

Abstract
This study aimed to identify PYGM mutations in patients with McArdle disease from Turkey by next generation sequencing (NGS). Genomic DNA was extracted from the blood of the McArdle patients (n=67) and unrelated healthy volunteers (n=53). The PYGM gene was sequenced with NGS and the observed mutations were validated by direct Sanger sequencing. A diagnostic algorithm was developed for patients with suspected McArdle disease. A total of 16 deleterious PYGM mutations were identified, of which 5 were novel, including 1 splice-site donor, 1 frame-shift, and 3 non-synonymous variants. The p.Met1Val (27-patients/11-families) was the most common PYGM mutation, followed by p.Arg576* (6/4), c.1827+7A>G (5/4), c.772+2_3delTG (5/3), p.Phe710del (4/2), p.Lys754Asnfs (2/1), and p.Arg50* (1/1). A molecular diagnostic flowchart is proposed for the McArdle patients in Turkey, covering the 6 most common PYGM mutations found in Turkey as well as the most common mutation in Europe. The diagnostic algorithm may alleviate the need for muscle biopsies in 77.6% of future patients. A prevalence of any of the mutations to a geographical region in Turkey was not identified. Furthermore, the NGS approach to sequence the entire PYGM gene was successful in detecting a common missense mutation and discovering novel mutations in this population study.
Keywords: glycogenosis, population specific, novel mutation, rare muscle disorders, molecular screening, genomics
Introduction
McArdle disease, also known as glycogen storage disease type V (OMIM 232600), is caused by a deficiency in muscle myophosphorylase (EC 2.4.1.1) activity and manifests with childhood to adult onset of exercise intolerance, fatigue, myalgia, and muscle cramps [1]. The serum creatine kinase (CK) activity is increased particularly following exercise and severe rhabdomyolysis may follow strenuous exercise [2]-[4]. In sporadic patients, the disease is diagnosed by muscle biopsy showing a complete lack of myophosphorylase activity and occasional subsarcolemmal glycogen storage in myocytes [5]-[7]. Identification of causative mutations in the PYGM gene can confirm the diagnosis, which then may be used diagnostic purposes for the relatives, e.g. related populations and geographical regions [8].
In humans, the PYGM gene is located on chromosome 11q.12-q13.2, its long transcript [NC_000011.9, Chr:11 Reference: GRCh37.p13 Primary Assembly, RNA_NM_005609.3, protein_NP_005600.1] is composed of 20 exons and the exonic sequence contains 18,625 bp, which is coding 842 amino acids [8].
McArdle disease is inherited by an autosomal recessive trait. According to the latest molecular update, 148 PYGM mutations causing McArdle disease and 39 additional polymorphisms with a minor allele frequency ≥1% have been reported [9]-[11]. Knowing the specific PYGM mutation(s) in particular populations could facilitate the screening of family members and future patients with suspicious symptoms by non-invasive techniques only requiring DNA obtained from peripheral blood, thereby reducing the need of muscle biopsies [12].
Furthermore, the accurate diagnosis of many myopathy patients may be merely hampered due to population specific mutations that are not included in the regular sequencing programs of several diagnostic laboratories. Hence, it is important to determine the PYGM mutation pool in specific populations. Therefore, the aim of this study was to identify the mutations of the McArdle disease in a cohort from Turkey by whole PYGM gene sequencing with next generation sequencing (NGS) techniques. Based upon this technology, a molecular diagnostic flowchart is proposed to ascertain precise diagnoses of most of the patients with McArdle disease and their families, from Turkey.
Material and Methods
1. Patients, relatives, and the control group
McArdle patients and relatives from 33 families living in various regions of Turkey that have been diagnosed from 1997–2013 (n=67) were admitted. The diagnosis was based upon the complete absence of myophosphorylase activity and glycogen accumulation in muscle biopsies, which were conducted at the 2 main neuromuscular clinics of Turkey, Istanbul University, Istanbul Medical Faculty and Hacettepe University, School of Medicine, Ankara. One additional University Hospital in Western Turkey could have participated with a very minor number of patients but could not provide a list of probands with complete myophosphorylase activity loss during the study sample collection period. Only families with at least one biopsy-confirmed McArdle patient (proband of family) were included in the study. Patients expressed the presence of consanguinity in their families during their initial clinical examination, which comprised 26 families and the remaining 7 families were not aware of consanguinity, however, at least 3 of them demonstrated a high penetrance of the disease.
The McArdle patients were assigned into 3 groups with respect to symptoms and muscle biopsy results; basically 56/67 studied patients (Group I and II) demonstrated characteristic signs of the disease, whereas Group III was only interviewed. Group I included the proband(s) of each family who were clinically evaluated and diagnosed by muscle biopsy (n=39). Group II was composed of relatives of proband(s), who appeared clinically affected by examination but were not evaluated by muscle biopsy (n=17). Group III refers to individuals with symptoms similar to a related proband, who were interviewed but had no clinical examination or a muscle biopsy during the study period (n=11). Blood samples of healthy parents and siblings were also collected if available. All studied patients demonstrated characteristic symptoms of the McArdle disease, including elevated serum CK levels, occasional myoglobinuria, and they referred second wind phenomenon, and some also showed rhabdomyolysis attacks. Family pedigrees were drawn manually and then with a software program (Progeny, Delray Beach, FL, USA).
Blood samples were collected in ethylenediaminetetraacetic acid (EDTA) containing tubes, in Ankara, Istanbul and in different geographic regions throughout Turkey. In addition, following a clinical evaluation at the Istanbul University Hospital, samples of a group of unrelated healthy volunteers from Turkey with no clinical evidence of myopathy (n=53) were selected as the control group. The age and sex distribution between the study and control groups were in accordance with the Hardy-Weinberg equilibrium (p=0.9 and p=0.6, respectively) (IBM, SPSS Statistics, 20). The Ethical Review Board of Istanbul Medical Faculty, Istanbul University approved the study (No: 2012/833-1073) and written informed consents were obtained from the patients, relatives, and controls before blood collection, examination and interview.
2. Molecular sequencing studies of PYGM gene
Genomic DNA was extracted from blood (MasterPure™ Epicentre DNA Purification Kit, Madison, WI, USA) then quantified (Nanodrop, Wilmington, DE, USA). The entire PYGM gene (RefSeqGene_NG_013018.1, EC 2.4.1.1) encompassing an area of 19,137 bp, including the 5′ upstream and 3′ downstream regions, were amplified by 3 long PCRs using sequence specific primer sets (Table-S1). The polymerase chain reaction (PCR) products were purified and quantified (Qubit, Invitrogen, CA, USA). The DNA for each sample was adjusted to 2 nmol and pooled to create the DNA library, prior to initiating the NGS sequencing process. Each DNA sample was equalized to 50 ng/μl and examined by NGS (Miseq sequencing system, Illumina, San Diego, CA, USA).
The results from bioinformatics of the NGS analysis were confirmed by Sanger sequencing (Applied Biosystems Genetic Analyzer 3500XL, ThermoFisher Scientific, Waltham, MA, USA) with new primer sets designed for each mutation (Table-S1). Additionally, haplotype effects to the PYGM gene were examined by whole exome sequencing (deep sequencing core facility of Advanced Genomics and Bioinformatics Research Center, IGBAM and TUBITAK, Gebze, Turkey) for 11 selected patients with the McArdle disease, including 6 individual probands from 6 consanguineous families, and 5 patients from 1 consanguineous family.
3. Whole exome sequencing and analysis
3.1. Massively parallel whole exome sequencing
Genomic DNA samples were prepared for massively parallel sequencing (Illumina TruSeq Sample Preparation kit, San Diego, CA, USA). Exonic regions were captured by NimbleGen SeqCap EZ Human Exome Library v3.0 kit. Illumina TruSeq PE Cluster kit v3-cBot-HS (San Diego, CA, USA) was used for paired-end cluster generation, and TruSeq SBS Kit v3-HS was used for sequencing the post-capture libraries. Initial clustering was performed on an Illumina cBot instrument (San Diego, CA, USA) and paired-end sequencing was performed on an Illumina HiSeq 2500 system in a private laboratory in Istanbul with a read length of 110 bp. All procedures were carried out according to the manufacturer’s instructions. Base calling and image analyses were done using Illumina’s Real Time Analysis software version 1.13 with default parameters (San Diego, CA, USA).
3.2. Targeted and exome sequencing analysis pipeline
The paired-end sequence reads were aligned to the human genome sequence (hg19) using Burrows-Wheeler Analysis software [13], and SAMtools were applied to remove any PCR duplicates [14]. The depth of coverage of targeted exome regions was calculated using BEDtools [15] and the realignments around insertions and deletions (indels) were performed using the Genome Analysis Toolkit v1.6 (GATK) IndelRealigner [16]. To discover single nucleotide substitutions (SNSs) and small indels, the GATK Unified Genotyper [16] was run on aligned reads with standard recommended parameters. The obtained variants were annotated for novelty by comparing them with dbSNP (build 135). Functional annotations of variants with an adequate quality score were performed using SnpEff and SnipSift [17]. Variants of interest were then evaluated using VarSifter [18], and the final sets of selected variants were visually inspected using Integrative Genomics Viewer (IGV) [19]. HomSI [20] was used for the homozygosity mapping analysis of the whole exome sequencing data.
Amino acid change predictions and protein modeling were conducted with SIFT BLink (La Jolla, CA) [21] and Pspired (London, UK) [22] online free software programs. Locations of all the variants were mapped using the National Center for Biotechnology Information (NCBI) and Ensemble databases using the NC_000011.9 GRCh37.p13 Primary Assembly. The listed variants were retrieved from the long transcript (mRNA: NM_005609.3, protein: NP_005600.1).
4. Development of the algorithm
The NGS and Sanger sequencing findings were compared by a hierarchal task analysis. The allelic frequency of each mutation, the number of families in which the mutation was observed, and the group distribution of the patients were the 3 criteria taken into consideration for the placement of the mutations in the diagnostic algorithm. An analysis was conducted to identify whether the disease and/or the observed mutations in this study cohort showed a clustering with respect to the family’s ancestral city, and was cross-referenced to the 7 geographical regions of Turkey. The ancestral city of each patient was defined as the city of birth of the ancestors of the father.
Results
1. Patient cohort
The overall mean age and standard deviation varied from 8 to 70 (33.4±13.7) years, and there were 30 females and 37 males (1:1.2). The control group had an age distribution of 18 to 62 (32.9±11.8), and there were 18 females and 35 males (1:1.9).
2. Mutations identified in a cohort from Turkey
Among the 67 patients and relatives with McArdle disease, 16 deleterious PYGM mutations were identified by whole PYGM gene NGS (Table-1). There were 11 previously described PYGM mutations including 1 start codon loss (SL) and 1 non-synonymous (NS) change; 2 codon deletions (CCD and CD), 2 stop gains (SG), 2 splice-site regions (SR), and 3 frame shifts (FS). In addition, 5 novel mutations were identified, including 1 splice-site donor (SD), 1 FS, and 3 NS changes. None of the controls (n=53) had any of the PYGM variants observed in this study. The p.Phe710del, p.Lys754Asnfs and p.Arg50* mutations were exclusively observed homozygous in the affected patients.
Table-1.
PYGM mutations validated by Sanger sequencing. The mutation positions are listed in the long transcript for both the RNA and protein codes, as well in respect to the exon- intron numbers.
| Mutation name and type* (rs#) | Long transcript | Exon | |||
|---|---|---|---|---|---|
| RNA (NM_005609.3) | Protein (NP_005600.1) | ||||
| 1 | SL | Start lost (rs267606993) | c.1A>G | p.Met1Val | Exon 1 |
| 2 | FS4 | Frame shift 4 (rs749548980) | c.144_145insCCACCGGTGTAACG | p.48Thr_49Profs | Exon 1 |
| 3 | SG2 | Stop gain 2 (rs116987552) | c.148C>T | p.Arg50* | Exon 1 |
| 4 | FS3 | Frame shift 3 (rs780193588) | c.164_166delGTCTC | p.55Ala_57Alafs | Exon 1 |
| 5 | CCD | Codon change + codon deletion (rs764823441) | c.509_511delAGA | p.Lys170del | Exon 4 |
| 6 | SR2 | Splice site region 2 (rs142234258) | c.660G>A | p.Gln220= | Exon 5 |
| 7 | SD | Splice site donor (Novel) | c.772+2_3delTG | – | Intron 6 |
| 8 | NS16 | Non synonymous coding 16 (rs116135678) | c.1094C>T | p.Ala365Val | Exon 10 |
| 9 | FS2 | Frame shift 2 (Novel) | c.1180_1181insG | p.394Glu_395Thrfs | Exon 10 |
| 10 | NS10 | Non synonymous coding 10 (Novel) | c.1369C>A | p.Ala457Glu | Exon 11 |
| 11 | NS8 | Non synonymous coding 8 (Novel) | c.1712T>G | p.Ile571Ser | Exon 14 |
| 12 | SG1 | Stop gain 1 (rs119103255) | c.1726C>T | p.Arg576* | Exon 14 |
| 13 | NS7 | Non synonymous coding 7 (Novel) | c.1733T>C | p.Leu579Pro | Exon 14 |
| 14 | SR1 | Splice site region 1 (rs532747) | c.1827+7A>G | - | Intron 15 |
| 15 | CD | Codon deletion (rs527236147) | c.2128_2130delTTC | p.Phe710del | Exon 17 |
| 16 | FS1 | Frame shift 1 (rs398124210) | c.2262delA | p.Lys754Asnfs | Exon 18 |
The annotations in italic are novel mutations,
The results of the NGS analyses of the 56/67 McArdle patients, covering 16 mutations including 5 novel mutations, were confirmed by Sanger sequencing of the PYGM gene including flanking regions. Homozygous affected (44/67, 64.6%), compound heterozygous (7/67, 10.4%) and heterozygous (5/67, 7.4%) patients, with a likely second unknown mutant allele, were all verified for their mutations (Table S-2). Furthermore, samples from healthy non-myopathic parents (5) and siblings (1) of patients with McArdle disease demonstrated heterozygosity for the mutant and wild-type alleles.
Interestingly the 5 patients (3 families), heterozygous for one mutation, were represented in different groups (Group I=2, Group II=2, Group III=1); all patients had relatives including siblings genotyped with the mutant allele found in the heterozygous patients. A representative family for this situation was observed in 2 siblings where the proband (patients 52 – Group I) was SR1-heterozygous, and the sibling (patient 53) from Group II was SR1-homozygous. Molecular testing of the consanguinous parents has been offered, however, the father was deceased and the mother was not available for this study. A second similar example was observed between 3 siblings, for which the proband (patients 63 – Group I) was homozygous for p.Met1Val mutation, one of the his diseased siblings (patient 64 – Group II) was heterozygous for a NS16 mutation but p.Met1Val-wild type. And the second sibling (patient 103 – Group II) was heterozygous for the p.Met1Val mutation but NS16-wild type. Despite the tedious and repetitive Sanger sequencing to discover the segregation of the mutations in each respective family, a mutation in the second allele could not be identified among the heterozygous patients. The last p.Met1Val-heterozygous patient (patient 110- Group III) with an undefined second mutant allele was part of a large consanguine family (Figure 1). The p.Met1Val mutation was homozygously present in all myopathic family members, except for this heterozygous patient (patient 110) and additional 3 young family members (Group II=1, Group III=2) with low NGS coverage and no identifiable mutant allele by direct Sanger sequencing. Samples that yielded low NGS sequencing coverage (11), also represented in each group (Group I=6, Group II=2, Group III=3), were included in the algorithm and sequenced by direct Sanger. Those in Groups I and II were previously examined and demonstrated typical McArdle symptoms, and those in Group I demonstrated complete lack of myophosphorylase activity. Although, patients from Groups II and III were related to at least one proband, the penetrance of the mutation could not be identified with NGS or direct sequencing.
Figure-1.
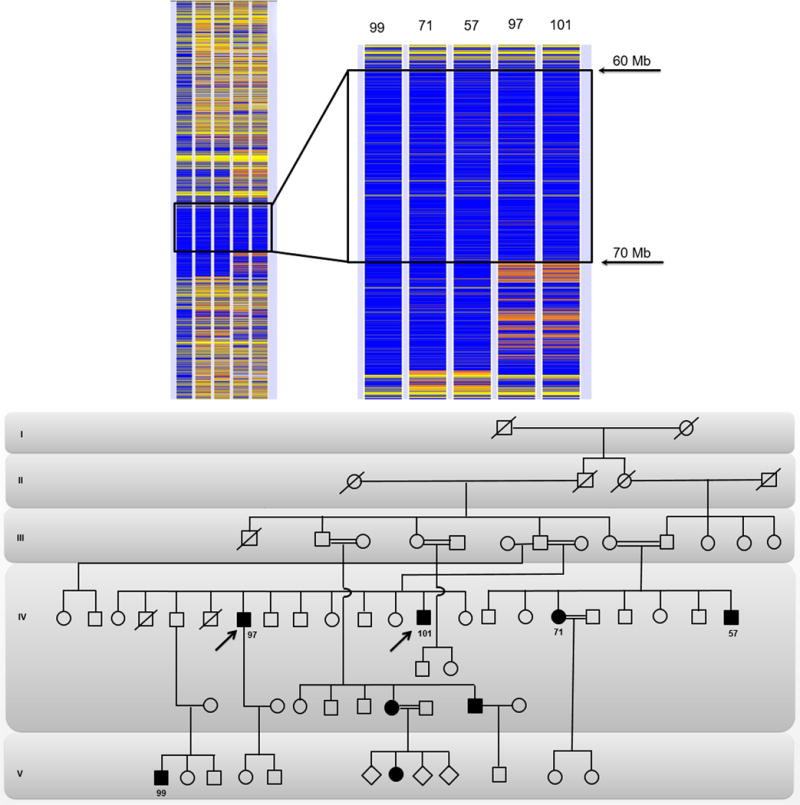
Homozygosity map of chromosome 11 and zoom-in shared homozygote region by the affected individuals with p.Met1Val mutation is encircled in a black rectangle. The patient 99 in column 1 was arbitrarily selected as an index case. The variants of the index case are colored in blue and yellow, displaying homozygote and heterozygote areas, respectively. For other patients (columns 2 to 5), while a homozygote variant is indicated with blue, the contrasting homozygote variant is represented with white and the heterozygote variant with orange. For all patients, gray areas indicate no call and yellow indicate non-informative single nucleotide polymorphisms, which are heterozygote for the index case. The penetrance of the p.Met1Val mutation between generations of the large consanguineous family can be observed in the pedigree drawing while the numbers of individuals included in the homozygosity analysis are depicted below the circles and squares. The arrows represent the probands, the white circles and squares represent the non-myopathic subjects, the dark circles and squares represent the affected patients, and the ones with an oblique strait line represent the deceased family members.
A homozygosity mapping analysis was conducted for an initially selected 11 patients, which showed a shared homozygous region of chromosome 11 from 60 Mb to 70 Mb stretch involving PYGM for all affected individuals (Figure 1).
All patients showed typical symptoms of the McArdle disease, and genotype-phenotype correlation was further evaluated for rhabdomyolysis (myoglobinuria, Tables-2 and S-2) and cross-referenced with the mutations observed in the study. The most common mutations observed in the study cohort are as follows, the p.Met1Val is a single nucleotide change (c.1A>G) (27/67, 40.3%) in the start codon substituting ATG with GGT, causing Met to be replaced by Val, triggering the loss of the start codon and abolishing the translation of the protein. The p.Arg576* (c.1726C>T) (6/67, 8.9%) nonsense mutation converts a CGA to TGA, leading to a premature stop codon (Figure-2). The c.1827+7A>G (5/67, 7.5%) splice site mutation, caused an A to G substitution, is located 7 bp downstream to exon 15 in intron 15. Moreover, the novel 2 bp deletion c.772+2_3delTG (5/67, 7.5 %), located 2 bp downstream to exon 6 in intron 6 predicts to disrupt the protein sequence, at the exon-exon boundary at amino acid position 170, by disrupting the link of the last nucleotide of exon 6 (T) and the first 2 nucleotides of exon 7 (TC) to code the TTC-Phe (Figure-3). The p.Phe710del (4/67, 5.9%) is an in-frame 3 bp (TTC) or codon deletion which eliminates a Phe (Figure-4), and the 1 bp deletion leading to a frame shift p.Lys754Asnfs (c.2262delA) (2/67, 2.9%) was also observed in the study cohort. Last but not least, the p.Arg50* (c.148C>T) (1/67, %) nonsense mutation that substitutes ACG for TGA - a stop codon, creating a premature termination with secondary RNA decay.
Table-2.
Mutations included in the molecular diagnostic algorithm, and their corresponding details, including their occurrence in the study group (n=67) and families (n=33), number of patients/groups and their allelic state.
| Diagnostic algorithm mutation placement | Total number of patients (%)/family (%) | Groups | Number of Homozygote/Heterozygote (Presence of rhabdomyolysis) | ||||
|---|---|---|---|---|---|---|---|
| I | II | III | |||||
| 1 | SL | p.Met1Val | 27 (40.3)/11 (33.3) | 13 | 10 | 4 | 22/5 (+) |
| 2 | SG1 | p.Arg576* | 6 (8.9)/4 (12.1) | 5 | – | 1 | 4/2 (+) |
| 3 | SR1 | c.1827+7A>G | 5 (7.5)/4 (12.1) | 4 | 1 | – | 3/2 (+) |
| 4 | SD | c.772+2_3delTG | 5 (7.5)/3 (9) | 3 | 1 | 1 | 3/2* (+) |
| 5 | CD | p.Phe710del | 4 (5.9)/2 (6) | 2 | 1 | 1 | 4/0 (+) |
| 6 | FS1 | p.Lys754Asnfs | 2 (2.9)/1 (3) | 2 | – | – | 2/0 (−) |
| 7 | SG2 | p.Arg50* | 1 (1.5)/1 (3) | 1 | – | – | 1/0 (−) |
Group I, proband(s) clinically evaluated and diagnosed confirmed by muscle biopsy; Group II, relative of proband, clinically affected without by muscle biopsy; Group III, relative of proband, without clinical examination or a muscle biopsy.
Two siblings compound heterozygote with p.Met1Val, which are also included in the p.Met1Val row.
Figure-2. Sequencing results of a consanguineous families with McArdle’s disease caused by NS8 and p.Arg576* (Stop gain 1-SG1).

The upper panel represents, a: heterozygote mother, b: heterozygote father, for NS8 and SG1 mutations respectively, c: the compound heterozygote affected son, and d: homozygote affected female relative only for the SG1 mutation. The nonsense mutation leads to premature termination of the myophosphorilase protein. The oblique arrows represent the probands in each family, the white circles and squares represent the non-myopathic subjects, the dark circles and squares represent the affected patients, and the ones with an oblique strait line represent the deceased family members. NS: non-synonymous, SG: stop gain, CD: codon deletion, Het: heterozygote, WT: wild type, M: mutant.
Figure-3. Pathology and sequencing results of patients homozygous and compound heterozygous for the novel c.772+2_3delTG splice site donor (SD) variant in PYGM gene.
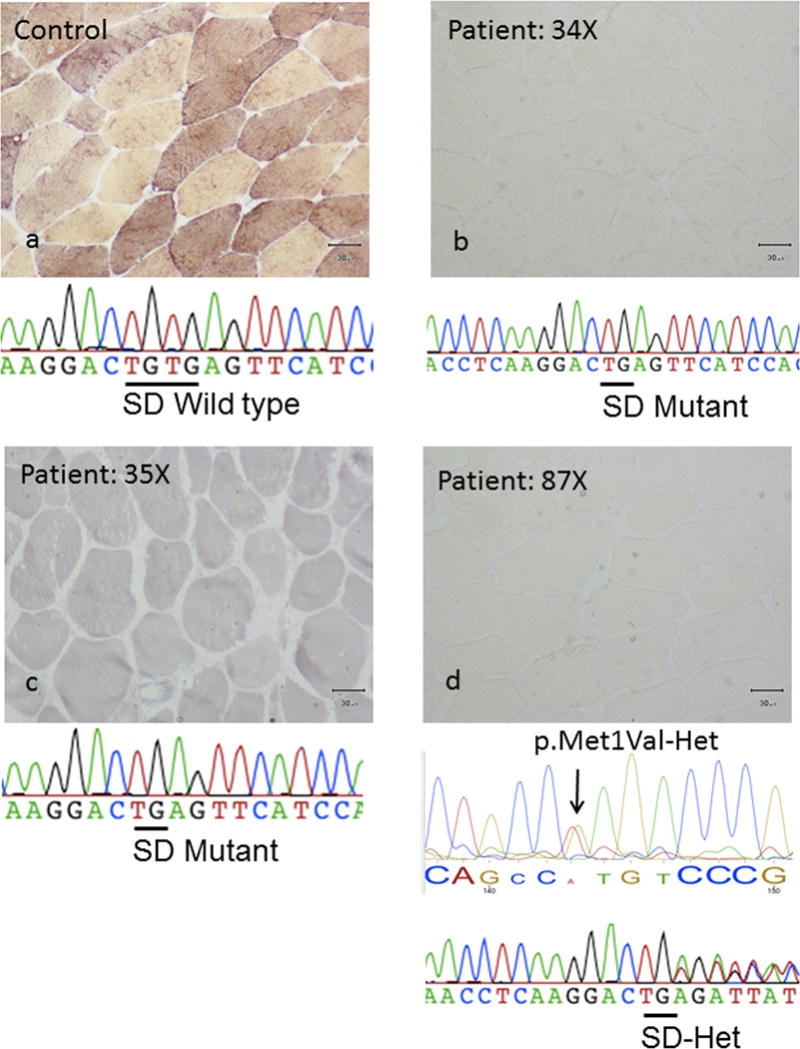
(a) Represents a wild type unrelated control pathology and sequence, (b) and (c) represents 2 unrelated homozygote mutant probands (patients 34 and 35), and (d) represents a third compound heterozgote proband (patient 87) for the novel SD mutation.
Figure-4.
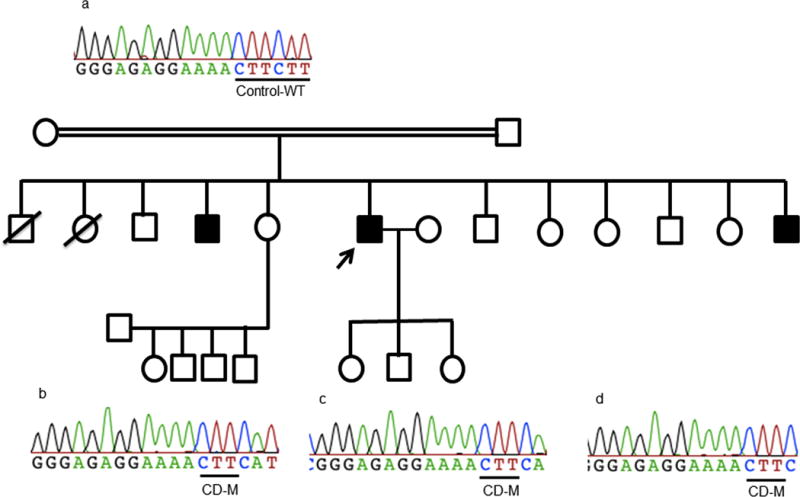
The p.Phe710del (codon deletion-CD) variants, the upper panel (a) represents a wild type unrelated control sequence, and (b), (c), and (d) represents 3 siblings homozygote for the p.Phe710del mutation. The arrows represent the probands in each family, the white circles and squares represent the non-myopathic subjects, the dark circles and squares represent the affected patients, and the ones with an oblique strait line represent the deceased family members. CD: codon deletion, WT: wild type, M: mutant.
3. Diagnostic molecular algorithm for McArdle disease
Based upon hierarchal task analysis the NGS technology for the discovery of novel mutations for rare disorders such as the McArdle disease was shown to be very successful, however, the verification by direct Sanger sequencing revealed a number of misreads. Hence, based upon the frequency of McArdle patients in Turkey with a specific PYGM mutation, a molecular diagnostic algorithm is proposed, including 7 mutations from the most (74.6%) to the least common (1.5%) (Figure-5, Table-2). Based upon this proposed algorithm, 74.6% (50/67) of the patients could be diagnosed molecularly including both homozygous and heterozygous patients for any of the mutations in the algorithm, and 58.2% (39/67) of the patients could be identified as homozygous for one of the mutations in the algorithm. A specific geographical clustering of the disease for this cohort and a hotspot for any of the mutations, with respect to cities or the 7 geographical regions of Turkey were not identified (Figure-6). For the initial implementation of the algorithm and the verification of NGS sequencing, regular PCR and direct Sanger sequencing was employed.
Figure 5. Hierarchal task analysis and molecular flowchart for the diagnosis of McArdle disease in patients from Turkey.
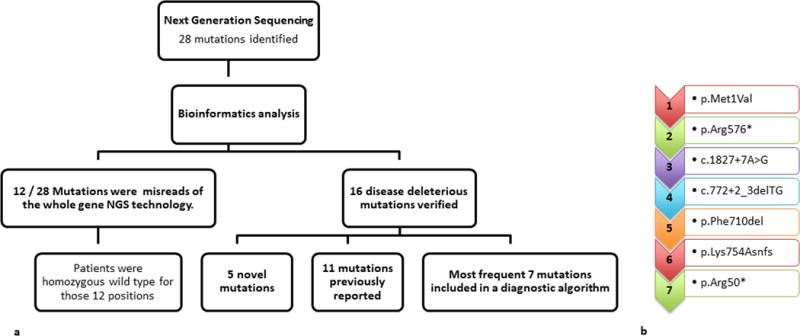
The NGS was successful in discovering 5 novel mutations in rare disorders, however also revealed the importance for cross-checking with the gold standard, the direct Sanger sequencing, as almost one third of the initially identified mutations by whole PYGM gene sequencing (12/28) were falsely called. Due to the high cost of NGS and the necessity to validate the mutations with direct sequencing a diagnostic flowchart was developed for future identification of patients with McArdle disease from Turkey. Based upon this proposed algorithm, 74.6% of patients (50/67) could be diagnosed molecularly including both homozygote and heterozygote patients for any of the mutations in the algorithm, and 58.2% of the patients (39/67) could be identified homozygous for 1 of the mutations in the algorithm. This algorithm is suggested for future patients from Turkey with suspected McArdle disease following a clinical examination.
Figure-6. The distribution of the families with McArdle disease who have volunteered to this study and a graphical analysis of the identified mutations in respect to the 7 geographical regions (1-7) of Turkey.
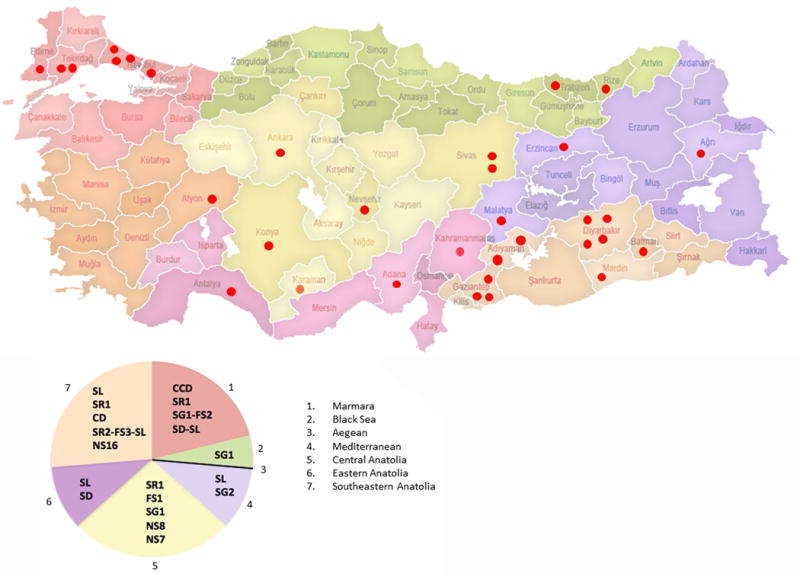
Each red dot indicates 1 family, and represents their ancestral city of birth and the region. The pie analysis demonstrates the distribution of the mutations observed in this study population. The mutation of the 1 family that volunteered from the Aegean region (3) could not be identified hence the region could only be represented with the black line.
Discussion
This is the first study to investigate both the molecular basis of McArdle disease in Turkey and the whole PYGM gene approach by NGS techniques. In this manner, 6 common and 10 additional PYGM variations were identified in the study cohort among 67 McArdle patients of 33 families. This study documents, a missense mutation (p.Met1Val) specific to this cohort, the occurrence of a codon deletion (p.Phe710del), which was previously reported only in the Japanese population, 5 additional novel PYGM mutations, and introduce the pathogenic nature of the SR1 and the CCD mutations. The latter 2 mutations were previously reported, SR1 as a benign allele and CCD with an unknown clinical significance, in respect to the 1000Genome Project, ClinVar and the dbSNP websites [23] [24]. Furthermore, we propose a molecular diagnostic flowchart for patients with McArdle disease from Turkey. By means of NGS, we were able to sequence not only the exons and exon–intron boundaries [25] but also the entire PYGM gene. Thus, we not only embraced the NGS technology and advanced bioinformatics, but also verified each discovered PYGM mutation with Sanger sequencing. Both, Nogales-Gadea et. al., (2015b) [9] and de Castro et al., (2015) [26] have stressed the importance of new techniques, with which still increased the heterogeneity of the PYGM gene, also underlining the value of regional studies.
The NGS and Sanger sequencing of PYGM were compared with a hierarchal task analysis, which shows a large number of variants initially called by NGS technology (28 mutations). The direct sequencing verification of all the variants revealed that 12/28 variants were actually miscalls by NGS. This is leaving 16 deleterious mutations, which 5 were novel and 11 had previously been reported. Clearly, NGS technology is effective in the discovery of novel mutations, which was also suggested by the FORGE study conducted in Canada, where NGS technology was suggested for the discovery of mutations of rare disorders [27]. Although whole gene sequencing by NGS is successful, it would be costly to implement to each patient. Hence, a diagnostic algorithm is proposed based on the more frequent mutations observed in this study cohort, that can be applicable to McArdle patients worldwide with ancestral origin from Turkey. The algorithm includes 7 mutations, which is estimated to reduce the use of invasive muscle biopsies by more then 70% in Turkey and will offer an economic alternative for a precise molecular diagnosis in comparison to whole gene sequencing by NGS or regular Sanger sequencing (Figure-5, Table-2). The method of choice implemented for the algorithm herein was simply regular PCR and direct Sanger sequencing, however, in respect to the addition of new patients in the future the method can be optimized into a multiplex PCR-RFLP assay. The first molecular diagnostic flowchart for McArdle disease was reported to decrease the need of muscle biopsies by 75% in Spain, also using conventional methods [12].
Rhabdomyolysis attacks were present in most of the mutations included to the algorithm (Table-2). Particularly the presence of rhabdomyolysis in probands and patients homozygous for the novel SD and NS7 mutations, and for those homozygote for CCD and SR1 mutations is of utmost importance, as they were previously considered as benign PYGM variants. These 4 mutations constituted 9/67 and were composed of all 3 groups (Group I=6, Group II=2, Group III=1), where patients in Groups II and III were first-degree relatives to probands, and family trios were investigated when possible.
The most common mutation among the study cohort was the p.Met1Val (Table-2). This mutation in the PYGM gene was found in 40% of all McArdle patients in this study cohort. This missense mutation has previously been described in 2 patients and indeed both were from Turkey, living in Germany [28] and in Spain [29]. Thus, this mutation likely arose in and is specific to Turkey. Other population specific mutations have been reported for McArdle disease in Finnish (p.Glu540X) [30], Japanese (p.Thr310X) [31], and Spanish cohorts (p.Try798Arg) [32].
Interestingly, the p.Arg50*, the most common PYGM mutation reported in Caucasians with European ancestry [9],[28],[33]-[37], which has never been reported in Japan [31], was one of the rarest mutations in our study cohort, being observed in only 1 patient (1.5%). Considering that it is the most common PYGM mutation in Caucasians, it is believed that it would be prudent to include it as the last mutation in the diagnostic algorithm for Turkey. Additionally, it could be readily introduced by marriage with non-Turks. It is worth noting that p.Gly205Ser [35], the second most common mutations observed in European Caucasians, was not observed in this cohort.
The second most common mutation in this cohort was the previously described c.1726C>T/p.Arg576* nonsense PYGM mutation in exon 14 [28]. Two of 6 patients with this mutation are compound heterozygotes with 2 novel variants on the other allele; one with NS8 (p.Ile571Ser; Figure-2) and the other with a frame-shift (FS2, p.394Glu_395Thrfs), which are both in conserved regions on the myophosphorylase protein. Both novel PYGM mutations were private and were not observed in the control group. The novel NS8 single nucleotide change is located 5 amino acids prior to the p.Arg576* nonsense mutation. The father (patient 113) and mother (patient 133) were heterozygous for the p.Arg576* and the NS8 mutation, respectively (Table-S2). The father firmly denied consanguinity between his spouse, however during the course of the study it became evident that another proband homozygous for the p.Arg576* nonsense mutation was his distant relative, explaining his heterozygous state. On the other hand, the carrier status of the non-consanguineous mother might imply a relatively high degree of carriers in the population, which supports the recent statements of the underestimation of the disease frequency due to regular PYGM screenings [9], [26]. The parents of the second compound heterozygous patient (p.Arg576*/p.394Glu_395Thrfs) were unavailable for further study. Hence, the segregation of the mutations could not be detected in this trio, alleged non-consanguineous marriage.
One of the novel missense mutations, discovered in 1 homozygous patient in this study cohort, was a leucine to proline substitution (NS7, p.Leu579Pro) located in a highly conserved helix in exon 14. Thereby increasing the number of mutations observed in this particular exon in this cohort to 3, with p.Arg576*, NS7 and NS8. The consanguine parents were deceased thus could not be investigated for the presence of heterozygocity. Exon 14 involves the active site, which explains the loss of function by a missense mutation [29].
The c.1827+7A>G splice site mutation, located in intron 15, 7 bp downstream of exon 15, is the third most common mutation in this cohort. This mutation was previously reported as a polymorphism in the PYGM gene with a minor frequency allele ≥1% with respect to the 1000 Genome Project (dbSNP build 141; [9]), and associated with a benign allele by ClinVar [10]. This study is the first to document its putative pathogenicity with 3 homozygous probands from 3 different families and 2 heterozygous patients with undefined second mutant allele. All showed classical symptoms of McArdle disease, and the probands of each family had a complete lack of myophosphorylase activity.
Similarly, the p.Lys170del mutation was previously reported as a benign polymorphism in exon 4 [24], however, here we report on its pathogenic effect for the first time. This 3 bp deletion was identified in 1 consanguine family, the father being the proband of the family (Group I) who has second wind and rhabdomyolosis, and daughter child (Group II) both homozygous affected for the codon deletion. The non-symptomatic mother volunteered for a molecular testing and revealed heterozygous for the deletion.
A novel splice site mutation at position c.772+2_3delTG is the fourth most common mutation identified in this study cohort. Three patients were homozygous for this splice site mutation (Figure-3) and 2 siblings were compound heterozygotes with the p.Met1Val mutation on the other allele. Splicing is known to usually begin with 5′-gt and end with ag-3′ [38]. The Phe at this position is highly conserved from all mammals to zebrafish, moreover, the SIFT BLink tool predicts tolerance only for Phe. This novel mutation is suggested to disrupt the protein sequence, as no other variants were identified in the PYGM gene of the 3 homozygous patients, and all 3 showed complete lack of myophosphorylase activity, classical symptoms, including referred second wind and rhabdomyolysis. The c.772+2_3delTG deletion predicts a disruption at the boundary between exon 5 and 6, and suggests erroneous splicing, or alternatively could lead to exon 6 skipping or deeper intronic cryptic site activation with intron retention or partial exon loss, ultimately, leading to a frame shift and premature stop codon 3′ to the mutation. The 3 families, in which the alternate splice site was discovered, are from different cities (Istanbul, Erzincan and Malatya) and geographical regions of Turkey and are unrelated based upon pedigree. Other donor splice sites have previously been identified in the choline kinase beta gene, for patients from Turkey (c.1031+1G>A) [39] and Europe (c.1031+3G>C), for congenital muscular dystrophy with the application of new sequencing generation technologies [40]. Although an effort was made to pursue RT-PCR PYGM-mRNA experiments from the family trio affected by the novel SD mutation, the analysis was not successful. And, no protein and functional studies were performed for the novel SD and SR1 mutations, however, future studies are planned to further investigate their effects [41].
The p.Phe710del in exon 17 is the fifth mutation in this study cohort. Since, 8 out of 16 McArdle patients from Japan were homozygous for this mutation, the codon deletion was thought to be specific for the Asian/Oriental populations [42] [43]. However, this survey expands its occurrence in Turkey with 4 patients from 2 unrelated families from different cities (Diyarbakır and Gaziantep) but the same geographic region. Further research from other Asian and Oriental countries and possibly new McArdle patients in Turkey, will be informative on the penetrance of this codon deletion mutation in the East Asian population. Interestingly, 3bp deletions reported so far, including the p.Phe710del observed in this study, are generally adjacent to small repeat regions and have been associated with genetic disorders [44]. In the case of the p.Phe710del mutation, the second “TTC” repeat is deleted. Triplicate deletions have been attributed to the DNA replication slippage process, which occurs more frequently in sequences that contain shorter repeat regions where the genetic material is exposed to instability [45]. The pathogenicity could be predicted by the originally suggested increased tendency to degradation due to the shorter protein length [42], however, the fact that the in-frame mutation is located in a highly conserved beta-helix location, the deletion of 1 amino acid could lead to conformational changes, which may also lead to poor packaging of the protein [46], and both situations could prime the protein’s impairment.
A single bp deletion p.Lys754Asnfs in exon 18, is the sixth mutation of the diagnostic algorithm, leading to a 3′ frame-shift in the coding sequence and a premature stop codon -3′ downstream. This mutation was observed in only 1 family and could have been considered as a private mutation, if it had not previously been reported in a patient from Turkey, living in Germany [25]. Although, the family included in this study cohort, stated to have no relatives in Germany, it would be interesting to determine if the two families originated from the same city in Turkey. Originally, the mutation was described on the long transcript as c.2336_2338delA49* at codon 753, followed by a premature termination 49 bp downstream. However, based upon the current open reading frame [GRCh37.p13, RefSeq_ NM_005609.3, RefSeq_ NP_005600.1] the 1 bp deletion resulted in a frame shift (c.2262delA/p.Lys754Asnfs), and premature termination.
McArdle patients with unidentified mutations, whether due to low NGS coverage or DNA degradation (11 patients, represented in all 3 groups), could be subjected to clinical re-examination and the diagnostic algorithm with fresh DNA samples, especially the 3 patients in Group III, in order to identify the nature of their neuromuscular disease.
In conclusion, the application of a molecular genetic algorithm by PCR and direct sequencing, following a history and neurological examination of patients with suspected McArdle disease from Turkey, will simplify and offer a non-invasive and an economic option in comparison to muscle biopsy based histochemical and biochemical tests and whole gene sequencing. The algorithm is suggested to provide a molecular diagnosis for more then 70% of the McArdle patients in Turkey. The findings of this study revealed a range of mutations that substantially differ from the European Caucasian general mutations, and might bear a potential explanation for the many inconclusive reports of myopathy patients from Turkey tested in private laboratories elsewhere, where the test protocols are designed in accordance to Caucasians with European ancestry. Therefore, stressing the importance of regional molecular studies. This can also be expected for other hereditary myopathies.
The discovery of 5 novel mutations increases the heterogeneity of the PYGM gene mutations, and, in conjunction with others [28], [47], provides further evidence that McArdle disease can be caused by population specific PYGM mutations as well as common ones without a clustering to specific geographical regions. Furthermore, the presence of the p.Arg50* nonsense mutation and p.Phe710del PYGM deletion in McArdle patients from Turkey, which are commonly seen in the European Caucasian and Japanese populations, respectively, may suggest the global exchange of alleles in Turkey as the major passage site between the East and the West.
The drawbacks that have been encountered in this study are believed to originate from the technical difficulties of the NGS technology, which generated low coverage of the gene for some samples in which the genotype could not be accurately identified. The algorithm was also inconclusive for these patients, bringing about the possibility of unrecognized mutations.
Supplementary Material
Acknowledgments
The authors would like to thank the many doctors and clinical staff that helped in the collection of blood samples across Turkey and also the participants. This study was supported in part by the Scientific Research Projects Coordination Unit of Istanbul University (# 24789, # 28050, # 38973), the Republic of Turkey Ministry of Development Infrastructure (# 2011K120020), the Scientific and Technological Research Council of Turkey (BILGEM–TUBITAK # T439000), the Turkish Neurological Society (# 00005) and National Institutes of Health (OD #010939).
Footnotes
Conflict of interest: None
References
- 1.Darras BT, Friedman NR. Metabolic myopathies: a clinical approach; part II. Pediatr Neurol. 2000;22:171–81. doi: 10.1016/s0887-8994(99)00122-8. [DOI] [PubMed] [Google Scholar]
- 2.Chen YT. Glycogen storage diseases and other inherited disorders of carbohydrate metabolism. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, editors. Harrison’s Principle of Internal Medicine. 16th. The McGraw-Hill Companies, Inc.; Pennsylvania, USA: 2005. pp. 2319–23. [Google Scholar]
- 3.DiMauro S. Muscle glycogenoses: An overview. Acta Myol. 2007;26:35–41. [PMC free article] [PubMed] [Google Scholar]
- 4.Lucia A, Nogales-Gadea G, Pérez M, Martín MA, Andreu AL, Arenas J. McArdle disease: what do neurologists need to know? Nat Clin Pract Neurol. 2008;4:568–77. doi: 10.1038/ncpneuro0913. [DOI] [PubMed] [Google Scholar]
- 5.Deschauer M, Opalka JR, Lindner A, Zierz S. A novel nonsense mutation (R269X) in the myophosphorylase gene in a patient with McArdle disease. Mol Genet Metab. 2001;74:489–91. doi: 10.1006/mgme.2001.3252. [DOI] [PubMed] [Google Scholar]
- 6.Deschauer M, Hertel K, Zierz S. Two novel mutations in the myophosphorylase gene in a patient with McArdle disease. Muscle Nerve. 2003;27:105–7. doi: 10.1002/mus.10261. [DOI] [PubMed] [Google Scholar]
- 7.Martín MA, Rubio JC, Wevers RA, et al. Molecular analysis of myophosphorylase deficiency in Dutch patients with McArdle’s disease. Ann Hum Genet. 2003;68:17–22. doi: 10.1046/j.1529-8817.2003.00067.x. [DOI] [PubMed] [Google Scholar]
- 8.DiMauro S, Andreu AL, Bruno C, Hadjigeorgiou GM. Myophosphorylase deficiency (glycogenosis type V; McArdle disease) Curr Mol Med. 2002;2:189–96. doi: 10.2174/1566524024605770. [DOI] [PubMed] [Google Scholar]
- 9.Nogales-Gadea G, Brull A, Santalla A, et al. McArdle Disease: Update of Reported Mutations and Polymorphisms in the PYGM Gene. Hum Mutat. 2015a;36:669–78. doi: 10.1002/humu.22806. [DOI] [PubMed] [Google Scholar]
- 10.NCIB-NIH. Short genetic variations dbSNP. https://www.ncbi.nlm.nih.gov/projects/SNP/. Access date: 24.3.2017.
- 11.ExAC Browser. http://exac.broadinstitute.org/gene/ENSG00000068976. Access date: 13.10.2016.
- 12.Rubio JC, Garcia-Consuegra I, Nogales-Gadea G, et al. A proposed molecular diagnostic flowchart for myophosphorylase deficiency (McArdle disease) in blood samples from Spanish patients. Hum Mutat. 2007;28:203–4. doi: 10.1002/humu.9474. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009a;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009b;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gormez Z, Bakir-Gungor B, Sagirogl MS. HomSI: a homozygous stretch identifier from next-generation sequencing data. Bioinformatics. 2014;30:445–7. doi: 10.1093/bioinformatics/btt686. [DOI] [PubMed] [Google Scholar]
- 21.SIFT BLink. http://sift.jcvi.org/www/SIFT_BLink_submit.html. Access date: 24.3.2016.
- 22.Pspired (http://bioinf.cs.ucl.ac.uk/psipred/), Access date: October 10, 2016.
- 23.NCBI-NIH. ClinVar. https://www.ncbi.nlm.nih.gov/clinvar/variation/95294/
- 24.NCBI-NIH. Short Gentic variations. dbSNP; https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=764823441. [Google Scholar]
- 25.Kubisch C, Wicklein EM, Jentsch TJ. Molecular diagnosis of McArdle disease: revised genomic structure of the myophosphorylase gene and identification of a novel mutation. Hum Mutat. 1998;12:27–32. doi: 10.1002/(SICI)1098-1004(1998)12:1<27::AID-HUMU4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 26.De Castro M, Johnston J, Biesecker L. Determining the prevalence of McArdle disease from gene frequency by analysis of next-generation sequencing data. Genet Med. 2015;17:1002–6. doi: 10.1038/gim.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beaulieu CL, Majewski J, Schwartzentruber J, et al. FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet. 2014 Jun 5;94(6):809–17. doi: 10.1016/j.ajhg.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vorgerd M, Kubisch C, Burwinkel B, et al. Mutation analysis in myophosphorylase deficiency (McArdle’s disease) Ann Neurol. 1998;43:326–31. doi: 10.1002/ana.410430310. [DOI] [PubMed] [Google Scholar]
- 29.Deschauer M, Morgenroth A, Joshi PR, et al. Analysis of spectrum and frequencies of mutations in McArdle disease. Identification of 13 novel mutations. J Neurol. 2007;254:797–802. doi: 10.1007/s00415-006-0447-x. [DOI] [PubMed] [Google Scholar]
- 30.Bruno C, Löfberg M, Tamburino L, et al. Molecular characterization of McArdle’s disease in two large Finnish families. J Neurol Sci. 1999a;165:121–5. doi: 10.1016/s0022-510x(99)00091-x. [DOI] [PubMed] [Google Scholar]
- 31.Bruno C, Tamburino L, Kawashima N, et al. A nonsense mutation in the myophosphorylase gene in a Japanese family with McArdle’s disease. Neuromuscul Disord. 1999b;9:34–7. doi: 10.1016/s0960-8966(98)00096-0. [DOI] [PubMed] [Google Scholar]
- 32.Fernández R, Navarro C, Andreu AL, et al. A novel missense mutation (W797R) in the myophosphorylase gene in Spanish patients with McArdle disease. Arch Neurol. 2000;57:217–9. doi: 10.1001/archneur.57.2.217. [DOI] [PubMed] [Google Scholar]
- 33.Lucia A, Ruiz JR, Santalla A, et al. Genotypic and phenotypic features of McArdle disease: insights from the Spanish national registry. J Neurol Neurosurg Psychiatry. 2012;83(3):322–8. doi: 10.1136/jnnp-2011-301593. [DOI] [PubMed] [Google Scholar]
- 34.Bartram C, Edwards RH, Clague J, Beynon RJ. McArdle’s disease: a nonsense mutation in exon 1 of the muscle glycogen phosphorylase gene explains some but not all cases. Hum Mol Genet. 1993;2:1291–93. doi: 10.1093/hmg/2.8.1291. [DOI] [PubMed] [Google Scholar]
- 35.Tsujino S, Shanske S, DiMauro S. Molecular genetic heterogeneity of myophosphorylase deficiency (McArdle’s disease) N Engl J Med. 1993;329:241–5. doi: 10.1056/NEJM199307223290404. [DOI] [PubMed] [Google Scholar]
- 36.El-Schahawi M, Tsujino S, Shanske S, DiMauro S. Diagnosis of McArdle’s disease by molecular genetic analysis of blood. Neurology. 1996;47:579–80. doi: 10.1212/wnl.47.2.579. [DOI] [PubMed] [Google Scholar]
- 37.Martinuzzi A, Tsujino S, Vergani L, et al. Molecular characterization of myophosphorylase deficiency in a group of patients from northern Italy. J Neurol Sci. 1996;137:14–9. doi: 10.1016/0022-510x(95)00298-g. [DOI] [PubMed] [Google Scholar]
- 38.Clancy S. RNA splicing: introns, exons and splicesome. Nature Education. 2008;1:31. [Google Scholar]
- 39.Mitsuhashi S, Ohkuma A, Talim B, et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am J Hum Genet. 2011;88:845–51. doi: 10.1016/j.ajhg.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oliveira J, Negrão L, Fineza I, et al. New splicing mutation in the choline kinase beta (CHKB) gene causing a muscular dystrophy detected by whole-exome sequencing. J Hum Genet. 2015;60:305–12. doi: 10.1038/jhg.2015.20. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Consuegra I, Blázquez A, Rubio JC, et al. Taking advantage of an old concept, “illegitimate transcription”, for a proposed novel method of genetic diagnosis of McArdle disease. Genet Med. 2016;18(11):1128–1135. doi: 10.1038/gim.2015.219. [DOI] [PubMed] [Google Scholar]
- 42.Tsujino S, Shanske S, Nonaka I, et al. Three new mutations in patients with myophosphorylase deficiency (McArdle disease) Am J Hum Genet. 1994;54:44–52. [PMC free article] [PubMed] [Google Scholar]
- 43.Sugie H, Sugie Y, Ito M, Fukuda T, Nonaka I, Igarashi Y. Genetic analysis of Japanese patients with myophosphorylase deficiency (McArdle’s disease): single-codon deletion in exon 17 is the predominant mutation. Clin Chim Acta. 1995;236:81–6. doi: 10.1016/0009-8981(95)06044-x. [DOI] [PubMed] [Google Scholar]
- 44.Richards RI, Sutherland GR. Dynamic mutations: a new class of mutations causing human disease. Cell. 1992;70:709–12. doi: 10.1016/0092-8674(92)90302-s. [DOI] [PubMed] [Google Scholar]
- 45.Michel B. Replication for arrest and DNA recombination. Trens Biochem Sci. 2000;25:173–8. doi: 10.1016/s0968-0004(00)01560-7. [DOI] [PubMed] [Google Scholar]
- 46.Martín MA, Rubio JC, Buchbinder J, et al. Molecular heterogeneity of myophosphorylase deficiency (McArdle’s disease): a genotype-phenotype correlation study. Ann Neurol. 2001;50:574–81. [PubMed] [Google Scholar]
- 47.Wu Y, Weber JL, Vladutiu GD, Tarnopolsky MA. Six novel mutations in the myophosphorylase gene in patients with McArdle disease and a family with pseudo-dominant inheritance pattern. Mol Genet Metab Mol Genet Metab. 2011;104:587–91. doi: 10.1016/j.ymgme.2011.08.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.