood analyses. We measured serum DHA levels and ELOVL5 expression, as previously published.5
Statistical Analysis
Comparison of clinical characteristics between groups (placebo vs DHA treatment) was carried out using Mann–Whitney test or chi‐square test, as appropriate. To assess the effect of DHA treatment on clinical scores over time, in the double‐blind phase we used 2‐way mixed analysis of variance (ANOVA) with TIME (T0 vs T1) as within‐subject factor and TREATMENT (placebo vs DHA) as between‐subjects factor; in the open‐label phase, we applied 1‐way mixed ANOVA with TIME (baseline vs T2) as within‐subject factor. Mauchly test was used to test for assumption of sphericity, whereas Greenhouse–Geisser epsilon determination was used to correct in case of sphericity violation.
Correlation between functional scores and demographic or clinical characteristics was assessed using Spearman rank‐order correlations. Statistical analyses were performed using SPSS version 21 (SPSS, Chicago, IL).
Results
Randomized Double‐Blind Placebo‐Controlled Phase
No significant differences in demographic characteristics or SARA and ICARS scores between patients who received DHA or placebo were found. Clinical evaluation by SARA scores of the DHA versus placebo groups after 16 weeks (T0 vs T1) showed a statistically significant TIME × TREATMENT interaction (T0 vs T1, mean ± standard error, DHA group: 10.8 ± 1.0 vs 7.8 ± 0.9; placebo group: 9.7 ± 3.2 vs 9.4 ± 4.0; F 1;8 = 5.88, p = 0.042; Fig 2A).
Figure 2.
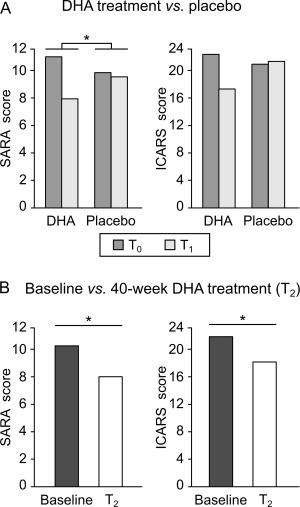
Clinical assessment in the double‐blind randomized placebo‐controlled phase and the open‐label phase. (A) Scale for the Assessment and Rating of Ataxia (SARA) and International Cooperative Ataxia Rating Scale (ICARS) scores in the docosahexaenoic acid (DHA)‐treated group and in the placebo‐treated group before (T0, dark gray bars) and after 16‐week DHA/placebo treatment (T1, light gray bars) in spinocerebellar ataxia 38 (SCA38) patients. (B) SARA scores and ICARS scores at baseline (dark bars) and after 40‐week DHA treatment (T2, white bars) in SCA38 patients. *p < 0.05.
The pre–post effect (T0 vs T1) of the DHA group exhibited a non‐null mean difference (MD) in SARA scores (MD = +3.00, 95% confidence interval [CI] = +1.46 to + 4.65) as compared to the placebo group (MD = +0.30, 95% CI = −1.25 to + 1.84). Accordingly, the TIME × TREATMENT mean difference was non‐null (3.00 − 0.30 = +2.70, 95% CI = +0.13 to + 5.27).
On ICARS scores, there was also an improvement, but not statistically significant TIME × TREATMENT interaction (T0 vs T1, DHA group: 22.0 ± 3.3 vs 17.0 ± 2.5; placebo group: 20.6 ± 8.1 vs 21.0 ± 9.2; F 1;8 = 4.25, p = 0.073; see Fig 2A). The coverage interval of the TIME × TREATMENT mean difference included the null value (5.0−[−0.4] = +5.4, 95% CI = ‐0.64 to + 11.4), although the DHA group had a non‐null pre–post effect (MD = +5.0, 95% CI = +1.37 to + 8.63) as compared to the placebo group (MD = −0.4, 95% CI = −4.03 to + 3.23).
In both DHA and placebo groups, no significant differences of cerebellar metabolism at the preestablished threshold between T0 and T1 (T0 < T1) were reported. EMG/ENG parameters were unchanged in both groups. Serum DHA levels and ELOVL5 expression did not show any significant TIME × TREATMENT interaction.
Open‐Label Phase
Each subject underwent DHA treatment (600mg/day) in the open‐label phase for 40 weeks, and the differences between baseline and 40‐week follow‐up (T2) were evaluated. We found a significant improvement in clinical symptoms, with significantly reduced SARA scores at T2 compared to baseline (baseline: 10.1 ± 1.9, T2: 7.9 ± 1.7; MD = +2.2, 95% CI = +0.93 to + 3.46; F 1;9 = 11.4, p = 0.008; see Fig 2B). The same pre–post effect was shown for ICARS scores (baseline: 21.5 ± 4.6, T2: 17.9 ± 4.2; MD = +3.8, 95% CI = +1.39 to + 6.41; F 1;9 = 7.96, p = 0.020; see Fig 2B).
A significant difference in cerebellar metabolism between baseline and T2 was observed, with an increase in cerebellar metabolism at T2 as compared to baseline in the left posterior cerebellar lobe (x, y, z = 32, −79, −23; T = 8.56; p = 0.03, cluster size = 558) and in the right posterior cerebellar lobe (x, y, z = 22, −84, −17; T = 7.74; p = 0.03, cluster size = 475; Fig 3). No significant differences in the opposite contrast (baseline > T2) were found at the preestablished threshold.
Figure 3.
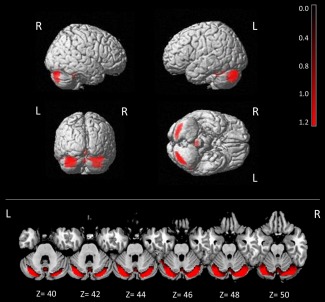
Improvement of cerebellar hypometabolism after 40‐week docosahexaenoic acid (DHA) treatment in spinocerebellar ataxia 38 (SCA38) patients. Pattern of cerebellar metabolism in SCA38 patients at baseline versus 40‐week DHA treatment (baseline < 40‐week DHA treatment, p < 0.05, false discovery rate corrected). See Results for details. The results are superimposed on a 3‐dimensional standardized template and axial magnetic resonance imaging template. L = left; R = right; Z = Montreal coordinates.
On EMG/ENG, motor and sensory conduction velocities did not worsen during 40‐week DHA treatment.
No differences in serum DHA levels before and after 40‐week treatment were found. We showed a slight but significant reduction of ELOVL5 expression in blood comparing T2 with baseline (reduction to 88% from baseline, 95% CI = 74% to 94%; F 1;9 = 5.48, p = 0.044).
There was no significant correlation between the change in SARA and ICARS scores (T2 minus baseline scores) and age, gender, age at disease onset, duration of disease, SARA and ICARS scores at baseline, change of ELOVL5 expression, and change of cerebellar metabolism as measured by brain FDG‐PET (Spearman rank‐order correlation, all p > 0.05).
Safety Assessment
No side effects or adverse events were reported during DHA supplementation in either the double‐blind or the open‐label phase.
Discussion
No effective treatment is currently available for most hereditary ataxias, and management remains supportive and symptomatic.12, 13 The rationale of this study stemmed from the observation that SCA38 is characterized by an increased amount of ELOVL5 protein with a mislocalization of the aberrant form in the Golgi apparatus and by a decrease of its final products, in particular DHA, in patients’ serum.5
Because ELOVL5 is strictly regulated by the amount of arachidonic acid and DHA via a transcriptional feedback loop,14 we reasoned that the administration of DHA might have exerted a double goal: compensating the decrease of very long chain fatty acids and lowering ELOVL5 aberrant protein.5 The endogenous synthesis of DHA within the brain is low compared with its uptake from dietary and/or liver sources.15, 16 DHA is a well‐known dietary supplement, well tolerated at high dosage and with the ability to cross the blood–brain barrier by passive and active transport.17, 18, 19
In recent years, polyunsaturated fatty acids like DHA have gained much attention due to promising results in a number of neurodegenerative conditions.20, 21 Moreover, polyunsaturated fatty acids are required for the normal development of the central nervous system and their deficiency can impair cerebral function in mice.22
In this study, we showed that DHA supplementation ameliorates clinical symptoms and cerebellar metabolism in SCA38 patients. Our data are in agreement with the hypothesis that DHA intake, acting directly on ELOVL5 expression, reduces mutant ELOVL5 cellular levels, as indicated by a decrease of ELOVL5 expression in patients’ blood after 40‐week DHA treatment. Furthermore, a general neuroprotective effect of DHA may occur, by promoting brain cell survival and repair through neurotrophic, antiapoptotic, and anti‐inflammatory signaling.23 As expected, DHA administration was safe and no side effect was reported.
The rationale of our study was similar to that described for X‐linked adrenoleukodystrophy and Lorenzo oil treatment, the administration of which was able to normalize very long chain fatty acids in plasma and to provide a clinical benefit.24
We conducted the present phase II study in 2 stages. We had already demonstrated a clinical improvement in the double‐blind randomized placebo controlled phase, although the low number of subjects (5 DHA vs 5 placebo) and the short‐term follow‐up prevented us from reporting significant changes of cerebellar metabolism and ELOVL5 expression. In the open‐label phase presented here, in which we considered the 10 patients longitudinally, we reported significant clinical improvement, especially of posture and gait, along with a marked increase in cerebellar hypometabolism and restored ELOVL5 expression.
We acknowledge that the small number of patients and the lack of one of primary efficacy measure outcome (ie, ICARS) in the double‐blind placebo‐controlled phase are limitations of the study. Moreover, we did not find any significant change of serum DHA levels before and after treatment, and this might give rise to concerns regarding the robustness of the effect. Larger phase III studies addressing long‐term efficacy and administration in still asymptomatic subjects with ELOVL5 mutations are warranted to prove DHA supplementation to be an effective therapy for SCA38. All patients included in this study carry the same mutation in ELOVL5. Therefore, only the effect of DHA on this single mutation has been assessed in this trial and generalization to all SCA38 patients is not possible, requiring further studies.
Possible benefits of DHA supplementation in other SCAs with reduced brain fatty acids and phospholipids, such as SCA1 and Friedreich ataxia, also need to be considered.25 Based on this observation, we may speculate that supplementation with DHA, the main component of brain phospholipids, might be beneficial in the early stage of such diseases.
The treatment is a relatively inexpensive (approximately $500 per patient per year), well tolerated, and easy to administer as a dietary intervention. We propose that this evidence‐based strategy started early in life might delay disease onset and slow the progression of symptoms in SCA38. Thanks to replacement treatment, a delayed dependency in these patients may sensibly reduce direct and indirect costs on national health systems.
Author Contributions
E.D.G., A.B., and B.B. contributed to the concept and study design. All authors contributed to data acquisition and analysis. M.M., A.A., A.B, and B.B. drafted the manuscript and figures, and all authors approved the final version.
Potential Conflicts of Interest
Nothing to report.
Table 1.
Demographic and Clinical Characteristics of Enrolled Patients according to Treatment Group
| DHA Group | Placebo Group | SCA38 Overall | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Group, n = 10a |
| Gender | M | M | F | F | F | F | M | M | F | F | 40% M |
| Age at onset, yr | 38 | 46 | 35 | 38 | 36 | 50 | 44 | 34 | 37 | 26 | 38.4 ± 6.8 |
| Age at evaluation, yr | 46 | 49 | 51 | 49 | 39 | 73 | 47 | 40 | 58 | 35 | 48.7 ± 10.8 |
| SARA score, T0 | 13.5 | 9.0 | 13.0 | 10.0 | 8.5 | 17.5 | 5.0 | 6.0 | 17.5 | 2.5 | 10.2 ± 5.1 |
| ICARS score, T0 | 32.0 | 18.0 | 28.0 | 17.0 | 15.0 | 36.0 | 6.0 | 13.0 | 44.0 | 4.0 | 21.3 ± 13.2 |
| First symptom | Gait ataxia | Gait ataxia | Gait ataxia | Gait ataxia | Gait ataxia | Gait ataxia | Gait ataxia | Gait ataxia | Gait ataxia | Gait ataxia | — |
Mean ± standard deviations, otherwise specified.
DHA = docosahexaenoic acid; F = female; ICARS = International Cooperative Ataxia Rating Scale; M = male; SARA = Scale for the Assessment and Rating of Ataxia; SCA38 = spinocerebellar ataxia 38; T0 = time at enrollment.
Acknowledgment
This work was supported by Fondazione Telethon (GGP14225).
We thank the patients and their families for taking part in the study, Dr S. Gazzina for imaging analysis, and Dr M. Cosseddu for technical support.
Trial registration: ClinicalTrials.gov NCT03109626.
References
- 1. Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014;42:174–183. [DOI] [PubMed] [Google Scholar]
- 2. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–894. [DOI] [PubMed] [Google Scholar]
- 3. van Gaalen J, Giunti P, van de Warrenburg BP. Movement disorders in spinocerebellar ataxias. Mov Disord 2011;26:792–800. [DOI] [PubMed] [Google Scholar]
- 4. Sun YM, Lu C, Wu ZY. Spinocerebellar ataxia: relationship between phenotype and genotype—a review. Clin Genet 2016;90:305–314. [DOI] [PubMed] [Google Scholar]
- 5. Di Gregorio E, Borroni B, Giorgio E, et al. ELOVL5 mutations cause spinocerebellar ataxia 38. Am J Hum Genet 2014;95:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Borroni B, Di Gregorio E, Orsi L, et al. Clinical and neuroradiological features of spinocerebellar ataxia 38 (SCA38). Parkinsonism Relat Disord 2016;28:80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lien EL. Toxicology and safety of DHA. Prostaglandins Leukot Essent Fatty Acids 2009;81:125–132. [DOI] [PubMed] [Google Scholar]
- 8. Yabe I, Matsushima M, Soma H, et al. Usefulness of the Scale for Assessment and Rating of Ataxia (SARA). J Neurol Sci 2008;266:164–166. [DOI] [PubMed] [Google Scholar]
- 9. Trouillas P, Takayanagi T, Hallett M, et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. J Neurol Sci 1997;145:205–211. [DOI] [PubMed] [Google Scholar]
- 10. Della Rosa PA, Cerami C, Gallivanone F, et al. A standardized [18F]‐FDG‐PET template for spatial normalization in statistical parametric mapping of dementia. Neuroinformatics 2014;12:575–593. [DOI] [PubMed] [Google Scholar]
- 11. Nichols TE, Holmes AP. Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp 2002;15:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooper JM, Korlipara LV, Hart PE, et al. Coenzyme Q10 and vitamin E deficiency in Friedreich's ataxia: predictor of efficacy of vitamin E and coenzyme Q10 therapy. Eur J Neurol 2008;15:1371–1379. [DOI] [PubMed] [Google Scholar]
- 13. Cooper JM, Schapira AH. Friedreich's ataxia: coenzyme Q10 and vitamin E therapy. Mitochondrion 2007;7(suppl):S127–S135. [DOI] [PubMed] [Google Scholar]
- 14. Moon YA, Hammer RE, Horton JD. Deletion of ELOVL5 leads to fatty liver through activation of SREBP‐1c in mice. J Lipid Res 2009;50:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Demar JC Jr, Ma K, Chang L, et al. alpha‐Linolenic acid does not contribute appreciably to docosahexaenoic acid within brain phospholipids of adult rats fed a diet enriched in docosahexaenoic acid. J Neurochem 2005;94:1063–1076. [DOI] [PubMed] [Google Scholar]
- 16. Igarashi M, Ma K, Chang L, et al. Low liver conversion rate of alpha‐linolenic to docosahexaenoic acid in awake rats on a high‐docosahexaenoate‐containing diet. J Lipid Res 2006;47:1812–1822. [DOI] [PubMed] [Google Scholar]
- 17. Rapoport SI, Chang MC, Spector AA. Delivery and turnover of plasma‐derived essential PUFAs in mammalian brain. J Lipid Res 2001;42:678–685. [PubMed] [Google Scholar]
- 18. Rapoport SI, Rao JS, Igarashi M. Brain metabolism of nutritionally essential polyunsaturated fatty acids depends on both the diet and the liver. Prostaglandins Leukot Essent Fatty Acids 2007;77:251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nguyen LN, Ma D, Shui G, et al. Mfsd2a is a transporter for the essential omega‐3 fatty acid docosahexaenoic acid. Nature 2014;509:503–506. [DOI] [PubMed] [Google Scholar]
- 20. Hacioglu G, Seval‐Celik Y, Tanriover G, et al. Docosahexaenoic acid provides protective mechanism in bilaterally MPTP‐lesioned rat model of Parkinson's disease. Folia Histochem Cytobiol 2012;50:228–238. [DOI] [PubMed] [Google Scholar]
- 21. Yassine HN, Braskie MN, Mack WJ, et al. Association of docosahexaenoic acid supplementation with Alzheimer disease stage in apolipoprotein E epsilon4 carriers: a review. JAMA Neurol 2017;74:339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bourre JM. Effects of nutrients (in food) on the structure and function of the nervous system: update on dietary requirements for brain. Part 1: micronutrients. J Nutr Health Aging 2006;10:377–385. [PubMed] [Google Scholar]
- 23. Bazan NG. Neuroprotectin D1‐mediated anti‐inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer's disease. J Lipid Res 2009;50(suppl):S400–S405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Senior K. Lorenzo's oil may help to prevent ALD symptoms. Lancet Neurol 2002;1:468. [DOI] [PubMed] [Google Scholar]
- 25. Eder K, Kish SJ, Kirchgessner M, Ross BM. Brain phospholipids and fatty acids in Friedreich's ataxia and spinocerebellar atrophy type‐1. Mov Disord 1998;13:813–819. [DOI] [PubMed] [Google Scholar]