

Abstract
A series of 1,6-disubstituted indole derivatives was designed, synthesized and evaluated as inhibitors of human nitric oxide synthase (NOS). By varying the basic amine side chain at the 1-position of the indole ring, several potent and selective inhibitors of human neuronal NOS were identified. In general compounds with bulkier side chains displayed increased selectivity for nNOS over eNOS and iNOS isoforms. One of the compounds, (R)-8 was shown to reduce tactile hyperesthesia (allodynia) after oral administration (30 mg/kg) in an in vivo rat model of dural inflammation relevant to migraine pain.
Keywords: 1,6-Disubstitued indole derivatives; Nitric oxide; Nitric oxide synthase; Nitric oxide synthase inhibitors; Selective neuronal nitric oxide synthase inhibitors
Nitric oxide (NO) is an inorganic free radical that has diverse roles both in normal and pathological processes, including the regulation of blood pressure, neurotransmission and macrophage defense systems.1 NO is synthesized from the enzyme catalysis of L-arginine to L-citrulline by three isoforms of nitric oxide synthase (NOS): two constitutive forms in neuronal cells (nNOS) and endothelial cells (eNOS), and an inducible form in macrophage cells (iNOS). Overstimulation or overproduction of NO by nNOS and iNOS has been shown to play a key role in several disorders, including septic shock, arthritis, diabetes, ischemia-reperfusion injury, pain and various neurodegenerative diseases.2 However, any inhibitors to treat these conditions must avoid eNOS inhibition as this will lead to unwanted effects such as enhanced white cell and platelet activation, hypertension and atherogenesis.3 Therefore, the development of selective NOS inhibitors is of considerable interest, both from a therapeutic perspective and also as specific pharmacological tools.4
Although there is low homology among the three NOS primary sequences (~50%), the active sites of the enzymes appears to be relatively conserved with 16 out of 18 residues within 6 Å being identical, presumably explains the difficulty obtaining selective NOS inhibitors.5 Investigation into the synthesis and chemistry of novel isoform-selective NOS inhibitors has been an ongoing challenge, even though the general pharmacophore requirements are well established.6–12 Synthesis of substrate (L-arginine) based peptidomimetic non-selective as well as selective nNOS inhibitors have been extensively reported in the literature.13 In an effort to improve PK/PD properties by decreasing their peptidic nature, various small molecule selective nNOS inhibitors have also been reported.14
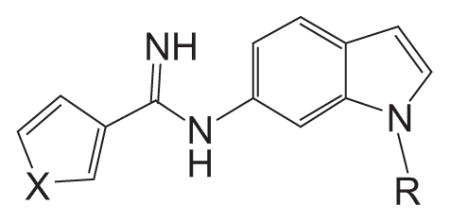
The pharmacophore model we adopted for the arginine binding site of the NOS enzyme includes a guanidine isosteric group (amidine group) and a basic amine group, both attached to a central aryl scaffold (indole core) as shown in Figure 1.4,15 The amidine group makes an important bidentate interaction with the conserved glutamic acid residue to achieve the necessary potency; whereas the basic amine is assumed to provide the nNOS isoform selectivity.15 Our design strategy is based on an indole core as an aryl scaffold and exploring various basic amine side chains for achieving the NOS isoform selectivity. As part of our ongoing efforts to find small molecule selective nNOS inhibitors for treating CNS disorders, herein we report the synthesis and biological activity evaluations of a series of 1,6-disubstituted indole derivatives and in vivo activity of (R)-8 in a rat model relevant to migraine pain.15
Figure 1.
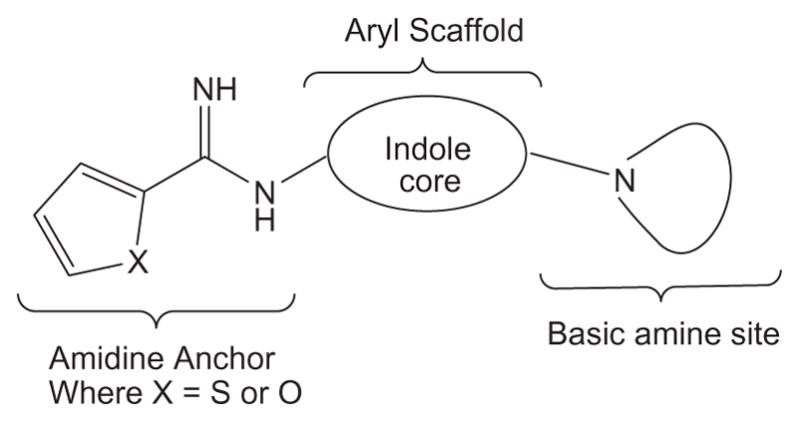
Pharmacophore model for selective nNOS inhibitor design.
Two general approaches were undertaken for the preparation of 1,6-disubstituted indole derivatives as shown in Schemes 1–5. 6-Nitro-1H-indole (1) was alkylated with various 2-chloro-ethanamine derivatives in the presence of potassium carbonate to obtain the alkylated nitro-intermediates 2–4 (Scheme 1). The nitro group in compounds 2–4 was reduced to the corresponding amine in the presence of palladium on carbon under an atmosphere of hydrogen. These anilines were coupled to the thiophene-2-carbimidothioate 5, resulting in the final compounds 6, 7 and (±)-8, respectively.16
Scheme 1.
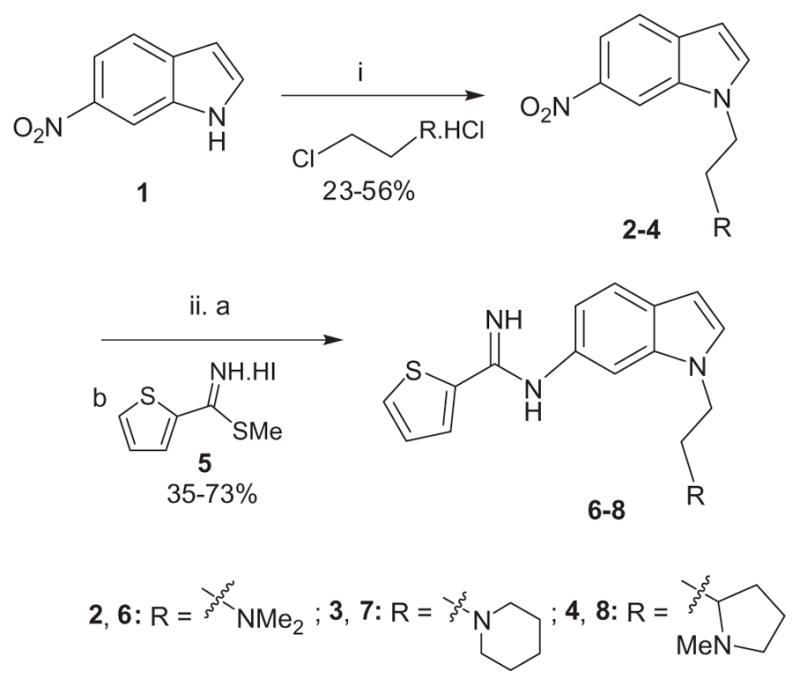
Reagents and conditions: (i) K2CO3, DMF, 80 °C; (ii) (a) Pd–C/H2, EtOH, rt, (b) 5, EtOH, rt.
Scheme 5.
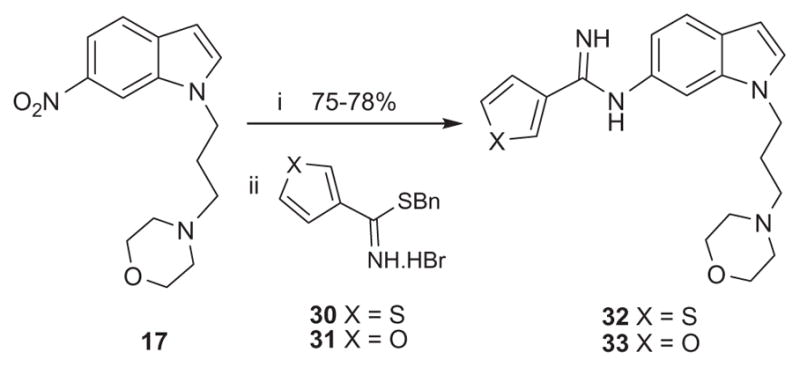
Reagents and conditions: (i) Pd–C/H2, EtOH, rt; (ii) 30 or 31, EtOH, rt.
During the synthesis of compound (±)-8, rearrangement through a ring opening (quarternization) reaction was observed (Scheme 2) under basic conditions.17 Two nitroindole derivatives, (±)-4 and the rearranged product 9 were easily separated by silica gel column chromatography. Following the same synthetic protocol and coupling to the thiophene-2-carbimidothioate 5 or the furan- 2-carbimidothioate 10 as outlined in Scheme 2 provided the target compounds 11 and 12, respectively.18 Compound (±)-4 was separated into its enantiomers (R)-4 and (S)-4 by resolution with dibenzoyl-L-tartaric acid in ethanol (Scheme 2). The separated enantiomers were converted into the final compounds (R)-8 and (S)-8 as described above. The stereochemistry of (S)-8 was determined by an independent chiral synthesis as shown in Scheme 3. (S)-2-(2-Chloroethyl)-1-methylpyrrolidine was reacted with 6-nitro- 1H-indole (1), followed by reduction and coupling with the thiophene- 2-carbimidothioate 5 under standard conditions described in Scheme 1, provided the pure enantiomer (S)-8.19
Scheme 2.

Reagents and conditions: (i) K2CO3, DMF, 80 °C; (ii) (a) Pd–C/H2, EtOH, rt, (b) 5 or 10, EtOH, rt; (iii) dibenzoyl-L-tartaric acid, EtOH; (iv) (a) Pd–C/H2, EtOH, rt, (b) 5, EtOH, rt.
Scheme 3.

Reagents and conditions: (i) K2CO3, DMF, 80 °C; (ii) (a) Pd–C/H2, EtOH, rt, (b) 5, EtOH, rt.
Compounds 22–29 were prepared as shown in Scheme 4. The 6- nitro-1H-indole (1) was alkylated in the presence of sodium hydride in DMF with appropriate 3 or 4-carbon chloroalkyl iodides to obtain intermediates 13 and 14, respectively. The chloride in 13 and 14 was then substituted with various amines in the presence of potassium carbonate and potassium iodide in refluxing acetonitrile to obtain the substituted amine intermediates 15–19, 20 and 21, respectively. Reduction of the nitro group and coupling with the thiophene-2-carbimidothioate 5 or the furan-2-carbimidothioate 10 was carried out as described above to obtain the target compounds 22–29. Target compounds 32 and 33 were prepared under similar conditions described above reacting with the thiophene-3-carbimidothioate 30 and furan-3-carbimidothioate 31, respectively, as shown in Scheme 5.20
Scheme 4.
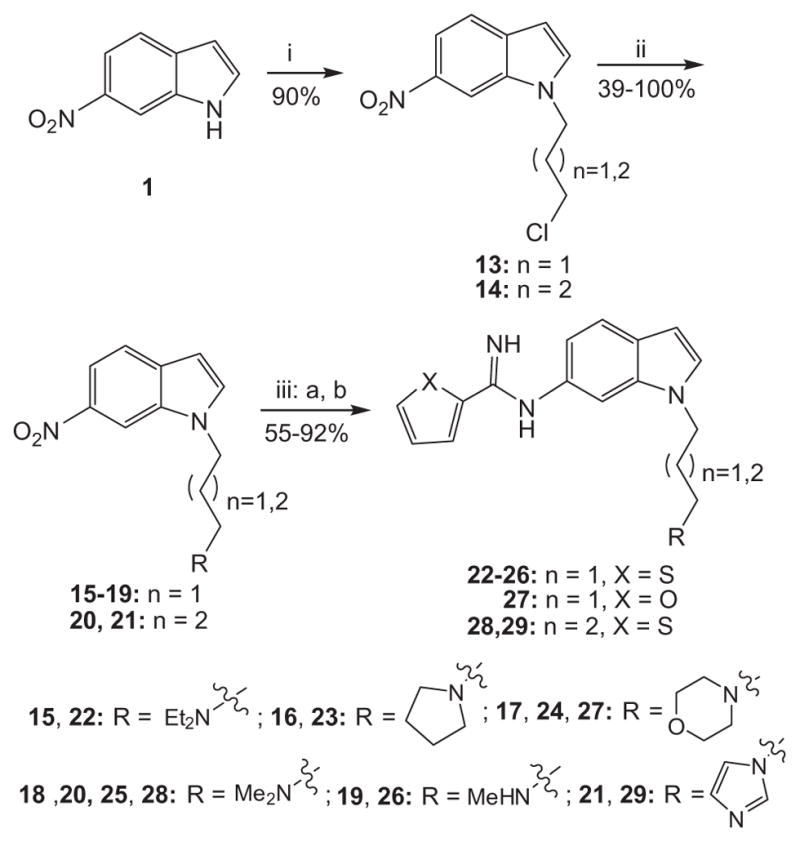
Reagents and conditions: (i) NaH, DMF, Cl(CH2)nl; (ii) K2CO3, CH3CN, Kl, R·HCl, reflux; (iii) (a) Pd–C/H2, EtOH, rt, (b) 5 or 10, EtOH, rt.
All compounds were converted into their dihydrochloride salts to improve their water solubility,21 and measured their inhibitory activities against all three human isoforms of NOS (Tables 1 and 2).22 In general, compounds with bulkier side chains displayed moderate nNOS inhibitory activities and good selectivities against eNOS and iNOS. Most of the tested compounds displayed poor inhibitory activities for the iNOS isoform. The most selective nNOS inhibitor from the series was compound (S)-8 with 120- and 360- fold selectivity for nNOS over eNOS and iNOS, respectively (Table 1). The most potent nNOS inhibitor was compound 22, which also displayed very good selectivity (68-fold) over eNOS. In general compounds with a cyclic side chain ((±)-8, (R)-8, (S)-8, 11, 23, 24 and 29) showed increased selectivity for nNOS over eNOS and iNOS isoforms compared to the compounds with an acyclic side chain (6, 25, 26 and 28). The diethyl amine group in compound 22 seems to fit the requirement of a bulky substituent. This is apparent when compared to dimethyl and monomethyl substituted amines (25 and 26) where reduction in nNOS selectivity was observed with the smaller substituents. Compound 29 has an imidazole group, which fits the requirement of a bulky substituent, however displayed poor activity for nNOS (IC50 = 1.22 μM). This may be due to the reduced basicity of the imidazole nitrogen and is consistent with our previous observation with 2-aminobenzothiazole compounds. 15 Compound 28 with a 4-carbon linker showed better activity for nNOS compared to the corresponding compounds with 2 and 3-carbon linkers (6 and 25). Furanyl amidines (12 and 27) did not show any significant difference in potency or selectivity when compared to their corresponding thiophene amidine compounds 11 and 24. At the same time, replacement of 2-thiophene and 2-furanyl amidine groups (24 and 27) with regioisomeric 3- thiophene and 3-furanyl amidines (32 and 33) did not show any significant differences in activity or selectivity among NOS isoforms (Table 2).
Table 1.
Inhibition of human NOS enzymes by 1,6-disubstituted indole derivatives
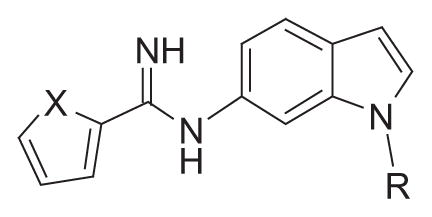
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | X | R | nNOS IC50 a (μM) | eNOS IC50 a (μM) | iNOS IC50 a (μM) | eNOS/nNOS | iNOS/nNOS |
| 6 | S |
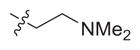
|
1.2 (0.99–1.3) | 15 (8.4–28) | 60 (37–98) | 13 | 50 |
| 7 | S |
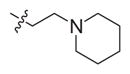
|
0.49 (0.39–0.6) | 3.8 (1.1–12.2) | 21 (6.1–72) | 8 | 43 |
| (±)-8 | S |
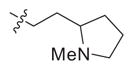
|
0.22 (0.08–0.5) | 19 (11–33.9) | 68 (17–268) | 86 | 309 |
| (R)-8 | S |
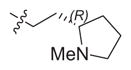
|
0.32 (0.12–0.7) | 16 (6.6–38.2) | 72 (20–255) | 50 | 225 |
| (S)-8 | S |
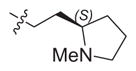
|
0.2 (0.12–0.3) | 24 (15–37.1) | 72 (19.7–267) | 120 | 360 |
| 11 | S |
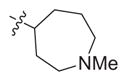
|
0.87 (0.46–1.65) | 37.7 (24.9–57.1) | NTb | 43 | NCc |
| 12 | O |
|
0.52 (0.36–0.77) | 30.6 (24.0–39.1) | 1.2 (0.83–1.9) | 59 | 2 |
| 22 | S |
|
0.15 (0.08–0.24) | 10.2 (5.6–18.3) | NTb | 68 | NCc |
| 23 | S |
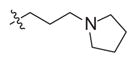
|
0.45 (0.29–0.69) | 11.2 (4.4–27.9) | NTb | 25 | NCc |
| 24 | S |
|
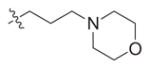
0.7 (0.5–0.9) | 28.3 (12.4–49.5) | 82 (49.9–82.1) | 40 | 117 |
| 25 | S |
|
2.73 (1.9–3.8) | 23.3 (16.8–32.3) | NTb | 8 | NCc |
| 26 | S |
|
1.78 (1.5–2.1) | 11.8 (9.2–15.2) | NTb | 6 | NCc |
| 27 | O |
|
2.4 (2.1–2.5) | 34 (24.1–49.6) | 36 (32–37) | 14 | 15 |
| 28 | S |
|
0.26 (0.2–0.3) | 2.53 (1.4–4.3) | 66 (41–105) | 10 | 254 |
| 29 | S |
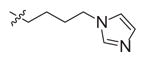
|
1.22 (0.6–2.2) | 23 (13–39) | >100d | 19 | NCc |
Values reported in parentheses are 95% confidence intervals.
Inhibitory activities were measured by the conversion of [3H]-L-arginine into [3H]-L-citrulline.
Not tested.
Not calculable.
Not active at the maximum test concentration of 100 μM.
Table 2.
Inhibition of human NOS enzymes by 1,6-disubstituted indole derivatives
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | X | R | nNOS IC50 a (μM) | eNOS IC50 a (μM) | iNOS IC50 a (μM) | eNOS/nNOS | iNOS/nNOS |
| 32 | S |
|
1.4 (1.1–1.6) | 17.1 (11.4–24.7) | 70 (31.8–155) | 12.1 | 50 |
| 33 | O |
|
1.0 (0.68–1.4) | 19.1 (15.9–24.7) | >100b | 19 | NCc |
Values reported in parentheses are 95% confidence intervals.
Inhibitory activities were measured by the conversion of [3H]-L-arginine into [3H]-L-citrulline.
Not active at the maximum test concentration of 100 μM.
Not calculable.
To investigate the potential therapeutic use of these selective nNOS inhibitors, compound (R)-8 was evaluated in a rat model of migraine pain.23 This model involves the application of an inflammatory soup (IS) onto the dura via a cannula with subsequent measurements in the development of cutaneous allodynia in the hind-paws, which peaks between 3 and 4 h. Oral administration of (R)-8 (30 mg/kg), 15 min prior to the IS attenuated the development of allodynia between 2 and 6 h post-dose with maximum effect showing at 4 h (Fig. 2).
Figure 2.
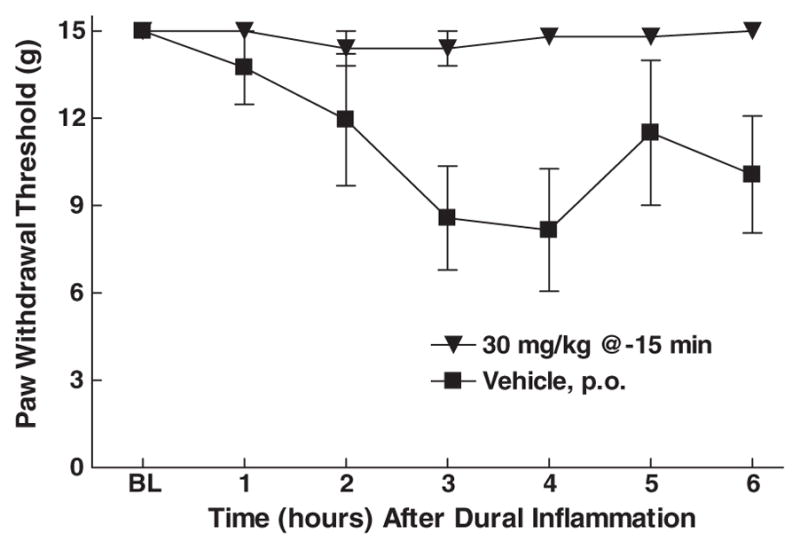
Oral administration of compound (R)-8 (30 mg/kg) attenuates the tactile hyperesthesia (allodynia) of the hindpaw in rats with inflammation of the dura in migraine pain model.
In summary, we have designed and synthesized a series of 1,6- disubstituted indole derivatives that are selective inhibitors of nNOS over both eNOS and iNOS isoforms. We have shown that by introducing a bulky amino-substituent at the 1-position of the indole core, the selectivity for nNOS over eNOS can increase over 100-fold. Replacement of 2-thiophene amidine group by 2-furanyl amidine or regioisomeric 3-thiophene and 3-furanyl amidine groups did not show any significant difference in potency or selectivity among NOS isoforms. Overall, compound (±)-8 and its enantiomers (R)-8 and (S)-8 showed superior potency for nNOS and increased selectivity over eNOS and iNOS isoforms. The in vivo results of compound (R)-8 in a rodent migraine pain model, suggests that these compounds may be useful for further evaluation in potential therapeutic applications of disease states in which nNOS inhibitors are thought to play a key role.
Acknowledgments
We are grateful to NoAb BioDiscoveries Inc. (Mississauga, ON, Canada) and Asinex Ltd (Moscow, Russia) for performing the human NOS inhibition assays. We thank the Natural Sciences and Engineering Research Council (NSERC) of Canada for providing an Industrial Research Fellowship to GM.
References and notes
- 1.Mustafa AK, Gadalla MM, Synder SH. Sci Signal. 2009;2:1. doi: 10.1126/scisignal.268re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vallance P, Leiper J. Nat Rev Drug Disc. 2002;1:939. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- 3.Cayatte AJ, Palacino JJ, Horten K, Cohen RA. Arterioscler Thromb. 1994;14:753. doi: 10.1161/01.atv.14.5.753. [DOI] [PubMed] [Google Scholar]
- 4.Maddaford S, Annedi SC, Ramnauth J, Rakhit S. Annu Rep Med Chem. 2009;44:27. [Google Scholar]
- 5.Haitao J, Huiying L, Martásek P, Roman LJ, Poulos TL, Silverman RB. J Med Chem. 2009;52:779. doi: 10.1021/jm801220a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Nat Struct Biol. 1999;6:233. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- 7.Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh DK, Getzoff ED, Stuehr DJ, Tainer JA. Science. 1997;278:425. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- 8.Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Science. 1998;279:2121. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 9.Raman CS, Li H, Martásek P, Kral V, Masters BSS, Poulos TL. Cell. 1998;95:939. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, Whitlow M, Poulos TL. J Biol Chem. 1999;274:21276. doi: 10.1074/jbc.274.30.21276. [DOI] [PubMed] [Google Scholar]
- 11.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. Biochemistry. 2002;41:13868. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 12.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Nat Chem Biol. 2008;4:700. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silverman RB. Acc Chem Res. 2009;42:439. doi: 10.1021/ar800201v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji H, Silverman RB. Curr Top Med Chem. 2005;5:603. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]
- 15.Patman J, Bhardwaj N, Ramnauth J, Annedi SC, Renton P, Maddaford SP, Rakhit S, Andrews JS. Bioorg Med Chem Lett. 2007;17:2540. doi: 10.1016/j.bmcl.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Bercot-Vatteroni M. Ann Chim. 1962;7:303. [Google Scholar]
- 17.DeVita RJ, Hollings DD, Goulet MT, Wyvratt MJ, Fisher MH, Lo JL, Yang YT, Cheng K, Smith RG. Bioorg Med Chem Lett. 1999;9:2615. doi: 10.1016/s0960-894x(99)00446-1. [DOI] [PubMed] [Google Scholar]
- 18.Collins JL, Shearer BG, Oplinger JA, Lee S, Garvey EP, Salter M, Duffy C, Burnette TC, Furfine ES. J Med Chem. 1998;41:2858. doi: 10.1021/jm980072p. [DOI] [PubMed] [Google Scholar]
- 19.Vernier JM, El-Abdellaoui H, Holsenback H, Cosford NDP, Bleicher L, Barker G, Bontempi B, Chavez-Noriega L, Menzaghi F, Rao TS, Reid R, Sacaan AI, Suto C, Washburn M, Lloyd GK, McDonald IA. J Med Chem. 1999;42:1684. doi: 10.1021/jm990035d. [DOI] [PubMed] [Google Scholar]
- 20.Baati R, Gouverneur V, Mioskowski C. Synthesis. 1999;6:927. [Google Scholar]
- 21.The free base in anhydrous methanol was treated with HCl (1 M solution in diethyl ether) and stirred at room temperature for 10 min. The solvent was evaporated and dried under reduced pressure to obtain the dihydrochloride salt as a solid.
- 22.Recombinant human iNOS, eNOS and nNOS were produced in Baculovirus-infected Sf9 cells (ALEXIS). In a radiometric method, inhibitory activities were measured by the conversion of [3H]-L-arginine into [3H]-L-citrulline. The enzymatic reaction was carried out in the presence or absence of varying concentrations of the inhibitor in water. The negatively charged [3H]-L-citrulline was separated from the positively charged [3H]-L-arginine using resin beads. Inhibition of enzyme activity by the inhibitor is measured by dividing the enzymatic conversion in the presence of inhibitor divided by the enzymatic conversion in the absence of inhibitor. IC50 value is the concentration of compound that gives rise to 50% inhibition. All assays were performed in duplicate. All assays were performed in duplicate.
- 23.Edelmayer RM, Vanderah TW, Majuta L, Zhang ET, Fioravanti B, De Felice M, Chichorro JG, Ossipov MH, King T, Lai J, Kori SH, Nelsen AC, Cannon KE, Heinricher MM, Porreca F. Ann Neurol. 2009;65:184. doi: 10.1002/ana.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]