

Abstract
Background
The Gram-positive bacterium Enterococcus faecium is a commensal of the human gastrointestinal tract and a frequent cause of bloodstream infections in hospitalized patients. The mechanisms by which E. faecium can survive and grow in blood during an infection have not yet been characterized. Here, we identify genes that contribute to growth of E. faecium in human serum through transcriptome profiling (RNA-seq) and a high-throughput transposon mutant library sequencing approach (Tn-seq).
Results
We first sequenced the genome of E. faecium E745, a vancomycin-resistant clinical isolate, using a combination of short- and long read sequencing, revealing a 2,765,010 nt chromosome and 6 plasmids, with sizes ranging between 9.3 kbp and 223.7 kbp. We then compared the transcriptome of E. faecium E745 during exponential growth in rich medium and in human serum by RNA-seq. This analysis revealed that 27.8% of genes on the E. faecium E745 genome were differentially expressed in these two conditions. A gene cluster with a role in purine biosynthesis was among the most upregulated genes in E. faecium E745 upon growth in serum. The E. faecium E745 transposon mutant library was then used to identify genes that were specifically required for growth of E. faecium in serum. Genes involved in de novo nucleotide biosynthesis (including pyrK_2, pyrF, purD, purH) and a gene encoding a phosphotransferase system subunit (manY_2) were thus identified to be contributing to E. faecium growth in human serum. Transposon mutants in pyrK_2, pyrF, purD, purH and manY_2 were isolated from the library and their impaired growth in human serum was confirmed. In addition, the pyrK_2 and manY_2 mutants were tested for their virulence in an intravenous zebrafish infection model and exhibited significantly attenuated virulence compared to E. faecium E745.
Conclusions
Genes involved in carbohydrate metabolism and nucleotide biosynthesis of E. faecium are essential for growth in human serum and contribute to the pathogenesis of this organism. These genes may serve as targets for the development of novel anti-infectives for the treatment of E. faecium bloodstream infections.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4299-9) contains supplementary material, which is available to authorized users.
Keywords: Enterococcus faecium, Transcriptome, Transposon mutant library screening, Nucleotide biosynthesis, Carbohydrate metabolism, Virulence, Zebrafish
Background
Enterococci are commensals of the gastrointestinal tract of humans and animals, but some enterococcal species, particularly E. faecium and E. faecalis, are also common causes of hospital-acquired infections in immunocompromised patients [1]. While E. faecalis has been recognized as an important nosocomial pathogen for over a century, E. faecium has emerged as a prominent cause of hospital-acquired infections over the last two decades [2]. Since the 1980s, E. faecium acquired resistance to multiple antibiotics, including β-lactams, aminoglycosides and finally, to the glycopeptide vancomycin [3]. Nosocomial infections are almost exclusively caused by a specific sub-population of E. faecium, termed clade A-1, which has emerged from a background of human commensal and animal E. faecium strains [4]. Strains in clade A-1 carry genetic elements that are absent from animal or human commensal isolates and which contribute to gut colonization or pathogenicity [5–9]. Clade A-1 E. faecium strains are rarely found in healthy individuals but can colonize the gut of immunosuppressed, hospitalized patients to high-levels. These strains can then cause infections by direct translocation from the gut into the bloodstream [10–12]. In addition, due to faecal contamination of the skin in hospitalized patients, the use of intravenous catheters is another risk factor for the introduction of E. faecium into the bloodstream [3, 13, 14]. Currently, E. faecium causes approximately 40% of enterococcal bacteremias. Due to the accumulation of antibiotic resistance determinants in clade A-1 strains, E. faecium infections are more difficult to treat than infections caused by E. faecalis or other enterococci [15–17]. To cause bloodstream infections, E. faecium needs to be able to survive and multiply in blood, but the mechanisms by which it can do so, have not yet been studied. To thrive in the bloodstream, an opportunistic pathogen has to evade host immune mechanisms and to adjust its metabolism to an environment that is relatively poor in nutrients [18].
To identify genes that are conditionally essential in bacteria, high-throughput screening methods for transposon mutant libraries have been developed and optimized for many different bacterial species [19, 20]. To perform high-throughput functional genomics in ampicillin-resistant, vancomycin-susceptible clinical E. faecium strains, we previously developed a microarray-based transposon mutagenesis screening method which was used to identify genes involved in the development of endocarditis [7], resistance to ampicillin [21], bile [22] and disinfectants [23]. However, microarray-based methods for transposon mutant library screening are limited in their accuracy and can only be used in strains for which the microarray was designed. To address these limitations, several methods, including Tn-seq [24] and TraDIS [25], which are based on high-throughput sequencing of the junctions of the transposon insertion sites and genomic DNA, have been developed [26].
In this study, we set-up Tn-seq in the clinical E. faecium isolate E745 to identify genes that contribute to survival and growth in human serum. In addition, we determined the transcriptional response of E. faecium E745 in that same environment. Finally, we substantiated the role of two E. faecium genes that contribute to growth in serum and in virulence, in a zebrafish model of infection. Collectively, our findings show that metabolic adaptations are key to E. faecium growth in serum and contribute to virulence.
Results
The complete genome sequence of E. faecium E745
In this study, we implemented RNA-seq and Tn-seq analyses in E. faecium strain E745, an ampicillin- and vancomycin-resistant clinical isolate. E. faecium E745 was isolated from a rectal swab of a hospitalized patient as part of routine surveillance during an outbreak of VRE in the nephrology ward of a Dutch hospital in 2000 [27, 28]. To allow the application of RNA-seq and Tn-seq in E. faecium E745, we first determined the complete genome sequence of this strain through a combination of short-read Illumina sequencing and long-read sequencing on the RSII Pacific Biosciences and Oxford NanoPore’s MinION systems. This resulted in a closed chromosomal sequence of 2,765,010 nt and 6 complete plasmids sequences, with sizes ranging between 9.3 kbp and 223.7 kbp (Additional file 1). Taken together, the chromosome and plasmids have 3095 predicted coding sequences. Phylogenetic analysis of the core genome of E745 and a set of 72 genomes representing global E. faecium diversity [4], showed that E. faecium E745 is a clade A-1 strain (Fig. 1). The E745 chromosome contains a pathogenicity island with the esp gene, which encodes a 207-kDa surface protein that is involved in biofilm formation and infection [6, 29, 30]. The vancomycin resistance genes of E. faecium E745 are of the vanA type [31] and are carried on the 32.4-kbp plasmid pE745-2. Additional antibiotic resistance genes in the E. faecium E745 genome are the trimethoprim resistance gene dfrG [32], which is located on plasmid pE745-6, and the chromosomally encoded macrolide resistance gene msrC [33].
Fig. 1.
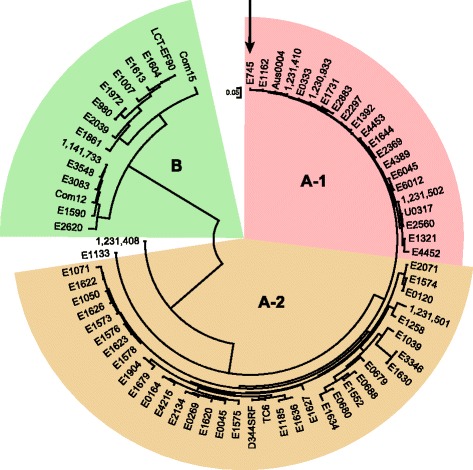
Maximum likelihood phylogenetic tree of E. faecium. The phylogenetic tree was based on a core genome alignment of 1,545,750 positions that was generated by ParSNP [61]. The tree includes the E. faecium E745 genome sequence generated in this study and the 72 E. faecium whole genome sequences described in Lebreton et al. [4]. The tree was visualized and mid-point rooted using MEGA 7.0.26 [62]. The different E. faecium clades are indicated. The position of E. faecium E745 in the phylogenetic tree is highlighted by an arrow
Transcriptome of E. faecium E745 during growth in rich medium and in human serum
After confirming that serum can support the growth of E. faecium E745, though at a lower growth rate than the rich medium BHI (Additional file 2), the transcriptional profile of E745 was determined by RNA-seq during exponential growth in BHI and in heat-inactivated human serum. A total of 99.9 million (15.6–17.6 million per sample) 100 bp paired-end reads were successfully aligned to the genome, allowing the quantification of rare transcripts (Fig. 2). A total of 3217 transcription units were identified, including 651 predicted multi-gene operons, of which the largest contains 22 genes (Fig. 2a and Additional file 3).
Fig. 2.
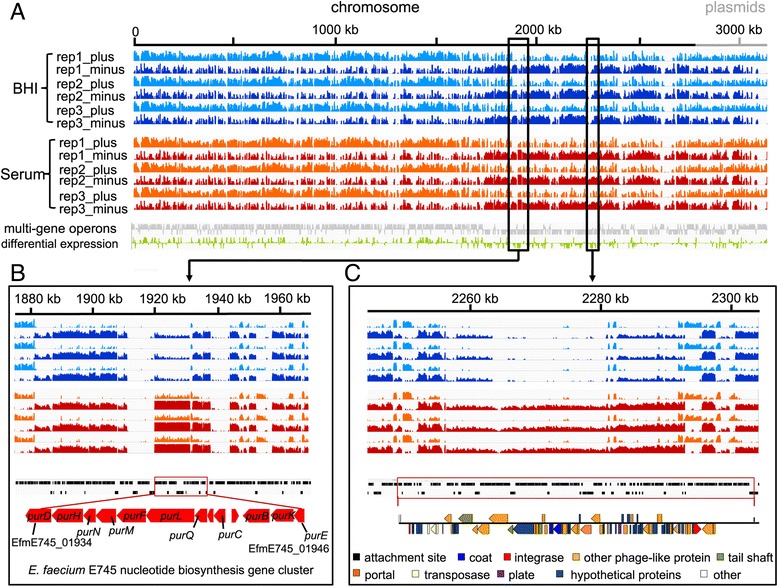
Transcriptome analysis of E. faecium E745. Coverage plots of RNA-seq data aligning to chromosome and plasmid DNA are shown in panel a. The y-axis of each track indicates reads coverage and is represented on a log scale, ranging from 0 to 10,000. The x-axis represents the genomic location. Light blue (BHI) or orange (serum) tracks correspond to sequencing reads aligned to the plus strand of the replicon, and dark blue (BHI) or dark red (serum) tracks correspond to sequencing reads aligned to the minus strand of the replicon. The grey track corresponds to multi-gene operons. The green track corresponds to differentially expressed genes (BHI vs serum), with the height of the green bars indicative of differential expression. In panels b and c, two serum-induced regions are shown, i.e. a gene cluster involved in nucleotide biosynthesis (panel b) and a prophage (panel c). The RNA-seq experiments were performed using three biological replicates
A comparative analysis of the E745 transcriptome during growth in BHI and in human serum, showed that 860 genes exhibited significantly (q < 0.001 and a fold change in expression of >2 or <0.5 between cultures grown in BHI versus heat-inactivated serum) different expression between these conditions (Additional file 4). Among the genes with the highest difference in expression between growth in serum and in rich medium, we identified a gene cluster with a role in purine biosynthesis (Fig. 2b). In addition, we found a 58.4 kbp prophage-like gene cluster that exhibited higher expression in E745 during growth in serum (Fig. 2c). Analysis of this phage sequence against genome sequences deposited at NCBI Genbank revealed that an essentially identical element (with 99% nucleotide identity) could be identified in E. faecium ATCC 700221 but not in other genome sequences of E. faecium or other bacteria.
To confirm the RNA-seq analysis, we independently determined expression levels of eight genes during growth in serum versus growth in BHI by qPCR (Additional file 5). RNA-seq and qPCR data were highly concordant (r2 = 0.98).
E. faecium E745 genes required for growth in human serum
A mariner-based transposon mutant library was generated in E. faecium E745 and Tn-seq [24] was performed on ten replicate transposon mutant libraries (after overnight growth in BHI at 37 °C), resulting in an average of 15 million Tn-seq reads for each library. To analyze the Tn-seq data, we divided the E745 genome in 25-nt windows. Of a total of 110,601 25-nt windows, 49,984 (45%) contained one or more sequence reads. No positional bias was observed in the transposon insertion sites in the chromosome and plasmids of E. faecium E745 (Additional file 6).
In order to identify genes that contribute to growth of E. faecium E745 in human serum, we performed Tn-seq on cultures of the E. faecium E745 transposon mutant libraries upon growth in rich medium (BHI) and in human serum. The human serum was either used natively, or was heat-treated to inactivate the complement system [34]. Minor differences were observed among conditionally essential genes between the experiments performed in native human serum or heat-inactivated human serum (Additional file 7) and the following results correspond to the experiments obtained with heat-inactivated serum. This condition was chosen because it may be a more reproducible in vitro environment, particularly since the interaction between the complement system and Gram-positive bacteria remains to be fully elucidated [35, 36].
We identified 37 genes that significantly contributed to growth of E745 in human serum (Fig. 3 and Additional file 8): twenty-nine genes were located on the chromosome and eight genes were present on plasmids (six genes on pE745–5, two genes on plasmid pE745–6). The relatively large number of genes identified indicates that growth of E. faecium in human serum is a multifactorial process. The genes that conferred the most pronounced effect on growth of E. faecium in serum included genes that are involved in carbohydrate uptake (manZ_3, manY_2, ptsL), a putative transcriptional regulator (algB) and genes involved in the biosynthesis of purine and pyrimidine nucleotides (guaB, purA, pyrF, pyrK_2, purD, purH, purL, purQ, purC) (Fig. 3). Notably, the purD, purH, and purL genes were found to exhibit higher expression upon growth in human serum in the RNA-seq analysis (Fig. 2). Nine genes were identified as negatively contributing to growth in serum, i.e. the transposon mutants in these genes were significantly enriched upon growth in serum. The effects of these mutations were relatively limited (Additional file 8), compared to the major effects observed in the transposon mutants discussed above, but it is notable that five (clsA_1, ddcP, ldt fm , mgs, and lytA_2) of these genes have predicted roles in cell wall and cytoplasmic membrane biosynthesis.
Fig. 3.
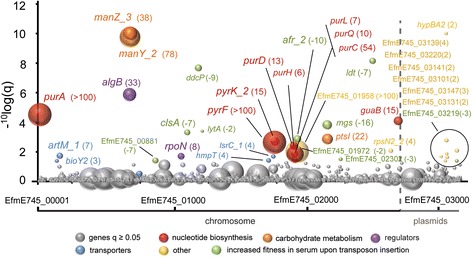
Tn-seq analysis to identify E. faecium genes required for growth in human serum. Bubbles represent genes, and bubble size corresponds to the fold-changes (for visual reasons, a 100-fold change in transposon mutant abundance is set as a maximum) derived from the read-count ratio of libraries grown in BHI to libraries grown in human serum. On the x-axis genes are shown in order of their genomic location and the chromosome and plasmids are indicated. The outcome of statistical analysis of the Tn-seq data is indicated on the y-axis. Genes with a significant change (q < 0.05) in fitness in serum versus BHI are grouped by function and are labelled with different colors, and the name or locus tag and the change in abundance between the control condition and growth in serum is indicated next to the bubbles in parentheses. Negative values indicate that mutants in these genes outgrow other mutants in serum, suggesting that these mutants, compared to the wild-type strain E. faecium E745, have a higher fitness in serum
We developed a PCR-based method (Additional file 9) to selectively isolate five transposon mutants (in the purine metabolism genes purD and purH, the pyrimidine metabolism genes pyrF and pyrK_2 and the phosphotransferase system (PTS) gene manY_2 from the transposon library. Growth in rich medium of these transposon insertion mutants was equal to the parental strain. However, all mutants were significantly impaired in their growth in human serum (Fig. 4a), confirming the results of the Tn-seq experiments.
Fig. 4.
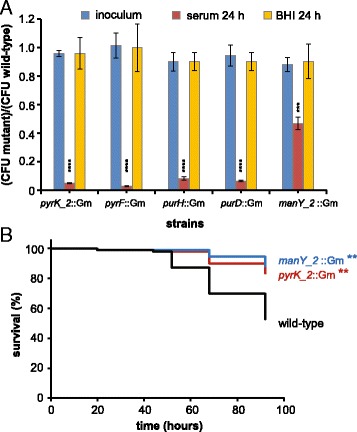
E. faecium transposon mutants with a growth defect in human serum and an attenuated phenotype in a zebrafish infection model. a Ratios of the viable counts of five mutants compared to wild-type E. faecium before (blue bars) and after 24 h of growth in human serum (red bars) or BHI (yellow bars). The viable counts of wild-type E. faecium E745 were (3.52 ± 0.07) × 105/ml in the inocula, (2.92 ± 0.14) × 108/ml after 24 h of growth in serum and (1.20 ± 0.20) × 109/ml after 24 h of growth in BHI, respectively. Error bars represent the standard deviation of the mean of three independent experiments. Asterisks represent significant differences (***: p < 0.001, ****: p < 0.0001) as determined by a two-tailed Student’s t-test) between the mutant strains and wild-type. b Kaplan-Meier survival curves of zebrafish embryos upon infection with E. faecium. Infection was initiated by the injection of 1.2 × 104 CFUs of the manY_2::Gm and pyrK::Gm transposon mutants and the wild-type E. faecium E745 into the circulation of zebrafish embryos 30 h post fertilisation. The experiment was performed three times and the mutants were significantly different (**: p < 0.01) from the wild-type in each experiment as determined by the Log-rank (Mantel-Cox) test with Bonferroni correction for multiple comparisons. This figure represents the combined results of the three replicates for E. faecium E745 (n = 93 zebrafish embryos), manY_2::Gm (n = 92) and pyrK::Gm (n = 90)
E. faecium pyrK_2 and manY_2 contribute to intravenous infection of zebrafish
Finally, we investigated whether the transposon insertion mutants in the manY_2 and pyrK_2 genes were attenuated in vivo (Fig. 4b). The mutants in these genes were selected because they represent the mutants in nucleotide and carbohydrate metabolism genes that were previously shown to contribute to the growth of E. faecium in human serum. As a model for intravenous infection, we used a recently described model in which E. faecium was injected into the circulation of zebrafish embryos to mimic systemic infections [37]. We showed that both the manY_2 and the pyrK_2 mutant were significantly less virulent than the parental strain. At 92 h post infection, survival of zebrafish embryos infected with the WT strain was 53%, as compared to 88% and 83% for zebrafish embryos that were infected with the transposon insertion mutants in manY_2 and pyrK_2, respectively.
Discussion
E. faecium can contaminate the skin and from there colonize indwelling devices such as intravenous catheters, or it can translocate from the gastrointestinal tract in immunosuppressed patients, leading to the development of bacteremia and endocarditis. E. faecium infections are often difficult to treat, due to the multi-drug resistant character of the strains causing nosocomial infections [3, 4]. However, the bloodstream poses challenges for the proliferation and survival of E. faecium, including a scarcity of nutrients.
In the present study, we sequenced the complete genome of a vancomycin-resistant E. faecium strain, and identified E. faecium genes that were essential for growth in human serum. A total of 37 genes, including genes with roles in carbohydrate uptake and nucleotide biosynthesis, were found to be required for fitness of E. faecium E745 in serum. Previously, fitness determinants for growth in human serum have been identified through large-scale screening of mutant libraries in both a Gram-negative (Escherichia coli) and a Gram-positive (Streptococcus pyogenes) pathogen [38, 39]. Notably, these studies have also identified the ability for de novo synthesis of purines and pyrimidines as a crucial factor for growth in serum. In addition, in diverse pathogenic bacteria (including Burkholderia cepacia, Pasteurella multocida, Acinetobacter baumannii, Salmonella enterica serovar Typhimurium, Bacillus anthracis, and Streptococcus pneumoniae), nucleotide biosynthesis contributes importantly to virulence [40–45]. The ability to synthesize nucleotides de novo thus appears to be an essential trait for the success of a pathogen that spreads through the bloodstream [38]. The data presented here indicate that de novo biosynthesis of nucleotides is also required for E. faecium growth in serum and virulence. The nucleotide biosynthesis pathway of E. faecium may be a promising target for the development of novel antimicrobials for the treatment of E. faecium bloodstream infections. Indeed, compounds that target guanine riboswitches, thereby inhibiting nucleotide biosynthesis, have already shown their efficacy in a Staphylococcus aureus infection model [46].
Three genes, ptsL, manY_2 and manZ_3, encoding subunits of E. faecium PTSs were found to contribute to growth in serum in our Tn-seq experiments. The ptsL gene is predicted to encode an enzyme that confers a phosphate group from phosphoenolpyruvate to Enzyme I of PTS, while manY_2 and manZ_3 are predicted to form the IIA and IIBC components of a permease system that is homologous (64% and 69% amino acid identity, respectively) to the PtnAB PTS permease of Lactococcus lactis MG1363. PtnAB is one of the glucose uptake systems of L. lactis [47] and the E. faecium homolog may have a similar function, which could explain its essential role during growth in serum, as glucose is the only carbohydrate that occurs in the free state in appreciable amounts in serum [18].
It is notable that among the nine genes that exhibited increased fitness upon inactivation by transposon insertion, five genes are predicted to have a role in cell wall or cytoplasmic membrane biosynthesis. The protein encoded by ddcP was previously characterized as a low-molecular-weight penicillin-binding protein with D-alanyl-D-alanine carboxypeptidase activity [21], while ldt fm acts as a peptidoglycan L,D transpeptidase [48]. The predicted α-monoglucosyldiacylglycerol synthase gene mgs is orthologous (73% amino acid identity) to bgsB in E. faecalis, which is required for the biosynthesis of membrane glycolipids [49]. The clsA_1 gene is predicted to be responsible for the synthesis of cardiolipin (bisphosphatidylglycerol) and its inactivation may modulate the physical properties of the cytoplasmic membrane [50]. Finally, lytA_2 is predicted to encode an autolysin, which may be involved in the turnover of peptidoglycan in the cell wall [51]. The transposon mutants in these genes were not further characterized in this study, but our findings suggest that non-essential pathways of cell wall or cytoplasmic membrane remodelling may confer subtle fitness defects to E. faecium when growing in a nutrient-poor environment, like serum.
Our RNA-seq-based transcriptional profiling of E. faecium E745 during mid-exponential growth in serum showed pervasive changes in gene expression compared to exponential growth in rich medium. The large number of differentially expressed genes may not all reflect the different growth conditions (serum and BHI) per se, but could also be influenced by the difference in growth rate during mid-exponential growth in serum and BHI (Additional file 2). The purine metabolism genes purL, purH, purD, which were found to be required for growth in serum in our Tn-seq experiments, were among those that were significantly upregulated during growth in serum compared to growth in rich medium. Notably, a single prophage was expressed at higher levels during growth in serum than in rich medium. The abundance of prophage elements in the genome of E. faecium has been noted before [4, 52]. Interestingly, in the related bacterium Enterococcus faecalis prophages encode platelet-binding proteins [53] and may have a role in intestinal colonization [54]. The contribution of E. faecium prophages to traits that are important for colonization and infection may provide important insights into the success of E. faecium as a nosocomial pathogen.
Conclusions
Our data indicate that nucleotide biosynthesis and carbohydrate metabolism are critical metabolic pathways for the proliferation and survival of E. faecium in the bloodstream. The proteins encoded by the genes required for growth in human serum that were identified in this study, could serve as candidates for the development of novel anti-infectives for the treatment of bloodstream-infections by multi-drug resistant E. faecium.
Methods
Bacterial strains, plasmids, growth conditions, and oligonucleotides
The vancomycin-resistant E. faecium strain E745 was used throughout this study. This strain was isolated from a rectal swab of a hospitalized patient, during routine surveillance of a VRE outbreak in a Dutch hospital [27, 28]. Unless otherwise mentioned, E. faecium was grown in brain heart infusion broth (BHI; Oxoid) at 37 °C. The E. coli strains DH5α (Invitrogen) was grown in Luria-Bertani (LB) medium. When necessary, antibiotics were used at the following concentrations: chloramphenicol 4 μg ml−1 for E. faecium and 10 μg ml−1 for E. coli, and gentamicin 300 μg ml−1 for E. faecium and 25 μg ml−1 for E. coli. All antibiotics were obtained from Sigma-Aldrich. Growth was determined by measuring the optical density at 660 nm (OD660). The sequences of all oligonucleotides used in this study are listed in Additional file 10.
Genome sequencing, assembly and bioinformatic analysis
E. faecium E745 was sequenced using a combination of Illumina HiSeq 100 bp paired-end sequencing, long-read sequencing using the Pacific Biosciences RS II SMRT technology and the MinION system with R7 flowcell chemistry (Oxford Nanopore Technologies). Corrected PacBio reads were assembled using the Celera assembler (version 8.1) [55] and assembled contigs were then corrected by aligning Illumina reads using BWA (version 0.7.9a), with default parameters for index creation and the BWA-MEM algorithm with the -M option for the alignment [56]. This approach resulted in 15 contigs, including one contig covering the entire 2.77 Mbp chromosome. After discarding contigs with low-coverage, the remaining contigs constituted 5 circular plasmid sequences and 5 non-overlapping contigs. These 5 contigs were aligned against the NCBI Genbank database and all were found to be part of the E. faecium plasmid pMG1 [57]. Based on this alignment the presumed order of contigs was determined and confirmed by gap-spanning PCRs and sequencing of the products. A single gap between two contigs, could not be closed by PCR. Thus, we assembled Illumina reads together with MinION 2D reads using the SPAdes assembler (version 3.0) [58], which produced a contig that closed the gap, resulting in a complete assembly of this plasmid. Sequence coverage of chromosomal and plasmid sequences was determined with SAMtools (version 0.1.18) using short read alignments to the assembly, which were generated using BWA (version 0.7.9a). SAMtools was also used to identify possible base-calling and assembly errors, by aligning short reads to assembled contigs. A base was corrected using the consensus of aligned reads [59]. The corrected sequences were annotated using Prokka (version 1.10) [60]. A maximum likelihood phylogenetic tree based on the core genome of E. faecium E745 and an additional 72 E. faecium strains representing the global diversity of the species [4], was generated using ParSNP [61] with settings -c (forcing inclusion of all genome sequences) and -x (enabling recombination detection and filtering). The resulting phylogenetic tree was visualized using MEGA 7.0.26 [62]. Antibiotic resistance genes in the assembled genome sequence of E. faecium E745 were identified using ResFinder [63]. The annotated genome of E. faecium E745 is available from NCBI Genbank database under accession numbers CP014529 – CP014535.
RNA-seq
Approximately 3 × 107 cfu of E. faecium E745 were inoculated into 14 ml of BHI broth and heat-inactivated serum, and grown at 37 °C until exponential phase. Cultures were centrifuged at room temperature (15 s; 21.380 g), and pellets were flash frozen in liquid N2 prior to RNA extraction, which was performed as described previously [21]. The ScriptSeq Complete Kit (Bacteria) (Epicentre Biotechnologies, WI) was used for rRNA removal and strand-specific library construction. Briefly, rRNA was removed from 2.5 μg of total RNA. To generate strand specific RNA-seq data, approximately 100 ng of rRNA-depleted RNA was fragmented and reverse transcribed using random primers containing a 5′ tagging sequence, followed by 3′ end tagging with a terminal-tagging oligo to yield di-tagged, single-stranded cDNA. Following magnetic-bead based purification, the di-tagged cDNA was amplified by PCR (15 cycles) using ScriptSeq Index PCR Primers (Epicentre Biotechnologies, WI). Amplified RNA-seq libraries were purified using AMPure XP System (Beckman Coulter) and sequenced by a 100 bp paired end reads sequencing run using the Illumina HiSeq 2500 platform (University of Edinburgh, United Kingdom). Data analysis was performed using Rockhopper [64] using the default settings for strand specific analysis.
Validation of RNA-seq results by quantitative real-time RT-PCR (qRT-PCR).
Total RNA isolated as described previously was used to confirm the transcriptome analysis by qRT-PCR. cDNA was synthesized as described above and qRT-PCR on these cDNAs was performed using the Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific, Breda, The Netherlands) and a StepOnePlus instrument (Life Technologies). The expression of tufA was used as a housekeeping control. Ct values were calculated using the StepOne analysis software v2.2. Transcript levels, relative to tufA, of the assayed genes were calculated using REST 2009 V2.0.13 (Qiagen, Venlo, the Netherlands). This experiment was performed with three biological replicates.
Generation of mariner transposon mutant library in E. faecium
To create a transposon mutant library in E. faecium E745 suitable for Tn-seq, the mariner transposon cassette (carrying a gentamicin resistance gene) in the transposon delivery plasmid pZXL5 [21] was adapted as follows. The transposon from pZXL5 was amplified by PCR using the set of primers: pZXL5_MmeI_SacII_Fw and pZXL5_MmeI_SacII_Rv. These primers introduced MmeI restriction sites in the inverted repeats on both sides of the transposon. The modified transposon delivery vector, termed pGPA1, was generated by the digestion of pZXL5 with SacII, followed by the insertion of the SacII-digested mariner transposon that contained MmeI restriction sites at its extreme ends. pGPA1 was electroporated into E. faecium E745 and the transposon mutant library was generated by selecting for gentamicin-resistant transposon mutants as described previously [21].
Tn-seq analysis of conditionally essential genes in E. faecium E745
The transposon mutant library created in E745 was prepared for Tn-seq analysis, similar to previously described procedures [65]. To identify genes that are essential for the viability of E. faecium in BHI, we used ten experimental replicates of the mutant library. Aliquots (20 μl) of the transposon mutant library, containing approximately 107 cfu, were used to inoculate 20 ml BHI broth and grown overnight at 37 °C. Subsequently, 1 ml aliquots of the cultures were spun down (15 s, 21.380 g) and used for the extraction of genomic DNA (Wizard genomic DNA purification kit, Promega Benelux). 2 μg of the extracted DNA was digested for 4 h at 37 °C using 10 U MmeI (New England Biolabs) and immediately dephosphorylated with 1 U of calf intestine alkaline phosphatase (Invitrogen) during 30 min at 50 °C. DNA was isolated using phenol-chloroform extraction and subsequently precipitated using ethanol. The DNA pellets were then dissolved in 20 μl water. The samples were barcoded and prepared for Tn-seq sequencing as described previously [65]. The sequence reads from all ten experimental replicates were mapped to the genome, and the mapped read-counts were then tallied for the analysis of the essentiality of the genes in the E. faecium E745 genome (further described below).
To identify genes that are required for growth in human serum, 20 μl aliquots of the frozen mutant library in E745 were inoculated in BHI broth and grown overnight as described above. Subsequently, bacterial cells were washed with physiological saline solution. Approximately 3 × 107 cfu were inoculated into 14 ml BHI broth, and approximately 3 × 106 cfu were inoculated into 14 ml human serum obtained from Sigma (Cat. No. H4522; Sterile filtered type-AB human serum) or heat-inactivated human serum (the same, after incubation for 30 min at 56 °C). The different inoculum-sized were used in order for a similar number of divisions to occur during the experiment. Cells were incubated at 37 °C for 24 h without shaking and then further processed for Tn-seq [65]. This experiment was performed in triplicate.
Tn-seq samples were sequenced (50 nt, single-end) on one lane of a Illumina Hiseq 2500 (Baseclear, Leiden, the Netherlands and Sequencing facility University Medical Center, Utrecht, The Netherlands), generating an average of 15 million high quality reads per sample.
Tn-seq data analysis
Raw Illumina sequence reads from Illumina sequencing were split, based on their barcode, using the Galaxy platform [66], and 16-nucleotide fragments of each read that corresponded to E745 sequences, were mapped to the E745 genome using Bowtie 2 [67]. The results of the alignment were sorted and counted by IGV [68] using a 25-nucleotide window size and then summed over the gene. Read mapping to the final 10% of a gene were discarded as these insertions may not inactivate gene function. Read counts per gene were then normalized to the total number of reads that mapped to the genome in each replicate, by calculating the normalized read-count RPKM (Reads Per Kilobase per Million input reads) via the following formula: RPKM = (number of reads mapped to a gene × 106) / (total mapped input reads in the sample x gene length in kbp). Statistical analysis of the RPKM-values between the experimental conditions was performed using Cyber-T [69]. Genes were determined to be significantly contributing to growth in human serum when the Benjamini-Hochberg corrected P-value was <0.05 and the difference in abundance of the transposon mutant during growth in BHI and serum was >2.
Isolation of mutants from the transposon mutant library pool
To recover a targeted transposon mutant from the complete mutant pool, a PCR-based screening strategy was developed (Additional data file 9). 40 μl of the transposon mutant library was inoculated into 40 ml of BHI broth with gentamicin and grown overnight at 37 °C with shaking (200 rpm). The overnight culture, containing approximately 109 cfu/ml, was then diluted to approximately 20 cfu/ml in 500 ml of BHI with gentamicin and kept on ice. Subsequently, 200 μl aliquots were transferred to wells of sterile 96 wells plates (n = 12, Corning Inc.). After overnight incubation at 37 °C without shaking, aliquots (15 μl) of each one of the 96 wells, were further pooled into a single new 96 well plate, as described in Additional file 9.
PCRs were performed on the final plate in which the transposon mutants were pooled, to check for the presence of the Tn-mutants of interest, using the primer ftp_tn_both_ends_MmeI, which is complementary to the repeats flanking the transposon sequence, in combination with a gene-specific primer. When a PCR was found to be positive in one of the wells of this plate, the location of the Tn-mutant was tracked backwards to the wells containing approximately 4 independent transposon mutants, by performing PCRs mapping the presence of the transposon mutant in each step. Cells from the final positive well were plated onto BHI with gentamicin and colony PCR was performed to identify the desired transposon mutant.
Growth of E. faecium E745 and individual mutants in human serum
Wild-type E745 and the mutant strains were grown overnight at 37 °C in BHI broth. Subsequently, bacterial cells were washed with physiological saline and approximately 3 × 105 cfu were inoculated into 1.4 ml BHI broth or heat-inactivated serum. Cells were grown in 1.5 ml tubes (Eppendorf) in triplicate for each condition and incubated at 37 °C for 24 h without shaking. Bacterial growth was determined by assessing viable counts, for which the cultures were serially diluted using physiological saline solution and plated onto BHI agar followed by overnight incubation at 37 °C.
Intravenous infection of zebrafish embryos
London wild-type (LWT) inbred zebrafish embryos, provided by the aquarium staff of The Bateson Center (University of Sheffield), were used for infection experiments. The parental E745 strain and its pyrK_2 and manY_2 transposon mutants were grown in BHI broth until they reached an optical density at 600 nm of approximately 0.5 and were then harvested by centrifugation (5500 g, 10 min). Bacteria were microinjected into the circulation of dechorionated zebrafish embryos at 30 h post fertilization, as previously described [70]. Briefly, anesthetized embryos were embedded in 3% (w/v) methylcellulose and injected individually with approximately 1.2 × 104 cfu using microcapillary pipettes. For each strain, 29 to 32 infected embryos were observed for survival up to 90 h post infection (hpi). This experiment was performed in triplicate.
Additional files
Genome sequence information for E. faecium E745. (XLSX 10 kb)
Growth of E. faecium E745 in BHI and serum. (PDF 962 kb)
Operons identified by RNA-seq in E. faecium E745. (XLSX 40 kb)
E. faecium E745 genes that exhibited significant (q < 0.001 and fold-change > 2) differential expression in human serum, as determined by RNA-seq. (XLSX 70 kb)
qRT-PCR validation of RNA-seq experiments. Correlation of RNA-seq and qRT-PCR expression ratios for the seven genes with various expression levels and genomic locations. The gene expression ratios obtained from both qRT-PCR and RNA-seq were normalized by a housekeeping control gene Efm745_00056 (tufA). The experiment was performed with three biological replicates. (PDF 121 kb)
Characterization of the E. faecium E745 transposon mutant library, showing the number of reads that were mapped to the E. faecium E745 chromosome and plasmids. The height of each peak represents the read abundance at a specific insertion site. On the y-axis, the number of mapped reads is shown on a log scale. (PDF 1427 kb)
Tn-seq data: comparison of heat-inactivated and native serum (XLSX 139 kb)
E. faecium E745 genes that significantly (q < 0.05 and fold-change <−2 or >2) contribute to growth in human serum, as identified by Tn-seq. (XLSX 13 kb)
Isolation of mutants from the transposon mutant library pool. (A) Schematic representation of the PCR reaction designed to find a particular Tn-mutant within the transposon mutant library. This PCR uses a combination of a gene-specific primer (blue arrow) and a transposon specific primer primer (yellow arrow). Positive PCR products, indicated by the green check marks, should occur when the transposon (depicted as a yellow triangle) is inserted in the gene of interest (depicted in blue). If the transposons inserted in adjacent genes or intergenic regions, no PCR product can be amplified (red crosses). (B) Schematic workflow to isolate Tn-mutants from the mutant library. The transposon mutant library is split into 12 plates (A1 - A12) of 96 wells, with each 200 μl well containing an average of 4 mutants. Plates were incubated overnight (Step 2). Plate A1 was then pooled into the first column of a new 96 well plate, denominated plate B1 (Step 3) and the same was done for plates A2 to A12. Subsequently, plate B1 was pooled again into the first column of a third plate, denominated C1 (Step 4). PCR using the gene-specific primer and the transposon specific primer was performed on the 8 wells of plate C1 (Step 5). A positive PCR was suggestive of the presence of a particular transposon-mutant (depicted as a red dot). The presence of the transposon mutant was then confirmed by PCR in plate B1 (step 6) and the corresponding plate A (step 7). Once a transposon-mutant was located to a particular well in plate A, the well was plated on BHI plates containing gentamicin, and the colonies were screened for the presence of the transposon mutant by PCR (step 8). (PDF 277 kb)
Oligonucleotides used in this study. (XLSX 10 kb)
Acknowledgements
Not applicable.
Funding
This work was supported by the European Union Seventh Framework Programme (FP7-HEALTH-2011-single-stage) “Evolution and Transfer of Antibiotic Resistance” (EvoTAR) under grant agreement number 282004 and by a grant from the Netherlands Organization for Scientific Research (VIDI: 917.13.357) to W.v.S. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
Sequence reads generated in this study have been made available at the European Nucleotide Archive under accession number PRJEB19025. The core genome alignment and Newick-formatted tree file that were generated for Fig. 1, are available in the Figshare repository (10.6084/m9.figshare.5545327.v1). Strains and vectors can be requested by contacting the corresponding author.
Abbreviations
- BHI
Brain Heart Infusion broth
- cDNA
complementary DNA
- cfu
colony forming units
- hpi
hours post-infection
- kbp
kilo-base pair
- LB
Luria-Bertani
- LWT
London wild-type
- NCBI
National Center for Biotechnology Information
- nt
nucleotides
- PTS
phosphotransferase system
- qPCR
quantitative polymerase chain reaction
- RNA-seq
RNA sequencing
- RPKM
reads per kilobase per million input reads
- rRNA
ribosomal RNA
- Tn-seq
transposon sequencing
- TraDIS
transposon directed insertion sequencing
- U
unit(s)
Authors’ contributions
XZ, VdM, AMGP, and TKP performed experiments. JRB, MdB and MRCR contributed bioinformatic analyses. XZ, MJMB, SM, RW and WvS designed the study. XZ, AMGP, JRB and WvS drafted the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Strain E745 was isolated as part of routine diagnostic procedures during a VRE outbreak. This aspect of the study did not require consent or ethical approval by an institutional review board. Zebrafish work was performed according to guidelines and legislation set out in United Kingdom law under the Animals (Scientific Procedures) Act 1986. Ethical approval was given by the University of Sheffield Local Ethical Review Panel. All zebrafish experiments in this study were performed on larvae before the free feeding stage (5.2 days post fertilization) and consequently did not fall under the animal experimentation law according to the Act.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4299-9) contains supplementary material, which is available to authorized users.
Contributor Information
Xinglin Zhang, Email: xinglinzhang@zju.edu.cn.
Vincent de Maat, Email: v.demaat@umcutrecht.nl.
Ana M. Guzmán Prieto, Email: ana.guzman.prieto@gmail.com
Tomasz K. Prajsnar, Email: t.prajsnar@sheffield.ac.uk
Jumamurat R. Bayjanov, Email: jumamurat@gmail.com
Mark de Been, Email: markdebeen@gmail.com.
Malbert R. C. Rogers, Email: m.r.c.rogers-2@umcutrecht.nl
Marc J. M. Bonten, Email: mbonten@umcutrecht.nl
Stéphane Mesnage, Email: s.mesnage@sheffield.ac.uk.
Rob J. L. Willems, Email: rwillems@umcutrecht.nl
Willem van Schaik, Email: w.vanschaik@bham.ac.uk.
References
- 1.Dupont H, Friggeri A, Touzeau J, Airapetian N, Tinturier F, Lobjoie E, et al. Enterococci increase the morbidity and mortality associated with severe intra-abdominal infections in elderly patients hospitalized in the intensive care unit. J Antimicrob Chemother. 2011;66:2379–2385. doi: 10.1093/jac/dkr308. [DOI] [PubMed] [Google Scholar]
- 2.Guzman Prieto AM, van Schaik W, Rogers MRC, Coque TM, Baquero F, Corander J, et al. Global emergence and dissemination of enterococci as nosocomial pathogens: attack of the clones? Front Microbiol. 2016;7:788. doi: 10.3389/fmicb.2016.00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10:266–278. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lebreton F, van Schaik W, Manson McGuire A, Godfrey P, Griggs A, Mazumdar V, et al. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. MBio. 2013;4:e00534–e00513. doi: 10.1128/mBio.00534-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X, Top J, de Been M, Bierschenk D, Rogers M, Leendertse M, et al. Identification of a genetic determinant in clinical Enterococcus faecium strains that contributes to intestinal colonization during antibiotic treatment. J Infect Dis. 2013;207:1780–6. [DOI] [PubMed]
- 6.Heikens E, Singh KV, Jacques-Palaz KD, Van Luit-Asbroek M, Oostdijk EAN, Bonten MJM, et al. Contribution of the enterococcal surface protein Esp to pathogenesis of Enterococcus faecium endocarditis. Microbes Infect. 2011;13:1185–90. [DOI] [PMC free article] [PubMed]
- 7.Paganelli FL, Huebner J, Singh KV, Zhang X, van Schaik W, Wobser D, et al. Genome-wide screening identifies phosphotransferase system permease BepA to be involved in Enterococcus faecium endocarditis and biofilm formation. J Infect Dis. 2016;214:189–195. doi: 10.1093/infdis/jiw108. [DOI] [PubMed] [Google Scholar]
- 8.Sillanpää J, Prakash VP, Nallapareddy SR, Murray BE. Distribution of genes encoding MSCRAMMs and pili in clinical and natural populations of Enterococcus faecium. J Clin Microbiol. 2009;47:896–901. doi: 10.1128/JCM.02283-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montealegre MC, Singh KV, Somarajan SR, Yadav P, Chang C, Spencer R, et al. Role of the Emp pilus subunits of Enterococcus faecium in biofilm formation, adherence to host extracellular matrix components, and experimental infection. Infect Immun. 2016;84:1491–1500. doi: 10.1128/IAI.01396-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamboj M, Blair R, Bell N, Sun J, Eagan J, Sepkowitz K. What is the source of bloodstream infection due to vancomycin-resistant enterococci in persons with mucosal barrier injury? Infect Control Hosp Epidemiol. 2014;35:99–101. doi: 10.1086/674406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Regt MJA, van Schaik W, van Luit-Asbroek M, Dekker HAT, van Duijkeren E, Koning CJM, et al. Hospital and community ampicillin-resistant Enterococcus faecium are evolutionarily closely linked but have diversified through niche adaptation. PLoS One. 2012;7:e30319. [DOI] [PMC free article] [PubMed]
- 12.Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest. 2010;120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bouza E, Kestler M, Beca T, Mariscal G, Rodríguez-Créixems M, Bermejo J, et al. The NOVA score: a proposal to reduce the need for transesophageal echocardiography in patients with enterococcal bacteremia. Clin Infect Dis. 2015;60:528–535. doi: 10.1093/cid/ciu872. [DOI] [PubMed] [Google Scholar]
- 14.Arias CA, Murray BE. Emergence and management of drug-resistant enterococcal infections. Expert Rev Anti-Infect Ther. 2008;6:637–655. doi: 10.1586/14787210.6.5.637. [DOI] [PubMed] [Google Scholar]
- 15.Coombs GW, Pearson JC, Daly DA, Le TT, Robinson JO, Gottlieb T, et al. Australian Enterococcal sepsis outcome Programme annual report, 2013. Commun Dis Intell Q Rep. 2014;38:E320–E326. doi: 10.33321/cdi.2014.38.52. [DOI] [PubMed] [Google Scholar]
- 16.de Kraker MEA, Jarlier V, Monen JCM, Heuer OE, van de Sande N, Grundmann H. The changing epidemiology of bacteraemias in Europe: trends from the European antimicrobial resistance surveillance system. Clin Microbiol Infect. 2013;19:860–868. doi: 10.1111/1469-0691.12028. [DOI] [PubMed] [Google Scholar]
- 17.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, et al. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control Hosp Epidemiol. 2008;29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 18.Krebs HA. Chemical composition of blood plasma and serum. Annu Rev Biochem. 1950;19:409–430. doi: 10.1146/annurev.bi.19.070150.002205. [DOI] [PubMed] [Google Scholar]
- 19.van Opijnen T, Camilli A. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat Rev Microbiol. 2013;11:435–442. doi: 10.1038/nrmicro3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barquist L, Boinett CJ, Cain AK. Approaches to querying bacterial genomes with transposon-insertion sequencing. RNA Biol. 2013;10:1161–1169. doi: 10.4161/rna.24765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X, Paganelli FL, Bierschenk D, Kuipers A, Bonten MJM, Willems RJL, et al. Genome-wide identification of ampicillin resistance determinants in Enterococcus faecium. PLoS Genet. 2012;8:e1002804. doi: 10.1371/journal.pgen.1002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Bierschenk D, Top J, Anastasiou I, Bonten MJ, Willems RJ, et al. Functional genomic analysis of bile salt resistance in Enterococcus faecium. BMC Genomics. 2013;14:299. doi: 10.1186/1471-2164-14-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prieto AMG, Wijngaarden J, Braat JC, Rogers MRC, Majoor E, Brouwer EC, et al. The two component system ChtRS contributes to chlorhexidine tolerance in Enterococcus faecium. Antimicrob Agents Chemother. 2017;61:e02122–e02116. doi: 10.1128/AAC.02122-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langridge GC, Phan M-D, Turner DJ, Perkins TT, Parts L, Haase J, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 2009;19:2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Opijnen T, Lazinski DW, Camilli A. Genome-wide fitness and genetic interactions determined by Tn-seq, a high-throughput massively parallel sequencing method for microorganisms. Curr Protoc Microbiol. 2015;36:1E.3.1–1E.3.24. doi: 10.1002/9780471729259.mc01e03s36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leavis HL, Willems RJL, van Wamel WJB, Schuren FH, Caspers MPM, Bonten MJM. Insertion sequence-driven diversification creates a globally dispersed emerging multiresistant subspecies of E faecium. PLoS Pathog. 2007;3:e7. [DOI] [PMC free article] [PubMed]
- 28.Mascini EM, Troelstra A, Beitsma M, Blok HEM, Jalink KP, Hopmans TEM, et al. Genotyping and preemptive isolation to control an outbreak of vancomycin-resistant Enterococcus faecium. Clin Infect Dis. 2006;42:739–746. doi: 10.1086/500322. [DOI] [PubMed] [Google Scholar]
- 29.van Schaik W, Top J, Riley DR, Boekhorst J, Vrijenhoek JEP, Schapendonk CME, et al. Pyrosequencing-based comparative genome analysis of the nosocomial pathogen Enterococcus faecium and identification of a large transferable pathogenicity island. BMC Genomics. 2010;11:239. doi: 10.1186/1471-2164-11-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heikens E, Bonten MJ, Willems RJ. Enterococcal surface protein Esp is important for biofilm formation of enterococcus faecium E1162. J Bacteriol. 2007;189:8233–8240. doi: 10.1128/JB.01205-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Courvalin P. Vancomycin resistance in Gram-positive cocci. Clin Infect Dis. 2006;42:S25–S34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 32.Sekiguchi J, Tharavichitkul P, Miyoshi-Akiyama T, Chupia V, Fujino T, Araake M, et al. Cloning and characterization of a novel trimethoprim-resistant dihydrofolate reductase from a nosocomial isolate of Staphylococcus aureus CM.S2 (IMCJ1454) Antimicrob Agents Chemother. 2005;49:3948–3951. doi: 10.1128/AAC.49.9.3948-3951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh KV, Malathum K, Murray BE. Disruption of an Enterococcus faecium species-specific gene, a homologue of acquired macrolide resistance genes of staphylococci, is associated with an increase in macrolide susceptibility. Antimicrob Agents Chemother. 2001;45:263–266. doi: 10.1128/AAC.45.1.263-266.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joisel F, Leroux-Nicollet I, Lebreton JP, Fontaine MA. Hemolytic assay for clinical investigation of human C2. J Immunol Methods. 1983;59:229–235. doi: 10.1016/0022-1759(83)90035-2. [DOI] [PubMed] [Google Scholar]
- 35.Berends ETM, Dekkers JF, Nijland R, Kuipers A, Soppe JA, van Strijp JAG, et al. Distinct localization of the complement C5b-9 complex on gram-positive bacteria. Cell Microbiol. 2013;15:1955–1968. doi: 10.1111/cmi.12170. [DOI] [PubMed] [Google Scholar]
- 36.Pence MA, Rooijakkers SHM, Cogen AL, Cole JN, Hollands A, Gallo RL, et al. Streptococcal inhibitor of complement promotes innate immune resistance phenotypes of invasive M1T1 group a Streptococcus. J Innate Immun. 2010;2:587–595. doi: 10.1159/000317672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prajsnar TK, Renshaw SA, Ogryzko NV, Foster SJ, Serror P, Mesnage S. Zebrafish as a novel vertebrate model to dissect enterococcal pathogenesis. Infect Immun. 2013;81:4271–4279. doi: 10.1128/IAI.00976-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samant S, Lee H, Ghassemi M, Chen J, Cook JL, Mankin AS, et al. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008;4:e37. doi: 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Breton Y, Mistry P, Valdes KM, Quigley J, Kumar N, Tettelin H, et al. Genome-wide identification of genes required for fitness of group a Streptococcus in human blood. Infect Immun. 2013;81:862–875. doi: 10.1128/IAI.00837-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jenkins A, Cote C, Twenhafel N, Merkel T, Bozue J, Welkos S. Role of purine biosynthesis in Bacillus anthracis pathogenesis and virulence. Infect Immun. 2011;79:153–166. doi: 10.1128/IAI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polissi A, Pontiggia A, Feger G, Altieri M, Mottl H, Ferrari L, et al. Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect Immun. 1998;66:5620–5629. doi: 10.1128/iai.66.12.5620-5629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang N, Ozer EA, Mandel MJ, Hauser AR. Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. MBio. 2014;5:e01163–e01114. doi: 10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuller TE, Kennedy MJ, Lowery DE. Identification of Pasteurella multocida virulence genes in a septicemic mouse model using signature-tagged mutagenesis. Microb Pathog. 2000;29:25–38. doi: 10.1006/mpat.2000.0365. [DOI] [PubMed] [Google Scholar]
- 44.Schwager S, Agnoli K, Kothe M, Feldmann F, Givskov M, Carlier A, et al. Identification of Burkholderia cenocepacia strain H111 virulence factors using nonmammalian infection hosts. Infect Immun. 2013;81:143–153. doi: 10.1128/IAI.00768-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaudhuri RR, Peters SE, Pleasance SJ, Northen H, Willers C, Paterson GK, et al. Comprehensive identification of Salmonella enterica serovar Typhimurium genes required for infection of BALB/c mice. PLoS Pathog. 2009;5:e1000529. doi: 10.1371/journal.ppat.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mulhbacher J, Brouillette E, Allard M, Fortier L-C, Malouin F, Lafontaine DA. Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog. 2010;6:e1000865. doi: 10.1371/journal.ppat.1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Castro R, Neves AR, Fonseca LL, Pool WA, Kok J, Kuipers OP, et al. Characterization of the individual glucose uptake systems of Lactococcus lactis: mannose-PTS, cellobiose-PTS and the novel GlcU permease. Mol Microbiol. 2009;71:795–806. doi: 10.1111/j.1365-2958.2008.06564.x. [DOI] [PubMed] [Google Scholar]
- 48.Mainardi J-L, Fourgeaud M, Hugonnet J-E, Dubost L, Brouard J-P, Ouazzani J, et al. A novel peptidoglycan cross-linking enzyme for a beta-lactam-resistant transpeptidation pathway. J Biol Chem. 2005;280:38146–38152. doi: 10.1074/jbc.M507384200. [DOI] [PubMed] [Google Scholar]
- 49.Theilacker C, Sava I, Sanchez-Carballo P, Bao Y, Kropec A, Grohmann E, et al. Deletion of the glycosyltransferase bgsB of Enterococcus faecalis leads to a complete loss of glycolipids from the cell membrane and to impaired biofilm formation. BMC Microbiol. 2011;11:67. doi: 10.1186/1471-2180-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davlieva M, Zhang W, Arias CA, Shamoo Y. Biochemical characterization of cardiolipin synthase mutations associated with daptomycin resistance in enterococci. Antimicrob Agents Chemother. 2013;57:289–296. doi: 10.1128/AAC.01743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vollmer W, Joris B, Charlier P, Foster S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev. 2008;32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- 52.Mikalsen T, Pedersen T, Willems R, Coque TM, Werner G, Sadowy E, et al. Investigating the mobilome in clinically important lineages of Enterococcus faecium and Enterococcus faecalis. BMC Genomics. 2015;16:282. doi: 10.1186/s12864-015-1407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matos RC, Lapaque N, Rigottier-Gois L, Debarbieux L, Meylheuc T, Gonzalez-Zorn B, et al. Enterococcus faecalis prophage dynamics and contributions to pathogenic traits. Hughes D. PLoS Genet. 2013;9:e1003539. doi: 10.1371/journal.pgen.1003539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duerkop BA, Clements CV, Rollins D, Rodrigues JLM, Hooper LVA. Composite bacteriophage alters colonization by an intestinal commensal bacterium. Proc Natl Acad Sci. 2012;109:17621–17626. doi: 10.1073/pnas.1206136109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldberg SMD, Johnson J, Busam D, Feldblyum T, Ferriera S, Friedman R, et al. A Sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. Proc Natl Acad Sci U S A. 2006;103:11240–5. [DOI] [PMC free article] [PubMed]
- 56.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanimoto K, Ike Y. Complete nucleotide sequencing and analysis of the 65-kb highly conjugative Enterococcus faecium plasmid pMG1: identification of the transfer-related region and the minimum region required for replication. FEMS Microbiol Lett. 2008;288:186–195. doi: 10.1111/j.1574-6968.2008.01342.x. [DOI] [PubMed] [Google Scholar]
- 58.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 61.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524. [DOI] [PMC free article] [PubMed]
- 62.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, et al. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 2013;41:e140. doi: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burghout P, Zomer A, van der Gaast-de Jongh CE, Janssen-Megens EM, Françoijs K-J, Stunnenberg HG, et al. Streptococcus pneumoniae folate biosynthesis responds to environmental CO2 levels. J Bacteriol. 2013;195:1573–1582. doi: 10.1128/JB.01942-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goecks J, Nekrutenko A, Taylor J. Galaxy team. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baldi P, Long ADA. Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- 70.Prajsnar TK, Cunliffe VT, Foster SJ, Renshaw SAA. Novel vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of zebrafish to non-host specialized pathogens. Cell Microbiol. 2008;10:2312–2325. doi: 10.1111/j.1462-5822.2008.01213.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genome sequence information for E. faecium E745. (XLSX 10 kb)
Growth of E. faecium E745 in BHI and serum. (PDF 962 kb)
Operons identified by RNA-seq in E. faecium E745. (XLSX 40 kb)
E. faecium E745 genes that exhibited significant (q < 0.001 and fold-change > 2) differential expression in human serum, as determined by RNA-seq. (XLSX 70 kb)
qRT-PCR validation of RNA-seq experiments. Correlation of RNA-seq and qRT-PCR expression ratios for the seven genes with various expression levels and genomic locations. The gene expression ratios obtained from both qRT-PCR and RNA-seq were normalized by a housekeeping control gene Efm745_00056 (tufA). The experiment was performed with three biological replicates. (PDF 121 kb)
Characterization of the E. faecium E745 transposon mutant library, showing the number of reads that were mapped to the E. faecium E745 chromosome and plasmids. The height of each peak represents the read abundance at a specific insertion site. On the y-axis, the number of mapped reads is shown on a log scale. (PDF 1427 kb)
Tn-seq data: comparison of heat-inactivated and native serum (XLSX 139 kb)
E. faecium E745 genes that significantly (q < 0.05 and fold-change <−2 or >2) contribute to growth in human serum, as identified by Tn-seq. (XLSX 13 kb)
Isolation of mutants from the transposon mutant library pool. (A) Schematic representation of the PCR reaction designed to find a particular Tn-mutant within the transposon mutant library. This PCR uses a combination of a gene-specific primer (blue arrow) and a transposon specific primer primer (yellow arrow). Positive PCR products, indicated by the green check marks, should occur when the transposon (depicted as a yellow triangle) is inserted in the gene of interest (depicted in blue). If the transposons inserted in adjacent genes or intergenic regions, no PCR product can be amplified (red crosses). (B) Schematic workflow to isolate Tn-mutants from the mutant library. The transposon mutant library is split into 12 plates (A1 - A12) of 96 wells, with each 200 μl well containing an average of 4 mutants. Plates were incubated overnight (Step 2). Plate A1 was then pooled into the first column of a new 96 well plate, denominated plate B1 (Step 3) and the same was done for plates A2 to A12. Subsequently, plate B1 was pooled again into the first column of a third plate, denominated C1 (Step 4). PCR using the gene-specific primer and the transposon specific primer was performed on the 8 wells of plate C1 (Step 5). A positive PCR was suggestive of the presence of a particular transposon-mutant (depicted as a red dot). The presence of the transposon mutant was then confirmed by PCR in plate B1 (step 6) and the corresponding plate A (step 7). Once a transposon-mutant was located to a particular well in plate A, the well was plated on BHI plates containing gentamicin, and the colonies were screened for the presence of the transposon mutant by PCR (step 8). (PDF 277 kb)
Oligonucleotides used in this study. (XLSX 10 kb)
Data Availability Statement
Sequence reads generated in this study have been made available at the European Nucleotide Archive under accession number PRJEB19025. The core genome alignment and Newick-formatted tree file that were generated for Fig. 1, are available in the Figshare repository (10.6084/m9.figshare.5545327.v1). Strains and vectors can be requested by contacting the corresponding author.