

Abstract
The upregulation of kidney injury molecule-1 (KIM-1) has been extensively studied in various renal diseases and following acute injury; however, the initial mechanisms controlling KIM-1 expression remain limited. In this study, KIM-1 expression was examined in mouse renal cell cultures and in two different models of acute kidney injury (AKI), ischemia reperfusion (IR)–induced and lipopolysaccharide (LPS)-induced sepsis. KIM-1 mRNA increased in both AKI models, and pharmacological inhibition of extracellular signal–regulated kinase 1/2 (ERK1/2) signaling attenuated injury-induced KIM-1 expression in the renal cortex. Toll-like receptor 4 knockout (TLR4KO) mice exhibited reduced ERK1/2 phosphorylation and attenuated KIM-1 mRNA after LPS exposure. TLR4KO mice were not protected from IR-induced ERK1/2 phosphorylation and upregulation of KIM-1 mRNA. Following renal IR injury, phosphorylation of signal transducer and activator of transcription 3 (STAT3) at serine 727 and tyrosine 705 increased downstream from ERK1/2 activation. Because phosphorylated STAT3 is a transcriptional upregulator of KIM-1 and inhibition of ERK1/2 attenuated increases in STAT3 phosphorylation, we suggest an ERK1/2-STAT3-KIM-1 pathway following renal injury. Finally, ERK1/2 inhibition in naive mice decreased KIM-1 mRNA and nuclear STAT3 phosphorylation in the cortex, indicating homeostatic regulation of KIM-1. These findings reveal renal ERK1/2 as an important initial regulator of KIM-1 expression in IR and septic AKI and at a physiologic level.
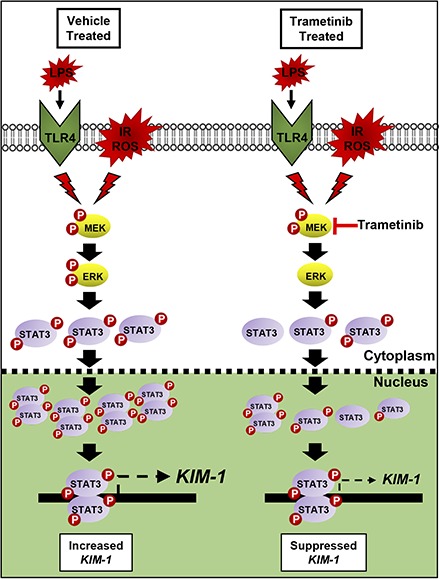
Visual Abstract.Proposed mechanism of IR, LPS, and ROS-induced renal damage that initiates ERK1/2 and STAT3 phosphorylation. STAT3 then binds to the KIM-1 promoter and increases KIM-1 mRNA. By preventing ERK1/2 phosphorylation following renal injury, STAT3 phosphorylation is decreased, leading to less phosphorylated STAT3 within the nucleus, and subsequently less KIM-1 mRNA increases post injury.
Introduction
Kidney injury molecule-1 (KIM-1) is a type I transmembrane glycoprotein that is rapidly upregulated at the gene and protein level following various types of injury to the kidney (Ichimura et al., 1998; Amin et al., 2004; Vaidya et al., 2010). For example, KIM-1 is increased in injuries induced by ischemia reperfusion (IR), cisplatin, sepsis, folic acid, aristolochic acid, and gentamicin, and occurs in disease states such as renal cell carcinoma, polycystic kidney disease, hyperoxaluria, and hypertension (Han et al., 2005; Ichimura and Mou, 2008; Khandrika et al., 2012; Tian et al., 2017).
The ectodomain, which contains an Ig-like domain and a glycosylated mucin domain, is cleaved by the metalloproteinase ADAM17 at the cellular membrane (Ichimura et al., 1998; Bailly et al., 2002; Gandhi et al., 2014). Membrane bound KIM-1 and cleaved KIM-1 increase with worsening renal injury (Zhang et al., 2007). KIM-1 is located on the apical surface of renal proximal tubule epithelial cells and recognizes phosphatidylserine and oxidized lipoprotein epitopes on the surface of apoptotic bodies (Ichimura et al., 2008).
Extracellular signal–regulated kinase 1/2 (ERK1/2) is a mitogen-activated protein kinase that is phosphorylated rapidly following renal injury (Masaki et al., 2003, 2004; Arany et al., 2004). We previously demonstrated ERK1/2 phosphorylation is elevated 3 hours post IR injury and 1 hour post lipopolysaccharide (LPS)-induced acute kidney injury (AKI) (Smith et al., 2015; Collier et al., 2016). Further exploration of the ERK1/2 pathway was performed utilizing a specific mitogen-activated protein kinase kinase 1/2 inhibitor, trametinib, which inhibits the increase in ERK1/2 phosphorylation since mitogen-activated protein kinase kinase 1/2 is directly upstream of ERK1/2 (Gilmartin et al., 2011; Yamaguchi et al., 2011). Interestingly, inhibiting LPS-induced ERK1/2 phosphorylation prevented KIM-1 mRNA upregulation after 3 and 18 hours post LPS administration. Smith et al. (2016) confirmed ERK1/2 inhibition attenuates the upregulation of KIM-1 mRNA following injury utilizing trametinib treatment 6 hours post cecal ligation and puncture surgery, a model of sepsis.
ERK1/2 and KIM-1 were studied in vitro using a porcine kidney epithelial cell line, LLC-PK1, transfected with human KIM-1 (Zhang and Cai, 2016). KIM-1 protein promoted cell migration and proliferation through activation of the ERK1/2 pathway, and inhibiting ERK1/2 attenuated these processes. Recently, KIM-1 was shown to modulate macrophages by promoting an M1 phenotype and macrophage migration by mitogen-activated protein kinase pathways, including ERK, p38, and c-Jun N-terminal kinase (Tian et al., 2017). It is clear that ERK1/2 and KIM-1 are activated and upregulated shortly after kidney injury; however, a direct connection between ERK1/2 and KIM-1 has received limited study.
Ajay et al. (2014) used a combination of bioinformatics, in vitro, and in vivo experiments to determine that phosphorylation of signal transducer and activator of transcription 3 (STAT3) at serine 727 (S727) and tyrosine 705 (Y705) increases KIM-1 transcription by binding to the KIM-1-specific promoter. Checkpoint kinase 1 (Chk1) was upregulated following IR injury due to DNA damage, which phosphorylated STAT3 and subsequently regulated KIM-1 expression (Ajay et al., 2014). Interestingly, ERK2 was the most enriched upstream kinase identified that was associated with the highest upregulated transcription factors following IR injury (Berger et al., 2007; Lachmann and Ma’ayan, 2009; Ajay et al., 2014). Given this information and our previous data demonstrating ERK1/2 is rapidly phosphorylated post IR injury, we hypothesized early ERK1/2 activation following injury preferentially regulates KIM-1 expression.
Materials and Methods
In Vitro.
A transgenic kidney proximal tubule (TKPT) cell line originally isolated from 8Tg(SV40E)Bri7 mice and developed by Ernest and Bello-Reuss (1995) was used in all in vitro experiments (MacKay et al., 1988). TKPT cells were grown in 35-mm tissue culture dishes. The culture medium was a 1:1 mixture of Dulbecco’s modified Eagle’s medium/F-12 (without phenol red or sodium pyruvate) supplemented with 5 mM glucose, 10% fetal bovine serum, 15 mM HEPES buffer, 2.5 mM L-glutamine, 1 μM pyridoxine HCl, 15 mM sodium bicarbonate, and 6 mM lactate. Hydrocortisone (50 nM), selenium (5 ng/ml), human transferrin (5 μg/ml), bovine insulin (10 nM), and L-ascorbic acid-2-phosphate (50 μM) were added to fresh culture medium. For all experiments, 90%–95% confluent TKPT cells were used, and fetal bovine serum was removed from culture media 24 hours before experimentation. TKPT cell monolayers were treated with hydroxyurea (HU) (AC151680050; Fisher Scientific, Hampton, NH), tert-butyl hydroperoxide (TBHP) (Sigma, St. Louis, MO), and H2O2 (H1009; Sigma) or vehicle (dimethylsulfoxide; Sigma) for time points indicated. Different TKPT cell passages used on different days represent the “N” value for the in vitro studies.
Naive and AKI Studies.
Trametinib (Gilmartin et al., 2011) was purchased from Selleckchem Chemicals (Houston, TX). Eight-to-nine week old male C57BL/6 mice (20–25 g) were acquired from Charles River Laboratories (Frederick, MD) and received an injection of trametinib (1 mg/kg i.p.) or vehicle control (NEOBEE M-5; Fisher Scientific). Four hours after injection kidneys were collected and flash frozen in liquid nitrogen for further analysis.
For IR AKI, mice were assigned to three groups: 1) sham, 2) IR + vehicle, and 3) IR + trametinib. Vehicle or trametinib was administered intraperitoneally 1 hour before surgery. For toll-like receptor 4 (TLR4) knockout [(KO), i.e., TLR4KO], IR AKI mice were assigned to four groups: 1) wild-type (WT) sham, 2) TLR4KO sham, 3) WT IR, and 4) TLR4KO IR. The IR grouped mice were subjected to IR surgery by bilateral renal pedicle clamping for 18.5 minutes as described previously (Funk and Schnellmann, 2012). Briefly, the renal artery and vein were isolated and blood flow was occluded with a vascular clamp for 18.5 minutes while maintaining a constant body temperature of 36 to 37°C. Sham mice were treated exactly the same as IR mice, except without the clamping of the renal pedicles. Mice were euthanized 3 or 24 hours after surgery, and blood and kidneys (flash frozen in liquid nitrogen) were collected for analysis.
For the LPS model of sepsis, 8-week-old male C57BL/6 mice (20–25 g) were given an intraperitoneal injection of 10 mg/kg LPS derived from Escherichia coli serotype O111:B4 (Sigma-Aldrich, St. Louis, MO). Control mice received an intraperitoneal injection of an equal volume of 0.9% normal saline. Mice received an injection of trametinib (1 mg/kg i.p.) or vehicle control 1 hour before administration of LPS. Mice were euthanized 18 hours after LPS exposure, and kidneys and blood were collected for analysis. Experiments with TLR4KO mice were graciously donated by Dr. Kenneth D. Chavin and generated by crossing C57BL/10ScN mice with the tlr4LPS-d mutation onto the C57BL/6 background for at least five generations (Ellett et al., 2009; Smith et al., 2015). All studies were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Animal use was approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina.
Serum Creatinine Measurement.
Blood was collected by retro-orbital bleeding puncture and allowed to sit at room temperature for 30 minutes. The samples were centrifuged at 10,000 rpm for 10 minutes to separate the serum. Serum creatinine (SCr) was determined using the Creatinine Enzymatic Reagent Assay kit (Pointe Scientific, Canton, MI) [c7548-120] based on the manufacturer’s directions. All values are expressed as SCr concentration in milligrams per deciliter.
Quantitative Reverse Transcription Polymerase Chain Reaction Analysis of mRNA Expression.
Total RNA was isolated from renal cortical tissue with TRIzol reagent (Life Technologies, Carlsbad, CA). The iScript Advanced cDNA Synthesis Kit (Bio-Rad, Hercules, CA) was used according to the manufacturer’s protocol. The generated cDNA was used with the SsoAdvanced Universal SYBR Green Supermix reagent (Bio-Rad). The relative mRNA expression of all genes was determined by the 2−ΔΔCt method, and mouse actin RNA and rabbit tubulin RNA were used as reference genes for normalization. The KIM-1 primer sequences used were forward: 5′-GCATCTCTAAGCGTGGTTGC-3′ and reverse: 5′-TCAGCTCGGGAATGCACAA-3′.
Immunoblot Analysis.
Protein was extracted from renal cortex using radioimmunoprecipitation assay buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, pH 7.4) with protease inhibitor cocktail (1:100), 1 mM sodium fluoride, and 1 mM sodium orthovanadate (Sigma-Aldrich).
Mouse tissue nuclear and cytosolic fraction lysates were prepared as previously described (Collier et al., 2016). Briefly, a piece of kidney cortex was homogenized in sucrose isolation buffer (250 mM sucrose, 1 mM EGTA, 10 mM HEPES, 1 mg/ml fatty acid-free bovine serum albumin, pH 7.4) added fresh with a dounce tissue grinder. Lysates were centrifuged at 1000g for 10 minutes. The first pellet was pushed through a 25G needle three times, rewashed three separate times in sucrose isolation buffer, and resuspended in radioimmunoprecipitation assay buffer. The first supernatant was centrifuged at 10,000g for 5 minutes for further purification of the cytosol fraction. Histone H3 and/or lamin B1 were used as the loading control for nuclear lysate immunoblots, and α-tubulin was used for the loading control of cytosolic lysates.
Equal protein quantities (10–60 μg) were loaded onto 4%–15% SDS-PAGE gels, resolved by gel electrophoresis, and transferred onto nitrocellulose membranes (Bio-Rad). Membranes were blocked in 5% bovine serum albumin or 5% milk in Tris-buffered saline/Tween 20 and incubated overnight with primary antibody at 4°C with gentle agitation. The primary antibodies used in these studies included phospho-ERK1/2 (1:1000), total ERK1/2 (1:1000), phospho-Chk1 (1:1000), Chk1 (1:1000), phospho-STAT3-Tyr705 (1:1000), phospho-STAT3-Ser727 (1:1000), total STAT3 (1:1000), and histone H3 (1:2000) from Cell Signaling Technology, Danvers, MA; lamin B1 (1:1000; Abcam, Cambridge, United Kingdom), phospho-Chk1 (1:800), and KIM-1 (1:1000) from R&D Systems, Minneapolis, MN; and β-actin (1:2000) from Santa Cruz Biotechnology, Dallas, TX.
Membranes were incubated with the appropriate horseradish peroxidase–conjugated secondary antibody before visualization using enhanced chemiluminescence (Thermo Scientific, Waltham, MA) and GE ImageQuant LAS4000 (GE Life Sciences; Marlborough, MA). Optical density was determined using the ImageJ software from the National Institutes of Health (Bethesda, MD) and Image Studio Lite from LI-COR (Lincoln, NE).
Statistical Analysis.
All data are shown as mean ± S.E.M. When comparing two experimental groups, an unpaired, two-tailed t test or Mann-Whitney U test was used to determine statistical differences. A one-way analysis of variance followed by Tukey’s post hoc test was performed for comparisons of multiple groups. P < 0.05 was considered statistically significant. All statistical tests were performed using GraphPad Prism software (GraphPad Software, San Diego, CA).
Results
ERK1/2 Increases KIM-1 mRNA Following Toxicant Exposure in Mouse Renal Cells.
To confirm that KIM-1 expression is upregulated following toxicant exposure, cultured mouse TKPT cells were treated with 1 and 10 mM HU, 0.1 and 1 mM hydrogen peroxide (H2O2), and 350 and 700 μM TBHP for 24 hours. HU causes cellular injury by inhibiting DNA synthesis and causing DNA damage (Singh and Xu, 2016), while H2O2 and TBHP are model oxidants that lead to oxidative stress causing protein, lipid, and DNA damage (Nowak et al., 2006). Each toxicant increased KIM-1 mRNA with increasing concentration (Fig. 1A). KIM-1 mRNA increased 8-, 3-, and 3-fold in response to 10 mM HU, 1 mM H2O2, and 700 μM TBHP, respectively (Fig. 1A). A 1-hour pretreatment with trametinib prevented HU-, H2O2-, and TBHP-induced KIM-1 mRNA increases (Fig. 1B). Interestingly, TKPT cells treated with trametinib alone showed a 54% decrease in KIM-1 mRNA compared with vehicle control cells after 24 hours (Fig. 1B). These data reveal a regulatory role for ERK1/2 on KIM-1 mRNA under control conditions and following DNA and oxidative damage.
Fig. 1.

ERK1/2 increases KIM-1 mRNA after toxicant exposure in mouse TKPT cells. (A) KIM-1 mRNA expression after 24 hours following exposure to HU, H2O2, and TBHP. (B) KIM-1 mRNA measured after 24 hours post toxicant administration with or without 1 hour trametinib (GSK) pretreatment. Data are represented as mean ± S.E.M., n ≥ 4. Superscripts indicate statistically significant differences (P < 0.05);*, significantly different than vehicle control; #, significance between same toxicant groups.
Early ERK1/2 Activation Mediates STAT3 Phosphorylation–Induced KIM-1 mRNA Upregulation Following IR AKI.
KIM-1 mRNA increased 4.7-fold 3 hours post IR AKI compared with sham controls (Fig. 2A). Trametinib attenuated the KIM-1 mRNA and SCr increases when administered 1 hour before IR surgery (Fig. 2, A and B). The phosphorylated ERK1/2 to total ERK1/2 ratio increased 4.5-fold in the IR groups compared with the sham groups (Fig. 2, C and D). Trametinib also completely inhibited ERK1/2 phosphorylation increases following AKI, as observed previously (Smith et al., 2015; Collier et al., 2016).
Fig. 2.

ERK1/2 mediates STAT3 phosphorylation and KIM-1 mRNA upregulation following 3 hours of IR AKI. (A) KIM-1 mRNA expression following IR AKI after 3 hours. (B) Serum creatinine was assessed 3 hours after IR AKI. (C) Representative immunoblot of phosphorylated ERK1/2, total ERK1/2, phosphorylated Chk1, and total Chk1 following 3 hours of IR AKI. (D) Densitometry analysis of phosphorylated ERK1/2 compared with total ERK1/2. (E) Densitometry analysis of phosphorylated Chk1 compared with total Chk1. (F) Representative immunoblot of phosphorylated STAT3 (S727 and Y705) and total STAT3 following 3 hours of IR AKI. (G and H) Densitometry analysis of phosphorylated STAT3 (S727 and Y705) compared with total STAT3. Data are represented as mean ± S.E.M., n = 6. Different superscripts indicate statistically significant differences (P < 0.05).
Ajay et al. (2014) reported that Chk1 phosphorylation is elevated 3 hours post IR AKI. In contrast, we did not observe any change in phosphorylated Chk1 to total Chk1 ratio following IR injury in the presence or absence of trametinib. However, transcription factor STAT3 phosphorylation at both activating sites, S727 and Y705, increased 2.5- and 3.5-fold after renal IR, respectively (Fig. 2, C and E–H) and pretreatment with trametinib partially blocked the increase in phosphorylation of STAT3 at S727 and Y705 (Fig. 2, G and H). These findings suggest that inhibiting ERK1/2 phosphorylation prevents KIM-1 mRNA upregulation 3 hours post IR AKI by preventing STAT3 phosphorylation at S727 and Y705.
ERK1/2 Inhibition Suppresses KIM-1 mRNA and Protein 24 Hours Following IR AKI.
After 24 hours, KIM-1 mRNA was ∼280-fold higher in the IR group compared with the sham controls (Fig. 3A). Trametinib partially blocked the increase in KIM-1 mRNA (∼120-fold higher compared with sham) compared with the IR mice (Fig. 3A). Serum creatinine increased to 1.1 mg/dl in the IR group and pretreatment with trametinib attenuated this rise in SCr after 24 hours (Fig. 3B). STAT3 phosphorylation at S727 and Y705 increased following renal IR injury and trametinib pretreatment blocked these increases (Fig. 3, D, G and H). Chk1 phosphorylation decreased in the IR group after 24 hours and ERK1/2 inhibition had no influence on Chk1 phosphorylation (Fig. 3, D and F). Trametinib inhibited ERK1/2 phosphorylation throughout the 24-hour period after IR surgery (Fig. 3, D and E). The IR group showed a large upregulation in KIM-1 kidney cortex protein that was markedly reduced in the presence of trametinib after 24 hours (Fig. 3C). Because KIM-1 is cleaved from the cell surface, KIM-1 protein was measured in the serum to detect any changes in cleavage regulation (Sabbisetti et al., 2014). Serum KIM-1 protein was high in the IR group and markedly reduced in the trametinib group (Fig. 3C). These findings reveal ERK1/2 remains activated 24 hours following IR AKI, which corresponds to increased phosphorylation of STAT3 at S727 and Y705, and leads to increases in KIM-1 mRNA and protein. This correlates with our observations of STAT3 phosphorylation at S727 and Y705 in the trametinib group compared with the IR group in our 3-hour study (Fig. 2, F–H). However, it is clear that ERK1/2 and STAT3 are not the only pathways responsible for the later increases in KIM-1 mRNA since the trametinib group is only partially attenuated after 24 hours (Fig. 3A).
Fig. 3.

ERK1/2 mediates KIM-1 mRNA and protein increases and STAT3 phosphorylation following 24 hours of IR AKI. (A) KIM-1 mRNA expression following IR AKI after 24 hours. (B) Serum creatinine was assessed 24 hours after IR AKI. (C) Representative immunoblot of renal KIM-1 and serum KIM-1 (n = 2 for serum KIM-1) following 24 hours of IR AKI. (D) Representative immunoblot of phosphorylated ERK1/2, total ERK1/2, phosphorylated STAT3 (S727 and Y705), total STAT3, phosphorylated Chk1, and total Chk1 following IR AKI. (E–H) Densitometry analysis of phosphorylated (E) ERK1/2 to total ERK1/2; (F) p-Chk1 to total Chk1; (G) p-STAT3 S727 to total STAT3; (H) p-STAT3 Y705 to total STAT3. Data are represented as mean ± S.E.M., n ≥ 5, except where indicated. Different superscripts indicate statistically significant differences (P < 0.05).
ERK1/2 Increases KIM-1 mRNA in Sepsis-Induced AKI and Requires TLR4.
Previously, we demonstrated LPS-induced AKI requires TLR4-mediated ERK1/2 activation at the initiation of injury to impair kidney function through mitochondrial dysfunction and inflammatory cytokines (Smith et al., 2015). Similarly, partial attenuation of KIM-1 mRNA upregulation through ERK1/2 inhibition prior to LPS administration after 18 hours was observed (Smith et al., 2015). Here, trametinib treatment prevented KIM-1 protein upregulation within the kidney cortex compared with LPS alone after 18 hours (Fig. 4A). ERK1/2 phosphorylation was inhibited in the trametinib + LPS group compared with the control + LPS group, whereas the LPS group was not different from controls (Fig. 4, A and B). To further illustrate that pharmacological ERK1/2 inhibition downregulates KIM-1 expression, ERK1/2 phosphorylation was studied following LPS in TLR4KO mice since TLR4 is required for LPS-induced kidney dysfunction (Smith et al., 2015). TLR4KO mice had reduced ERK1/2 phosphorylation compared with WT mice administered LPS (Fig. 4, D and F). LPS-induced increases in KIM-1 mRNA and KIM-1 protein were attenuated in LPS-treated TLR4KO mice, connecting ERK1/2 activation and KIM-1 to TLR4 (Fig. 4, D and E). For confirmation, TLR4 mRNA expression in the TLR4KO mice was negligible compared with WT TLR4 mRNA expression (Fig. 4C).
Fig. 4.

ERK1/2 increases KIM-1 mRNA and protein in LPS-induced AKI through TLR4 after 18 hours. (A) Representative immunoblot of KIM-1, phosphorylated ERK1/2, and total ERK1/2 following LPS-induced AKI. (B) Densitometry analysis of phosphorylated ERK1/2 compared with total ERK1/2. (C) TLR4 mRNA measured in WT mice kidney and TLR4KO mice kidney. (D) Representative immunoblot of KIM-1, phosphorylated ERK1/2, and total ERK1/2 following LPS-induced AKI in WT and TLR4KO mice. (E) KIM-1 mRNA measured in the kidneys of WT and TLR4KO mice following LPS-induced AKI after 18 hours. (F) Densitometry analysis of renal phosphorylated ERK1/2 compared with total ERK1/2 in the LPS-treated WT and TLR4KO mice. Data are represented as mean ± S.E.M., n ≥ 3. Different superscripts indicate statistically significant differences (P < 0.05).
TLR4 Is Not Required for IR-Induced ERK1/2 Phosphorylation and KIM-1 mRNA Upregulation.
To determine if ERK1/2 and KIM-1 are regulated by TLR4 during initiation of injury following IR AKI, TLR4KO mice were subjected to IR surgeries and KIM-1 alterations were observed after 3 hours. The WT and TLR4KO mice had similar increases in phosphorylated ERK1/2 following IR AKI compared with their sham controls (Fig. 5, A and C). Interestingly, KIM-1 mRNA was markedly elevated in the TLR4KO IR mice and WT IR mice trended upward (Fig. 5B). The aforementioned findings demonstrate that IR AKI–induced KIM-1 upregulation and ERK1/2 activation are TLR4 independent.
Fig. 5.

TLR4 is not required for IR-induced ERK1/2 phosphorylation and KIM-1 mRNA upregulation. (A) Representative immunoblot of phosphorylated ERK1/2 and total ERK1/2 following IR-induced AKI after 3 hours in WT and TLR4KO mice. (B) KIM-1 mRNA measured in the kidneys of WT and TLR4KO mice following IR-induced AKI after 3 hours. (C) Densitometry analysis of renal phosphorylated ERK1/2 compared with total ERK1/2 in the WT and TLR4KO mice following IR-induced AKI. Data are represented as mean ± S.E.M., n ≥ 3. Different superscripts indicate statistically significant differences (P < 0.05).
ERK1/2 Inhibition Decreases KIM-1 mRNA and Nuclear and Cytosolic STAT3 Phosphorylation under Physiologic Conditions.
To determine if ERK1/2 regulates KIM-1 at a physiologic level, naive mice were treated with trametinib or vehicle for 4 hours. Unexpectedly, trametinib-treated mice had ∼55% lower KIM-1 mRNA compared with vehicle control mice (Fig. 6A). The trametinib group had reduced ERK1/2 phosphorylation in the nucleus and cytosol (Fig. 6, G and H). Nuclear STAT3 phosphorylation at sites Y705 and S727 were down in the trametinib group by 55% and 46%, respectively, compared with the vehicle control group (Fig. 6, B–D). Phosphorylated Y705 and S727 on STAT3 in the cytosol were also reduced in the trametinib group (Fig. 6, E and F). To confirm that trametinib was inhibiting nuclear STAT3 phosphorylation, and subsequently KIM-1 mRNA downregulation, cytosol and nuclear fractions were isolated and confirmed for purity using α-tubulin and lamin B1 (Fig. 6H). The purified fractions demonstrated that inhibition of physiologic ERK1/2 phosphorylation results in a decrease in KIM-1 mRNA through reducing STAT3 phosphorylation at Y705 and S727 in the kidney. By decreasing active STAT3 within the nucleus there is potential for less KIM-1 promoter binding, resulting in lower KIM-1 mRNA expression.
Fig. 6.

ERK1/2 inhibition decreases KIM-1 mRNA and nuclear and cytosolic STAT3 phosphorylation under physiologic conditions in naive mice. (A) Representative immunoblot of nuclear phosphorylated STAT3 (S727 and Y705) and total STAT3 in naive mice after 4-hour treatment with trametinib. (B) KIM-1 mRNA measured at 4 hours post-treatment with trametinib. (C and D) Densitometry analysis of nuclear phosphorylated STAT3 (S727 and Y705) compared with total STAT3. (E) Representative immunoblot of cytosol phosphorylated STAT3 (S727 and Y705) and total STAT3 in naive mice after 4-hour treatment with trametinib. (F) Densitometry analysis of cytosol phosphorylated STAT3 (S727 and Y705) compared with total STAT3. (G) Densitometry analysis of nuclear and cytosol phosphorylated ERK1/2 compared with total ERK1/2. (H) Representative and verification renal immunoblot of nuclear and cytosol control proteins with and without trametinib treatment. Data are represented as mean ± S.E.M., n ≥ 6. Different superscripts indicate statistically significant differences (P < 0.05).
Discussion
In this study, ERK1/2 was identified as a regulator of KIM-1 expression. ERK1/2 inhibition prevented KIM-1 transcription in vitro following toxicant exposure, and attenuated increases in KIM-1 mRNA and protein in IR AKI. STAT3 phosphorylation at S727 and Y705 decreased as a result of ERK1/2 inhibition, leading to less KIM-1 expression. In sepsis-induced AKI, TLR4 was required for activation of ERK1/2 and KIM-1 upregulation. In contrast, ERK1/2 phosphorylation following IR AKI was not dependent on TLR4. Interestingly, in naive mice ERK1/2 regulates KIM-1 mRNA by physiologic STAT3 phosphorylation in the nucleus. We suggest ERK1/2 regulates renal KIM-1 expression at a physiologic level and initially during pathologic renal injury by phosphorylation of STAT3.
The discovery that KIM-1 is the most upregulated gene following AKI led to an increase in KIM-1 renal research (Ichimura et al., 1998; Vaidya et al., 2006, 2010). Ajay et al. (2014) explored the signaling pathway that results in upregulation of renal KIM-1 transcription. They reported KIM-1 transcriptional regulation by Chk1-STAT3 interaction following AKI-induced DNA damage. Chk1 was revealed as an upstream kinase of STAT3 and phosphorylation at S727 and Y705 on STAT3 was required for KIM-1 upregulation (Ajay et al., 2014). Phosphorylated STAT3 increased following AKI and was found to bind to the KIM-1 promoter, therefore increasing KIM-1 mRNA and protein (Ajay et al., 2014). While our studies support the work of Ajay et al. (2014), important differences were observed: 1) Chk1 phosphorylation did not increase following IR AKI after 3 and 24 hours, 2) ERK1/2 was responsible for the STAT3 phosphorylation after IR, and 3) our IR experimental design used mice rather than rats for AKI. Chk1 phosphorylation did not increase in the renal cortex in our experiments; however, KIM-1 mRNA and renal dysfunction was observed. Nevertheless, STAT3 phosphorylation was verified to increase following renal injury, and a reduction in phosphorylated STAT3 attenuated KIM-1 mRNA. However, ERK1/2 was demonstrated as the key initial regulator of STAT3 phosphorylation following renal injury, rather than Chk1.
We previously published work demonstrating that inhibiting the rapid activation of ERK1/2 following renal injury prevents and/or attenuates injury and dysfunction through restoration of mitochondrial function, renal microvascular perfusion, and attenuation of proinflammatory cytokines (Smith et al., 2015, 2016; Collier et al., 2016). Here, ERK1/2 involvement with STAT3 after renal injury was observed. STAT3 phosphorylation is activated early in tubule cells following injury, although the exact role in kidney dysfunction and recovery varies in different AKI models (Talbot et al., 2011; Brosius and He, 2015). For example, in HgCl2-, IR-, and aristolochic acid–induced AKI, early STAT3 phosphorylation was protective (Nechemia-Arbely et al., 2008; Zhou et al., 2010). In contrast, inhibition of STAT3 phosphorylation in polycystic kidney disease, unilateral ureteral obstruction, and human immunodeficiency virus–associated nephropathy has shown beneficial outcomes (Pang et al., 2010; Takakura et al., 2011; Gu et al., 2013). Our findings suggest that prevention of renal IR-induced STAT3 phosphorylation through ERK1/2 inhibition attenuates SCr and KIM-1 expression.
Previous experiments demonstrated activated ERK1/2 phosphorylates STAT3 at S727, while at the same time decreasing STAT3 Y705 phosphorylation (Booz et al., 2003; Kanai et al., 2003; Gough et al., 2013). However, in our study both S727 and Y705 phosphorylation increased following IR injury and decreased with pretreatment with trametinib, suggesting ERK1/2 is responsible for phosphorylating both sites. Twenty-four hours following IR AKI, a partial reduction of KIM-1 mRNA in the trametinib group was observed, along with complete attenuation of SCr elevation compared with the IR group. KIM-1 protein levels were substantially lower in the trametinib-treated mice, both in the kidney cortex and circulatory system since KIM-1 can be detected in the blood. This occurrence has been recognized due to the loss of cell polarity, microvascular permeability dysfunction, disruption of cell-cell adhesion junctions, and basement membrane cell detachment (Sabbisetti et al., 2014; Tian et al., 2017).
ERK1/2 phosphorylation was found to regulate KIM-1 expression in a second AKI model. LPS-induced AKI increased ERK1/2 phosphorylation and KIM-1 protein, whereas trametinib prevented ERK1/2 activation and attenuated the KIM-1 protein increase. Further characterization of the ERK1/2-KIM-1 pathway was demonstrated by utilization of TLR4KO mice. TLR4 is required for LPS-induced ERK1/2 phosphorylation and subsequently, KIM-1 mRNA upregulation (Smith et al., 2015). ERK1/2 is rapidly phosphorylated following LPS after 1 and 3 hours; however, in this study after 18 hours post-LPS phosphorylated ERK1/2 was not different than the control group, most likely stemming from a prompt compensatory mechanism. Nonetheless, 18 hours after cecal ligation and puncture, which is a slower developing sepsis model, ERK1/2 phosphorylation is still highly phosphorylated compared with controls (Smith et al., 2015, 2016). To verify that TLR4 signaling was not regulating KIM-1 expression directly, the TLR4KO mice underwent IR AKI and were compared with the WT mice. There were no differences in ERK1/2 phosphorylation between the TLR4KO IR group and the WT IR group, and the TLR4KO group had increased KIM-1 mRNA. Thus, TLR4 is required for LPS-induced ERK1/2 activation and increase in KIM-1 mRNA; however, in IR AKI TLR4 is not required since ERK1/2 is activated via other non-TLR4 upstream pathways. Therefore, we view ERK1/2 phosphorylation as a key event controlling KIM-1 regulation, which is activated through different pathways in different diseases.
To determine if ERK1/2 regulated KIM-1 at a physiologic level, trametinib was administered to naive mice. Although KIM-1 is expressed at very low levels in healthy noninjured kidneys, there is a physiologic basal level of detection (Ichimura et al., 1998; van Timmeren et al., 2006). In the absence of an injury, inhibiting physiologic ERK1/2 phosphorylation resulted in the downregulation of KIM-1 mRNA. This observation correlates with our in vitro results that demonstrated trametinib treatment in mouse TKPT cells decreased KIM-1 mRNA. Phosphorylated Chk1 was not altered in the trametinib-treated mice (data not shown). ERK1/2 phosphorylation was inhibited in the nuclear and cytosolic fractions of the kidney cortex, which decreased STAT3 Y705 and S727 phosphorylation in the nucleus. Therefore, less active STAT3 is able to bind to the KIM-1 promoter, corresponding to the KIM-1 mRNA reduction observed.
We have provided evidence that STAT3 phosphorylated at Y705 and S727 is partially responsible for renal KIM-1 upregulation, and that ERK1/2 involvement is upstream of STAT3. These findings provide further evidence that initial ERK1/2 activation is a key regulator of various pathways following renal injury, which may lead to the discovery of potential pharmacological-based therapies.
Abbreviations
- AKI
acute kidney injury
- Chk1
checkpoint kinase 1
- ERK1/2
extracellular signal–regulated kinase 1/2
- HU
hydroxyurea
- IR
ischemia reperfusion
- KIM-1
kidney injury molecule-1
- KO
knockout
- LPS
lipopolysaccharide
- SCr
serum creatinine
- STAT3
signal transducer and activator of transcription 3
- S727
serine 727
- TBHP
tert-butyl hydroperoxide
- TKPT
transgenic kidney proximal tubule
- TLR4
toll-like receptor 4
- WT
wild type
- Y705
tyrosine 705
Authorship Contributions
Participated in research design: Collier, Schnellmann.
Conducted experiments: Collier.
Performed data analysis: Collier.
Wrote or contributed to the writing of the manuscript: Collier, Schnellmann.
Footnotes
This study was funded by National Institutes of Health National Institute of General Medical Sciences [Grant R01GM084147] award to R.G.S.; the Biomedical Laboratory Research and Development Program of the Department of Veterans Affairs [Grant BX000851] awarded to R.G.S.; and the Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship through the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant F31DK105782] awarded to J.B.C.
References
- Ajay AK, Kim TM, Ramirez-Gonzalez V, Park PJ, Frank DA, Vaidya VS. (2014) A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J Am Soc Nephrol 25:105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin RP, Vickers AE, Sistare F, Thompson KL, Roman RJ, Lawton M, Kramer J, Hamadeh HK, Collins J, Grissom S, et al. (2004) Identification of putative gene based markers of renal toxicity. Environ Health Perspect 112:465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany I, Megyesi JK, Kaneto H, Price PM, Safirstein RL. (2004) Cisplatin-induced cell death is EGFR/src/ERK signaling dependent in mouse proximal tubule cells. Am J Physiol Renal Physiol 287:F543–F549. [DOI] [PubMed] [Google Scholar]
- Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV. (2002) Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J Biol Chem 277:39739–39748. [DOI] [PubMed] [Google Scholar]
- Berger SI, Posner JM, Ma’ayan A. (2007) Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics 8:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booz GW, Day JN, Baker KM. (2003) Angiotensin II effects on STAT3 phosphorylation in cardiomyocytes: evidence for Erk-dependent Tyr705 dephosphorylation. Basic Res Cardiol 98:33–38. [DOI] [PubMed] [Google Scholar]
- Brosius FC, III, He JC. (2015) JAK inhibition and progressive kidney disease. Curr Opin Nephrol Hypertens 24:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier JB, Whitaker RM, Eblen ST, Schnellmann RG. (2016) Rapid renal regulation of peroxisome proliferator-activated receptor γ coactivator-1α by extracellular signal-regulated kinase 1/2 in physiological and pathological conditions. J Biol Chem 291:26850–26859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellett JD, Evans ZP, Atkinson C, Schmidt MG, Schnellmann RG, Chavin KD. (2009) Toll-like receptor 4 is a key mediator of murine steatotic liver warm ischemia/reperfusion injury. Liver Transpl 15:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernest S, Bello-Reuss E. (1995) Expression and function of P-glycoprotein in a mouse kidney cell line. Am J Physiol 269:C323–C333. [DOI] [PubMed] [Google Scholar]
- Funk JA, Schnellmann RG. (2012) Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol 302:F853–F864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi R, Yi J, Ha J, Shi H, Ismail O, Nathoo S, Bonventre JV, Zhang X, Gunaratnam L. (2014) Accelerated receptor shedding inhibits kidney injury molecule-1 (KIM-1)-mediated efferocytosis. Am J Physiol Renal Physiol 307:F205–F221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, et al. (2011) GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res 17:989–1000. [DOI] [PubMed] [Google Scholar]
- Gough DJ, Koetz L, Levy DE. (2013) The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS One 8:e83395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Dai Y, Xu J, Mallipattu S, Kaufman L, Klotman PE, He JC, Chuang PY. (2013) Deletion of podocyte STAT3 mitigates the entire spectrum of HIV-1-associated nephropathy. AIDS 27:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han WK, Alinani A, Wu CL, Michaelson D, Loda M, McGovern FJ, Thadhani R, Bonventre JV. (2005) Human kidney injury molecule-1 is a tissue and urinary tumor marker of renal cell carcinoma. J Am Soc Nephrol 16:1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. (2008) Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118:1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. (1998) Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273:4135–4142. [DOI] [PubMed] [Google Scholar]
- Ichimura T, Mou S. (2008) Kidney injury molecule-1 in acute kidney injury and renal repair: a review. Zhong Xi Yi Jie He Xue Bao 6:533–538. [DOI] [PubMed] [Google Scholar]
- Kanai M, Konda Y, Nakajima T, Izumi Y, Kanda N, Nanakin A, Kubohara Y, Chiba T. (2003) Differentiation-inducing factor-1 (DIF-1) inhibits STAT3 activity involved in gastric cancer cell proliferation via MEK-ERK-dependent pathway. Oncogene 22:548–554. [DOI] [PubMed] [Google Scholar]
- Khandrika L, Koul S, Meacham RB, Koul HK. (2012) Kidney injury molecule-1 is up-regulated in renal epithelial cells in response to oxalate in vitro and in renal tissues in response to hyperoxaluria in vivo. PLoS One 7:e44174. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lachmann A, Ma’ayan A. (2009) KEA: kinase enrichment analysis. Bioinformatics 25:684–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay K, Striker LJ, Elliot S, Pinkert CA, Brinster RL, Striker GE. (1988) Glomerular epithelial, mesangial, and endothelial cell lines from transgenic mice. Kidney Int 33:677–684. [DOI] [PubMed] [Google Scholar]
- Masaki T, Foti R, Hill PA, Ikezumi Y, Atkins RC, Nikolic-Paterson DJ. (2003) Activation of the ERK pathway precedes tubular proliferation in the obstructed rat kidney. Kidney Int 63:1256–1264. [DOI] [PubMed] [Google Scholar]
- Masaki T, Stambe C, Hill PA, Dowling J, Atkins RC, Nikolic-Paterson DJ. (2004) Activation of the extracellular-signal regulated protein kinase pathway in human glomerulopathies. J Am Soc Nephrol 15:1835–1843. [DOI] [PubMed] [Google Scholar]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press (US); 2011. Available from: https://www.ncbi.nlm.nih.gov/books/NBK54050/ doi: 10.17226/12910. [PubMed] [Google Scholar]
- Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, Axelrod JH. (2008) IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol 19:1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak G, Clifton GL, Godwin ML, Bakajsova D. (2006) Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol Renal Physiol 291:F840–F855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang M, Ma L, Gong R, Tolbert E, Mao H, Ponnusamy M, Chin YE, Yan H, Dworkin LD, Zhuang S. (2010) A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int 78:257–268. [DOI] [PubMed] [Google Scholar]
- Sabbisetti VS, Waikar SS, Antoine DJ, Smiles A, Wang C, Ravisankar A, Ito K, Sharma S, Ramadesikan S, Lee M, et al. (2014) Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. J Am Soc Nephrol 25:2177–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Xu YJ. (2016) The cell killing mechanisms of hydroxyurea. Genes (Basel) 7:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Mayeux PR, Schnellmann RG. (2016) Delayed mitogen-activated protein kinase/extracellular signal-regulated kinase inhibition by trametinib attenuates systemic inflammatory responses and multiple organ injury in murine sepsis. Crit Care Med 44:e711–e720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Stallons LJ, Collier JB, Chavin KD, Schnellmann RG. (2015) Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J Pharmacol Exp Ther 352:346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura A, Nelson EA, Haque N, Humphreys BD, Zandi-Nejad K, Frank DA, Zhou J. (2011) Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet 20:4143–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot JJ, Shillingford JM, Vasanth S, Doerr N, Mukherjee S, Kinter MT, Watnick T, Weimbs T. (2011) Polycystin-1 regulates STAT activity by a dual mechanism. Proc Natl Acad Sci USA 108:7985–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Shao X, Xie Y, Wang Q, Che X, Zhang M, Xu W, Xu Y, Mou S, Ni Z. (2017) Kidney injury molecule-1 is elevated in nephropathy and mediates macrophage activation via the Mapk signalling pathway. Cell Physiol Biochem 41:769–783. [DOI] [PubMed] [Google Scholar]
- Vaidya VS, Ozer JS, Dieterle F, Collings FB, Ramirez V, Troth S, Muniappa N, Thudium D, Gerhold D, Holder DJ, et al. (2010) Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol 28:478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, Bonventre JV. (2006) Urinary kidney injury molecule-1: a sensitive quantitative biomarker for early detection of kidney tubular injury. Am J Physiol Renal Physiol 290:F517–F529. [DOI] [PubMed] [Google Scholar]
- van Timmeren MM, Bakker SJ, Vaidya VS, Bailly V, Schuurs TA, Damman J, Stegeman CA, Bonventre JV, van Goor H. (2006) Tubular kidney injury molecule-1 in protein-overload nephropathy. Am J Physiol Renal Physiol 291:F456–F464. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. (2011) Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol 39:23–31. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Cai CX. (2016) Kidney injury molecule-1 (KIM-1) mediates renal epithelial cell repair via ERK MAPK signaling pathway. Mol Cell Biochem 416:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Humphreys BD, Bonventre JV. (2007) Shedding of the urinary biomarker kidney injury molecule-1 (KIM-1) is regulated by MAP kinases and juxtamembrane region. J Am Soc Nephrol 18:2704–2714. [DOI] [PubMed] [Google Scholar]
- Zhou L, Fu P, Huang XR, Liu F, Lai KN, Lan HY. (2010) Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J Am Soc Nephrol 21:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]