

Abstract
Results of kinetic experiments and quantum chemical computations on a series of platinum-promoted polycyclization reactions are described. Analysis of these results reveal a reactivity model that reaches beyond the energetics of the cascade itself, incorporating an ensemble of pre-cyclization conformations of the platinum-alkene reactant complex, only a subset of which are productive for bi- (or larger) cyclization and lead to products. Similarities and differences between this scenario, including reaction coordinates for polycyclization, for platinum- and enzyme-promoted polycyclization are highlighted.
Graphical abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
INTRODUCTION
Polyene polycyclization reactions have a storied history. Nature has been promoting such reactions for millennia with enzymes that generate carbocations,1 while organic and organometallic chemists have been promoting such reactions for decades with reagents or catalysts that provide protons, act as Lewis acids, or generate radicals.2–4 The appeal of such reactions derives in part from the efficiency with which they can increase molecular complexity. In addition, the net conversion of a substrate π-bond to a product σ-bond is an inherently exothermic process, which thereby allows molecules with strained substructures to be produced—this is how terpenes containing, for example, cyclobutane groups are formed in nature.5
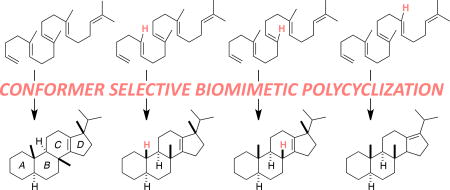
Here we describe competition experiments by which relative rates for the Pt(II)-promoted polycyclization of various methyl- edited polyenes,3c 1–4 (Scheme 1), were determined along with quantum chemical computations on their reaction coordinates. The latter are compared to previously reported reaction coordinates for the biologically relevant squalene and oxidosqualene cyclization reactions (Figure 1)6 in an attempt to assess to what degree the metal-promoted reactions actually mimic these biological processes. Note, however, that 1–3 have a methyl group at C12 rather than C13, allowing a 5-membered D ring to form directly.6 Ultimately, a model for reactivity emerged in which selection of productive reactant conformers out of a mixture of primarily nonproductive conformers is accomplished by an exergonic bicyclization process. This initial selection, coupled with slow termination events, enables down-stream cyclization steps to dominate the reactivity and efficiently provide tetracycles without significant premature termination of the cascade.
Scheme 1.

Polycyclizations described herein.
Figure 1.

Representative biological polycyclization reactions.
The series of polyenes shown in Scheme 1 was examined so that the influence of particular methyl groups on polycyclization rates could be assessed. The information obtained from these experiments sheds some light on the importance, or lack thereof, of methyl groups in terpene biosynthesis (are they key to the efficiency of biological polycyclizations or perhaps merely evolutionary leftovers?) and the viability of synthetic polycycyclizations with differently substituted substrates.
Previously, Johnson described a series of polycyclization reactions of 1,2-disubstituted alkene-containing substrates,7 which proceed through secondary carbocations upon cyclization. While comparison of vinyl and isopropenyl terminating groups resulted in cyclizations that struggled to engage the vinyl terminating group, direct comparison of substrates with selective placement of secondary alkenes throughout the cascade was not undertaken.7 In a similar vein, Corey described the effect of replacing the C-6 methyl group of 2,3-oxidosqualene with either a hydrogen or chloride (Figure 2),8 both of which were expected to reduce the nucleophilicity of the alkene. In that the rate of reaction was affected by these substitutions, the results of this study were consistent with oxirane opening and A ring formation being concerted in lanosterol polycyclization.6,8 Building on these seminal studies, we proposed the synthesis and cyclization of 1–4, in which selective replacement of trisubstituted alkenes with 1,2-disubstituted alkenes would reveal further insights into the nature of the polycyclization process.
Figure 2.

2,3-Oxidosqualene and analogs studied by Corey.8
SYNTHESIS OF POLYENES
We required a synthetic strategy that allowed for production of polyenes with specific methyl groups replaced by hydrogens. Previously reported substrate 1 was synthesized through Johnson’s classic three step sequence involving the addition of isopropenyl magnesium bromide to an aldehyde, mercury trifluoroacetate catalyzed vinylation and Claisen rearrangement.9 Although the analogous vinyl magnesium bromide protocol does not successfully yield the desired 1,2-alkene fragments, a one pot vinylation/Claisen rearrangement using (1,10-phenanthroline)Pd(OAc)2 and triethyleneglycol divinylether (TGDV) was effective.10 In combination with the Johnson Claisen reaction, this method provided the desired mix of stereochemically defined di- and tri-substituted alkene precursors for synthesis of 2–4 (see Supporting Information for details).
CATALYSIS
The electrophile (CNC)Pt2+ (Scheme 2) forms η2-alkene adducts that effectively activate terminal alkenes towards nucleophilic attack by other alkenes,3c,9,11 and thus serves as an excellent catalyst for poly-alkene cascade cyclization reactions. Its high electrophilicity enables the initiation of challenging alkene-terminated variants (Step A, Scheme 2)11b,c and when coupled to post cyclization proto-demetallation (Step B, Scheme 2), can mediate efficient cascade cycloisomerizations. In this scenario, the proto-demetallation turnover mechanism is triggered when the π-olefin adduct (a dication) converts to the monocationic alkyl (i.e., (CNC)Pt–R+) upon initiation (Scheme 2). It must additionally outcompete decomposition pathways arising from the inherent acid sensitivity of polyene substrates.
Scheme 2.

Overall catalytic cycle for the polycyclization of polyenes.
Cyclizations of 2–4 with the (CNC)Pt2+ catalyst proved, as expected, more challenging than their fully methylated counterpart 1, typically requiring significantly lengthened reaction times with multiple products being formed in several cases. The 5-demethylated substrate 2 proved to be the most challenging, and full consumption of substrate required 50 mol% catalyst loadings and long reaction times (~4 days). While the expected tetracycle 6 (eq 1) was formed as the major product, as observed by GC-MS, isolation and purification proved challenging. Removal of the minor products required multiple Ag+ impregnated silica gel columns,12 resulting in a net 18% isolated yield. The structure of this product was determined by comparison of the 1H NMR with that of known 5. While several other tetracyclic products were observed by GC-MS (tetracycles characteristically give M+–iso-propyl ions), they proved inseparable and could never be adequately characterized (see SI).
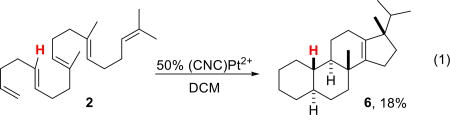 |
(1) |
Cyclization of the 9-demethylated substrate 3 yielded a tetracyclic product with a B/C ring junction lacking methyl groups, a structural motif that is characteristic of numerous natural sterols, but formed via a different route (eq 2).13 In contrast to the 10% catalyst loading needed for the conversion of 1 to 5, 3 required 15% catalyst to compensate for catalyst deactivation over the extended reaction time (4 days). Isolation of 7 in 54% yield was once again achieved using Ag+ impregnated silica gel,12 and the structure of the product was again assigned by comparison to 5.
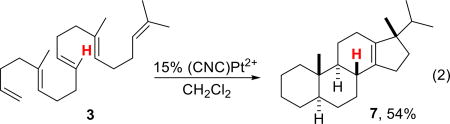 |
(2) |
Cycloisomerization of the 13-demethylated polyene 4 using 15 mol% (NCN)Pt2+ provided a mixture of two tetracyclic compounds (Scheme 3) that result from one and two hydride shifts following tetracyclization (8, 32% yield) and (9, 20% yield), respectively. The two alkene products were separable by Ag+ impregnated silica gel12 with 9 eluting first. The structure of 9 was again determined by comparison of its 1H NMR spectrum with that of 5, while 8 was characterized by 1H, 13C and selective 1D NMR experiments (see Supporting Information).
Scheme 3.

Polycyclization of polyene 4.
RELATIVE RATES OF POLYCYCLIZATION
The polyenes were paired for competition experiments by combining 1 equivalent of each substrate and 1 equivalent of (CNC)Pt2+ in dichloromethane. Reactions were checked hourly by GC-MS, and relative rates were determined from the evolution of the product ratios. In this way k1/k3 and k4/k3 were determined to be of 4.5 and 2.2, respectively. More substantial rate differences were observed on pairing 2 with 3 and 4 (k3/k2 = 10, and k4/k2 = 14). Taken together, these results indicate an order of polycyclization rates of: 1 > 4 > 3 > 2, with 2 being slowest by a significant margin. The overall range of relative rates determined by this ladder method is >40.
COMPUTATIONS ON REACTION COORDINATES
All computations were performed at the SMD(DCM)-M06/6-31+G(d,p):SDD level of theory (SDD for Pt, 6-31+G(d,p) for all other atoms),14 using Gaussian09.15 Methyl groups were used in place of mesityl groups on the catalyst to reduce computational time. Identities of all minima and transition state structures were verified by frequency analysis. Intrinsic reaction coordinate (IRC) calculations16 were carried out for cyclization transition state structures. All energies shown are free energies at 298K, except for those in IRC plots, which are electronic energies. In general, stationary points along reaction pathways were located in the reverse of the actual reaction direction, i.e., starting from products and lengthening bonds to arrive at initial guesses of stationary point structures.
Putative carbocations involved in the stepwise cascade of the fully methylated parent structure 1 are shown in Scheme 4 (R1 = R2 = R3 = Me). Not all of these structures were found to be minima, however. Relative free energies of stationary points are shown in Figure 3. First, note that carbocations B and C are not minima, i.e., the first three rings of the product (A/B/C) are formed in a concerted process. The remainder of the carbocations in Scheme 4 are predicted to be minima, although D and F are just barely so. Barriers for all reaction steps are predicted to be low and the overall reaction is predicted to be exergonic, as expected for a reaction in which π-bonds are traded for σ-bonds.5 A very similar reaction profile was found for polyene 2 (Figure 4). Again, three rings are predicted to form in concert and the fourth ring is predicted to form with a small barrier. However, in this case, carbocation B would be secondary, perhaps a factor that contributes to the higher barrier for cyclization of 2 compared to that for 1.
Scheme 4.

Putative intermediates in polyene polycyclization.
Figure 3.

Relative energies of stationary points in the cyclization/rearrangement of polyene 1 (R1 = R2 = R3 = Me). D is drawn explicitly.
Figure 4.

Relative energies of stationary points in the cyclization/rearrangement of polyene 2 (R1 = H, R2 = R3 = Me). D is drawn explicitly.
The reaction profile for polyene 3 (Figure 5) looks, at first glance, to differ significantly from those for 1 and 2, in that an intermediate with a single ring (B in Scheme 4) is predicted to form before the second and third rings form in concert. However, the barrier forward from B is negligible, <0.5 kcal/mol. The reaction profile for polyene 4 (Figure 6) differs in a more significant manner. For this system, the first two rings are predicted to form in concert, but an intermediate (C in Scheme 4) is predicted to precede formation of the third ring. While the barrier forward from C is small, approximately 2 kcal/mol, carbocation D is predicted to be slightly higher in energy than C and only 10 kcal/mol lower in energy than A. In contrast, carbocation D for polyenes 1–3 is predicted to be >20 kcal/mol lower in energy than A. The differences in stability of carbocation D stem from the substitution patterns of the polyenes; polyene 4 leads to a secondary carbocation, while polyenes 1–3 give tertiary carbocations.6,17
Figure 5.

Relative energies of stationary points involved in cyclization/rearrangement of polyene 3 (R1 = R3 = Me, R2 = H). D is drawn explicitly.
Figure 6.

Relative energies of stationary points involved in cyclization/rearrangement of polyene 4 (R1 = R2 = Me, R3 = H). D is drawn explicitly.
In general, the barrierless polycyclization cascades appear to pause (vide infra) prior to generating a 2° carbocation. However, despite the heightened challenge of a cation-olefin reaction with a 1,2-disubstituted alkene, the cyclization barriers are small and tetracycle formation (E, Scheme 4) is rapid. A minimum corresponding to a secondary cation for systems 2 and 3 is apparently not located due to better conformational preorganization, i.e., for system 4, the homoisoprenyl tail is not well positioned for formation of a 5-membered ring. In addition, the 1,2-hydride shift for 4 (F → G) is predicted to have a larger barrier and be less exergonic than the 1,2-methyl shift for the other systems. This prediction is consistent with relief of the 1,3-diaxial strain expected for systems with a shifting methyl group, which would be absent for 4. This prediction is also consistent with the experimental observation of a mixture of products for 4, resulting from quenching of both F and G (Scheme 3).
The predicted overall barriers for polycyclization of 1–4 are, respectively 11.2, 13.9, 12.4 and 11.5 kcal/mol. This leads to the prediction that 1 will cyclize most rapidly, followed by 4 (with a similar rate), then 3, and finally 2. These predicted relative rates match the experimentally determined relative rates (vide supra), but given the small range of energies that they span, the level of theory used and the conformational issues discussed below, we are hesitant to claim that such a match is more than coincidental.
The reaction coordinates for the productive cascades, however, do not tell the whole story. As shown in Figure 7 for 1, an assessment of the available conformers of the pre-cyclization Pt-π complex A (Scheme 4) indicate that numerous local minima are available at free energies considerably lower than that of the most productive cyclization conformer (arbitrarily assigned a relative energy of 0). The optimal geometry is thus present in minuscule quantities at thermal equilibrium. Histogram plots of the available conformers for 2–4, which display similar behavior, are available in the Supporting Information. This analysis thus leads to a picture of a [Pt2+]-π complex that samples a broad range of conformational minima. The most plentiful conformers do not adopt productive arrangements of the alkenes and thus do not generate carbocations capable of initiating the polyene cascade. Only when the polyene adopts the comparatively unfavorable nascent ring conformation can the highly exergonic cascade proceed.
Figure 7.

Histogram plot of local minimum free energies for A (R1 = R2 = R3 = Me). The dark blue bars represent structures with a C1–C6 (atom numbers are shown in Scheme 1) distance of <5 Å. The light blue bars represent structures containing C1–C6 and C5–C10 distances <5 Å. The orange bars represent structures containing three C1–C6, C5–C10 and C9–C14 distances <5 Å. The red bar represents the most favorable conformer discovered by computationally unzipping the rings (assigned 0.0 kcal/mol). The green bars represent structures for which C1–C6 distances are >5 Å.
Using a threshold of 5 Å between C1 and C6, we determined that 10 conformers of the 47 found for A (R1 = R2 = R3 = Me) through conformational searching (see Supporting Information for details), could conceivably initiate the formation of one ring (colored dark blue in Figure 7). However, this monocyclization is predicted to be endergonic, allowing retrocyclization to dominate when the polyene is not able to quickly engage the positive charge that develops upon cyclization to form the B-ring.18 A similar analysis of conformers that could conceivably form both the A and B rings (light blue in Figure 7) indicated that a comparable number of conformers are arranged to bicyclize. Of course, in the most favorable conformers (i.e., preorganized for formation of three or four rings; red and orange in Figure 7), each successive alkene is positioned to engage the positive developing nearby due to cyclization and continue the concerted (but not synchronous) cascade to the ABC or ABCD rings.
Overall barriers for cyclization consequently involve an energy term to generate the reactive conformer in addition to the intrinsic barrier for initiating the cascade. Since explicit solvent was not taken into account in our calculations, and the catalyst was studied in a truncated form, an accurate measure of the former component was not ascertained. Nevertheless, the message is that the preorganization necessary to host a low barrier ionic cascade comes at a significant cost in free energy, one that presumably is mitigated by the enzyme active site in a biogenic cyclization cascade.
OVERALL REACTIVITY/SELECTIVITY MODEL
The following picture of the Pt-promoted cascade has thus emerged. First, the Pt2+ catalyst binds to the terminal alkene π-bond. Binding to other, more substituted, π-bonds is less favorable on steric grounds and is rapidly reversed. Second, the Pt-bound substrate samples many conformations. Some of these are productive for monocyclization, but forming a single ring is predicted to be rapidly reversible.19 Third, the Pt-bound substrate must achieve a conformation that is productive for formation of at least two rings, since bicyclization is predicted to be exergonic and not readily reversible. Fourth, formation of the third and fourth rings ensue, each being exergonic. For some substrates in some conformations, multiple annulations may be combined into concerted but asynchronous processes to advance the reaction coordinate.20 Any minima/ intermediates encountered along the polycyclization pathway are predicted to have low barriers for subsequent cyclization, as long as productive conformers are formed. Once all four rings have formed, hydride and alkyl shifts may occur. The lack of byproducts arising from premature quenching of intermediate carbocations is likely a result of the relative slowness of the termination steps in the cascade coupled to the absence of competent external nucleophiles and bases in the reaction conditions employed. In other words, the rates of conformational change for each intermediate are sufficiently fast that productive (for cyclization) conformations can be generated more quickly than carbocation quenching. This model is consistent with the long reaction times necessary for reaction completion.
COMPARISON WITH (OXIDO)SQUALENE POLYCYCLIZATION
How similar are the shapes of the reaction coordinates described herein to those for the polycyclization of squalene and oxidosqualene? The result of an IRC calculation for system 2 is shown in Figure 8; this is the system for which the most complete IRC plot was generated (see Supporting information for plots for other systems). Note that two shoulders follow the initial transition state structure. These portions of the reaction coordinate correspond to structures resembling those expected for putative intermediates B and C,21 and represent the concerted but asynchronous formation of the A and B rings. For system 3, the generation of a secondary cation at C9 causes the second plateau to be just flat enough for a minimum to be located (Figure 5). IRC plots for the tricyclization of protonated squalene and protonated oxidosqualene, computed by Hess and Smentek,6 are shown in Figure 9. These reaction profiles are quite similar to that shown in Figure 8, except that monocyclized structures have a higher relative energy for system 2 than is observed for squalene or oxidosqualene, likely a result of metal complexation, whose effect is expected to be largest for the first-formed ring. Nonetheless, the similarity of the metal promoted reaction to the inherent reactivity22 predicted for the biologically relevant systems is clear. Results of recent calculations including oxidosqualene cyclase suggest that plateau regions may be transformed into very shallow minima in the presence of the enzyme,23 but such a transformation has little consequence for the rearrangement, i.e., lifetimes of species along the tricyclization reaction coordinate will not be significantly affected. Note also that the transition state structures for cyclization of the platinum-complexed polyenes are tighter than that found previously for a model of the carbocation derived from squalene (forming C–C bond lengths for rings A and B of 2.2 and 3.2–3.7 Å for the former and 4.0 and 4.2 Å for the latter).6
Figure 8.

Blue: IRC plot for tricyclization of polyene 2. Red: Dipole moment along the IRC. Approximate locations along the IRC of A–C ring formation are indicated.]
Figure 9.

IRC plots for tricyclization of squalene (top)6b and oxidosqualene (bottom).6c Reproduced with permission.
In terms of charge distributions, the enzymatic and platinum-promoted reactions do differ, however. While the dipole moment of platinum-bound cations decreases upon cyclization as shown in Figure 8 (red), charge is separated upon cyclization for the enzymatic reactions as the bulk of substrate positive charge moves away from the negatively charged carboxylate formed upon initial alkene protonation. Charge separation is generally unfavorable, but this is presumably compensated for by the enzyme active site microenvironment (and the inherent exergonicity of the polycyclization reaction). For the platinum- promoted reactions, however, charge is not separated. Since the catalyst is dicationic, polycyclization moves some of the positive charge concentrated initially in the region of carbons 1 and 2 to the far end of the hydrocarbon, reducing the molecular dipole and thereby enhancing the favorability of the reaction. Consistent with this assertion, the equilibrium constant for the bicyclization of Pt-coordinated dienesulfonamides is reduced in polar aprotic solvents able to stabilize the dicationic Pt-π complex.19
CONCLUSIONS
Overall, it seems to us fair to say that the platinum-promoted polycyclization reactions described herein are indeed biomimetic. Clearly, an enzymatic trigger for such a reaction is not necessary; other catalysts that generate reactive carbocations under mild conditions will do. In addition, an enzyme is not necessary to chaperone/template polycyclization. While cyclase enzymes enforce productive conformations and precisely position quenching agents, reducing the potential for premature termination, the platinum catalyst described here accomplishes the same feat by coupling an endergonic first cyclization with an exergonic second cyclization under conditions where carbocation termination is slow. If the remainder of the polyene is suitably oriented to continue the cascade, then a near barrierless path to product is available. If instead, the nascent C and D ring conformers are not perfect, then a rapid conformational exchange occurs to competitively provide a path to close all four rings before carbocation quenching occurs.24,26 While methyl groups do affect the rate of the polycyclization reaction, at least slightly, and sometimes allow for byproducts to form, their presence is not necessary for effective polycyclization.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the National Science Foundation (CHE-0957416 and CHE-030089 [via XSEDE] to DJT) and the National Institutes of Health (GM-60578 to MRG).
Footnotes
ASSOCIATED CONTENT
- Experimental and computational details (PDF)
- Coordinates of computed structures (mol2)
- Animation of progress along IRC in Figure 8 (gif)
References
- 1.(a) Cane DE. Chem Rev. 1990;90:1089–1103. [Google Scholar]; (b) Cane DE. In: Compr Nat Prod Chem. Meth-Cohn SDBN, editor. Pergamon; Oxford: 1999. pp. 155–200. [Google Scholar]; (c) Davis EM, Croteau R. Top Curr Chem. 2000;209:53–95. [Google Scholar]; (d) Christianson DW. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. (18) [DOI] [PubMed] [Google Scholar]; Christianson DW. Curr Opin Chem Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For leading references, see: van Tamelen EE. Acc. Chem. Res. 1975;8:152–158.Johnson WS. Angew. Chem. Int. Ed. Engl. 1976;15:9–17. doi: 10.1002/anie.197600091.Yoder RA, Johnston JN. Chem. Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l.
- 3.Selected examples of proton and Lewis acid promoted reactions with leading references to others: Pronin SV, Shenvi RA. Nature Chem. 2012;4:915–920. doi: 10.1038/nchem.1458.Ishibashi H, Ishihara K, Yamamoto H. J. Am. Chem. Soc. 2004;126:11122–11123. doi: 10.1021/ja0472026.Felix RJ, Munro-Leighton C, Gagne MR. Acc. Chem. Res. 2014;47:2319–2331. doi: 10.1021/ar500047j.
- 4.Selected examples of radical promoted reactions with leading references to others: Brill ZG, Grover HK, Maimone TJ. Science. 2016;352:1078–1082. doi: 10.1126/science.aaf6742.Justicia J, Jimenez T, Miguel D, Comtreras-Montoya R, Chahboun R, Alvarez-Manzaneda E, Collado-Sanz D, Cardenas DJ, Cuerva JM. Chem. Eur. J. 2013;19:14484–14495. doi: 10.1002/chem.201302575.
- 5.(a) Hong YJ, Tantillo DJ. Chem. Soc. Rev. 2014;43:5042–5050. doi: 10.1039/c3cs60452g. [DOI] [PubMed] [Google Scholar]; (b) Wang SC, Tantillo DJ. Org. Biomol. Chem. 2017;15:1976–1979. doi: 10.1039/c6ob02300b. [DOI] [PubMed] [Google Scholar]
- 6.A recent review: Hess BA., Jr Org. Biomol. Chem. 2017;15:2133–2145. doi: 10.1039/c7ob00222j.Smentek L, Hess BA., Jr J. Am. Chem. Soc. 2010;132:17111–17117. doi: 10.1021/ja1039133.Hess BA, Jr, Smentek L. Angew. Chem. Int. Ed. 2013;52:11029–11033. doi: 10.1002/anie.201302886.
- 7.Johnson WS. Bioorg. Chem. 1976;5:51–98. [Google Scholar]
- 8.Corey EJ, Cheng H, Baker CH, Matsuda SPT, Li D, Song X. J. Am. Chem. Soc. 1997;119:1277–1288. [Google Scholar]
- 9.Geier MJ, Gagné MR. J. Am. Chem. Soc. 2014;136:3032–3035. doi: 10.1021/ja500656k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei X, Lorenz JC, Kapadia S, Saha A, Haddad N, Busacca CA, Senanayake CH. J. Org. Chem. 2007;72:4250–4253. doi: 10.1021/jo062548f. [DOI] [PubMed] [Google Scholar]
- 11.(a) Serra D, Cao P, Cabrera J, Padilla R, Rominger F, Limbach M. Organometallics. 2011;30:1885–1895. [Google Scholar]; (b) Sokol JG, Korapala CS, White PS, Becker JJ, Gagne MR. Angew. Chem.-Int. Ed. 2011;50:5657–5660. doi: 10.1002/anie.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sokol JG, Cochrane NA, Becker JJ, Gagne MR. Chem. Comm. 2013;49:5046–5048. doi: 10.1039/c3cc41699b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li T-S, Li J-T, Li H-Z. J. Chrom. A. 1995;715:372–375. [Google Scholar]
- 13.Nes WD. Chem. Rev. 2011;111:6423–6451. doi: 10.1021/cr200021m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.SDD: Andrae D, Haeusserrmann U, Dolg M, Stoll H, Preuss H. Theor. Chem. Acc. 1990;77:123–141.SMD: Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n.M06: Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241.Zhao Y, Truhlar DG. Acc. Chem. Res. 2008;41:157–167. doi: 10.1021/ar700111a.
- 15.Frisch MJ, et al. Gaussian09 revision B.01. Gaussian, Inc; Pittsburgh, PA: 2010. see Supporting Information for full reference. [Google Scholar]
- 16.(a) Fukui K. Acc. Chem. Res. 1981;14:363–368. [Google Scholar]; (b) Chung LW, Sameera WMC, Ramozzi R, Page AJ, Hatanaka M, Petrova GP, Harris TV, Li X, Ke Z, Liu F, Li H-B, Ding L, Morokuma K. Chem. Rev. 2015;115:5678–5796. doi: 10.1021/cr5004419. [DOI] [PubMed] [Google Scholar]; (c) Maeda S, Harabuchi Y, Ono Y, Taketsugu T, Morokuma K. Int. J. Quantum Chem. 2015;115:258–269. [Google Scholar]
- 17.(a) Tantillo DJ. Nat. Prod. Rep. 2011;28:1035–1053. doi: 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]; (b) Tantillo DJ. Chem. Soc. Rev. 2010;39:2847–2854. doi: 10.1039/b917107j. [DOI] [PubMed] [Google Scholar]
- 18.Of the 47 conformers of [Pt]2+-1, 17 had no 1,6-relationships < 5Å, 10 had a 1,6-relationship < 5Å, 10 have 1,6- and 5,10-distances < 5Å, and 10 had 1,6-, 5–10-, and 9,14-distances < 5Å.
- 19.(a) For studies on the reversibility of diene-sulfonamide bicyclizations, see: Feducia JA, Gagné MR. J. Am. Chem. Soc. 2008;130:592–599. doi: 10.1021/ja075518i.(b) Note also that the Pt-promoted reactions are much more exothermic than the biological reaction examined in ref. 23.
- 20.(a) Tantillo DJ. J. Phys. Org. Chem. 2008;21:561–570. [Google Scholar]; (c) Williams A. Concerted Organic and Bioorganic Mechanisms. CRC Press; Boca Raton: 2000. [Google Scholar]; (d) Dewar MJS. J. Am. Chem. Soc. 1984;106:209–219. [Google Scholar]
- 21.(a) Kraka E, Cremer D. Acc. Chem. Res. 2009;43:591–601. doi: 10.1021/ar900013p. [DOI] [PubMed] [Google Scholar]; (b) Duarte F, Gronert S, Kamerlin SC. J. Org. Chem. 2014;79:1280–1288. doi: 10.1021/jo402702m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tantillo DJ. Angew. Chem. Int. Ed. 2017 doi: 10.1002/anie.201702363. [DOI] [Google Scholar]
- 23.Chen N, Wang S, Smentek L, Hess BA, Jr, Wu R. Angew. Chem. Int. Ed. 2015;54:8693–8686. doi: 10.1002/anie.201501986. [DOI] [PubMed] [Google Scholar]
- 24.Although not computed in this investigation, one reasonably surmises that the conformational diversity of the polyene side chains significantly decreases once the A and B rings have formed.
- 25.Similar observations have been made, through a combination of experiment and theory, for bismuth-promoted bicyclizations, see: Godeau J, Fontaine-Vibve F, Antoniotti S, Dunach E. Chem. Eur. J. 2012;18:16815–16822. doi: 10.1002/chem.201202263.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.