

Abstract
Antibiotic-resistant strains of Staphylococcus aureus pose a major threat to human health and there is an ongoing need for new antibiotics to treat resistant infections. In a high throughput screen (HTS) of 230,000 small molecules designed to identify bioactive wall teichoic acid (WTA) inhibitors, we identified one hit, which was expanded through chemical synthesis into a small panel of potent compounds. We showed that these compounds target TarG, the transmembrane component of the two-component ATP-binding cassette (ABC) transporter TarGH, which exports WTA precursors to the cell surface for attachment to peptidoglycan. We purified, for the first time, a WTA transporter and have reconstituted ATPase activity in proteoliposomes. We showed that this new compound series inhibits TarH-catalyzed ATP hydrolysis even though the binding site maps to TarG near the opposite side of the membrane. These are the first ABC transporter inhibitors shown to block ATPase activity by binding to the transmembrane domain. The compounds have potential as therapeutic agents to treat S. aureus infections, and purification of the transmembrane transporter will enable further development.
Graphical abstract
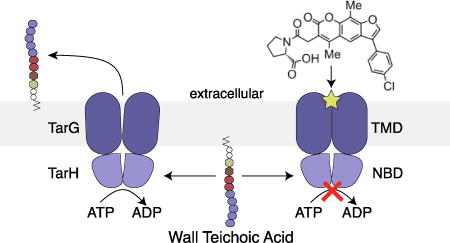
S. aureus has proven to be a highly adaptable pathogen, developing resistance almost as quickly as new antibiotics come to market.1 Maintaining a pipeline of antibiotics with activity against S. aureus is necessary to stay ahead of emerging resistance.2 The WTA pathway is a promising antibacterial target because WTAs, which are covalently attached to peptidoglycan, play crucial roles in cell division, antibiotic resistance, and pathogenesis.3 WTA precursors are synthesized on a lipid carrier on the inner leaflet of the plasma membrane and then exported to the cell surface by the two component ABC transporter TarGH (Figure 1).3b ABC transporters are found in all domains of life and use ATP binding and hydrolysis to power conformational changes to translocate molecules across the cell membrane.4 Although WTAs are required for infection,3a the first and second steps in the biosynthetic pathway, catalyzed by TarO and TarA, respectively, can be blocked genetically or pharmacologically without loss of viability; however, inhibiting subsequent steps is lethal and inhibitors of these late steps have potential as antibiotics.5 We describe here the discovery of a promising small molecule that inhibits the wall teichoic acid pathway ABC transporter and we show that it blocks the ATPase activity of the nucleotide binding domain (NBD). Resistance mutations map the binding site to the transmembrane domain. Therefore, we propose that conformational coupling between ABC transporter subunits can be exploited to develop specific inhibitors that can block activity of the ATPase from a distance.
Figure 1.

Schematic of cell wall biosynthetic pathways showing the sites of action of inhibitors mentioned in the text. Blue arrows denote the peptidoglycan pathway and red arrows denote the WTA pathway; these pathways use the same undecaprenyl (UndP) carrier. Antibiotic structures and legend abbreviations are explained in Figure S1.
The lethal phenotype resulting from a late block in the WTA pathway, which is due to depletion of peptidoglycan precursors (see Figure 1),2c,6 inspired us to develop a pathway-specific, whole cell assay for WTA-targeted antibiotics that involved screening a wildtype strain for growth inhibition while counterscreening a WTA null (ΔtarO) strain for suppression of bioactivity.3b As previously discovered WTA inhibitors had poor physical properties,7 we screened 230,000 small molecules at a final concentration of ~15 µM against wildtype S. aureus and the ΔtarO knockout strain. The screen produced a single strong hit (1), which proved to be a furanocoumarin derivative (Figure 2A). Compound 1 was found to have a minimum inhibitory concentration (MIC) of 1 µg/mL against S. aureus (Figure 2), including several β-lactam resistant strains (MRSA; Table S1). A literature search revealed that compound 1 had been identified as a growth inhibitor in a 2,000,000-compound screen for S. aureus antibiotics, but its target was not identified.8 Based on structurally related compounds also reported in that large screen, we synthesized a panel of analogs. Two l-proline derivatives (2 and 4) were found to be especially potent inhibitors of wildtype S. aureus growth (0.125 µg/mL), but showed no activity against the ΔtarO strain (Figure 2B and Table S1). This MIC is eight-fold lower than that of targocil, a well-characterized WTA-active antibiotic.7a Moreover, the kinetic solubility of these compounds is two to three logs greater than targocil’s, the half-lives were found to be 20–40 times longer in mouse liver microsomes, and the compounds were not cytotoxic (Table S2, Figure S2). Based on the promising properties of the compound, we elucidated its mechanism of action.
Figure 2.
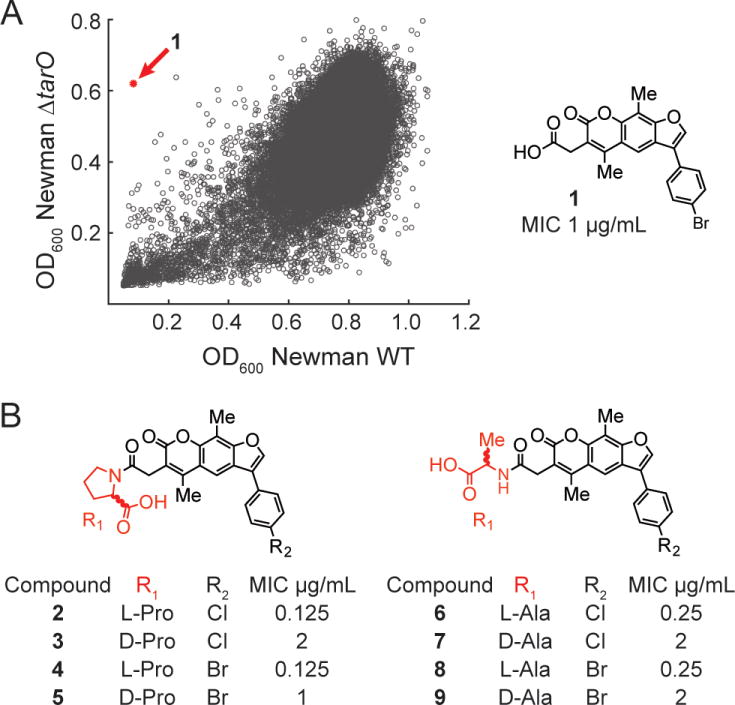
A HTS screening hit led to potent anti-MRSA compounds 2 and 4. (A) Plot of HTS results. Each circle represents the average OD600 of the strains in the presence of a library compound tested in duplicate. One compound (compound 1, red circle) inhibited growth of the WT Newman strain but not ΔtarO. (B) Synthesized analogs of 1 with activities against S. aureus Newman. MICs against MRSA strains are identical (Table S1).
We first assessed the effect of the compound on pool levels of the peptidoglycan precursor, Lipid II, using a previously developed assay.2c,6 Compounds that inhibit a late step in the wall teichoic acid pathway deplete Lipid II because this peptidoglycan precursor is biosynthesized on the same carrier lipid, undecaprenyl phosphate (UndP, Figure 1).2c,5–6 If the UndP carrier lipid is sequestered in WTA precursors, it is not available for peptidoglycan precursor synthesis. Cultures of S. aureus were treated for ten minutes with targocil, 2, or three peptidoglycan synthesis inhibitors with mechanisms of action that lead either to Lipid II depletion (bacitracin, which inhibits carrier lipid recycling) or Lipid II accumulation (moenomycin and vancomycin, which inhibit peptidoglycan assembly; Figure 1, Figure 3A). Cellular lipids were extracted and the Lipid II present therein was labeled with biotin to enable detection by streptavidin-HRP.2c,6 Like targocil and bacitracin, compound 2 depleted Lipid II. Combined with the suppression of bioactivity in the ΔtarO strain, this result confirmed inhibition of a late step in the WTA pathway.
Figure 3.
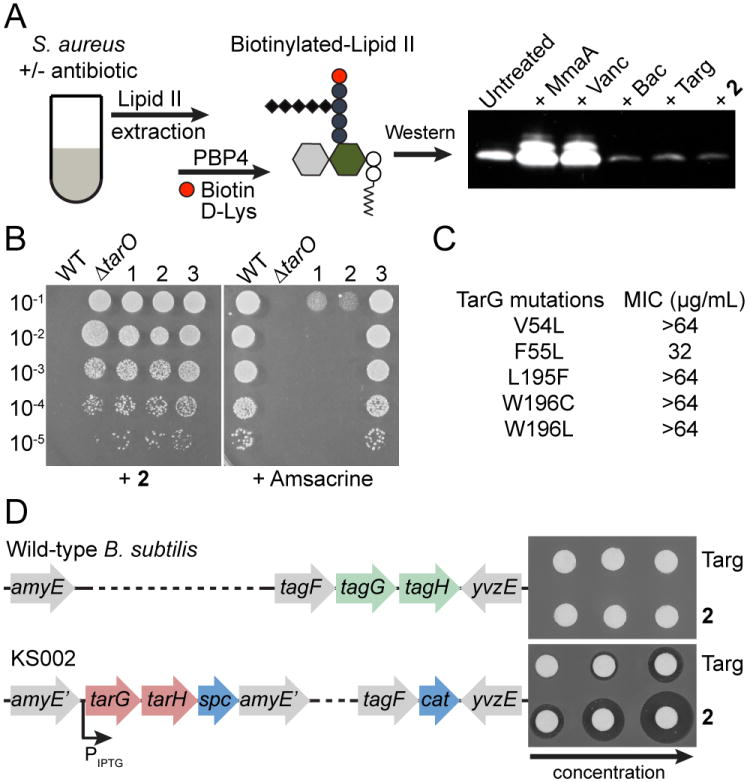
TarG is the target of 2. (A) Assay to detect Lipid II abundance after antibiotic treatment, with results for control antibiotics and 2 shown. Extracted Lipid II is labeled with biotin-d-Lys using S. aureus PBP4 to enable detection with HRP-streptavidin. (B) Mutants resistant to 2 (lanes 1–3) were sorted into two groups by plating on amsacrine. Susceptible mutants 1 and 2 had mutations in tarA while amsacrine-resistant mutant 3 had a mutation in tarG (see Table S3, S4 for full list and comparison to other TarG inhibitors). (C) Substitutions in TarG that conferred high level resistance to 2. (D) Disk diffusion assay shows that strain KS002, in which B. subtilis TagGH was replaced with S. aureus TarGH, is sensitive to 2.
To identify the molecular target within the WTA pathway, we selected resistant mutants on compound 2. Twenty-seven colonies from three independent cultures were selected for evaluation. We expected two classes of mutants: those with mutations in the molecular target and those with mutations that disrupted function of TarO or TarA.3b,5 To sort these mutants, we made use of the teichoic acid d-alanylation inhibitor, amsacrine, which prevents growth of WTA null strains.9 Seventeen mutants were unable to grow on the inhibitor (Figure 3B), and all of these were found to contain null mutations in tarO or tarA; all other mutants contained point mutations that resulted in amino acid substitutions in TarG, the transmembrane component of the ABC transporter that exports WTA precursors to the cell surface (Figure 3C; Table S3). These results validated the procedure used to classify mutants and suggested that TarG is the target of 2.
We used two different approaches to confirm TarG as the target. First, we expressed one of the resistant tarG alleles in a clean S. aureus background and found that expression conferred dominant resistance (Figure S3). Second, after verifying that 2 did not inhibit growth of B. subtilis (Figure S4), we made use of a previously engineered B. subtilis strain in which the endogenous WTA transporter genes (tagGH) were replaced with the S. aureus transporter genes at an ectopic locus.10 Compound 2 did not show a zone of inhibition in a disk diffusion assay against wildtype B. subtilis, but it showed a dose-dependent inhibition zone when tested against the strain expressing the S. aureus transporter (Figure 3D). This gain of sensitivity to compound 2 upon heterologous expression of S. aureus tarGH in B. subtilis confirmed the S. aureus wall teichoic acid transporter as its target.
Several classes of compounds that inhibit WTA export have now been identified, but the class reported here is the first with solubility properties that allow mechanistic characterization.3b,7 Elucidating how these compounds act may not only provide insight into how to improve them further, but could guide efforts to develop inhibitors of other ABC transporters. The ABC transporter family is very large and includes many possible therapeutic targets in both prokaryotes and eukaryotes, but few mechanistic studies on inhibitors have been reported.11 P-glycoprotein inhibitors have received the most attention due to the importance of this ABC transporter in multidrug resistance in cancer.12 Inhibitors that compete with ATP for binding to the nucleotide binding domain (NBD) or with exported substrates have been studied, but were abandoned due to lack of specificity and toxicity.12b The most promising P-glycoprotein inhibitors identified to date bind to the transmembrane (TM) domain in a manner that prevents substrate transport, but allows robust ATP hydrolysis.11c,13
To obtain information on how 2 inhibits TarGH, we co-expressed wildtype TarG with either TarH-His6 or an ATPase-inactive TarH-His6 mutant (E169Q), solubilized the complexes in dodecylmaltoside, purified them over an affinity column followed by size exclusion chromatography, and reconstituted them into proteoliposomes.14 The ATPase activity of the reconstituted transporter, measured using a continuous chromogenic assay, had kinetic parameters similar to those reported for other ABC transporters (Figure S5).15 The addition of compound 2 strongly inhibited ATPase activity with an IC50 of 137 nM, even though the ATP concentration was 1000-fold higher (Figure 4A, Figure S6). Additional experiments showed that the ATPase activity of the WT transporter in the presence of 1.0 µM 2 was comparable to that of a TarGH mutant containing a mutation that impairs ATP hydrolysis (TarH E169Q) (Figure S7; Figure S8).
Figure 4.

Compound 2 inhibits the ATPase activity of TarGH in proteoliposomes but binds in a remote location. (A) Averaged ATPase activity (n=3; error bars=SD) of reconstituted TarGH (200 nM) in the absence (black) and presence (red) of compound 2 (1 µM). Saturating levels of ATP (1 mM) were used. (B) Homology model of TarGH. TarH is cytoplasmic and much of TarG is embedded in the membrane. C) Top view of the TarG dimer. Mutations in residues shown in pink give high level resistance to 2.
To locate the binding site of 2 relative to the ATPase, we generated a homology model for TarGH using the human ABCG5/ABCG8 sterol transporter as the template and mapped the resistance mutations to the modeled structure (Figure 4B,C).16 In agreement with the topology of many other ABC exporters, each TarGH dimer has 12 TM helices,15b,17 which are grouped such that TM helices one and two from one monomer are in close proximity to TM helix five of the other monomer. The high-level resistance mutations selected with compound 2 map near the extracellular ends of TM helices one and five. While we cannot exclude the possibility that the resistance mutations affect the conformation of the ATPase from a distance such that it remains active but is incapable of binding inhibitor, we think it far more likely that the binding site is defined by the resistance mutations. We propose, therefore, that the binding site spans the dimer interface and, given the symmetry, that two molecules of 2 bind to the dimer. To inhibit ATP hydrolysis by binding to a remote site, the compound must lock the TM domain in a conformation that prevents the coupled inter-domain structural changes required for ongoing ATP hydrolysis by the NBDs.
Compound 2, hereafter to be called targocil-II, is the first known example of an ABC transporter inhibitor that prevents ATP hydrolysis by binding to an allosteric site in the TM domain. Given the sequence diversity of TM domains, this mode of binding would have clear advantages with respect to specificity over ATP-competitive inhibitors that bind to a very highly conserved binding pocket. Now that conditions have been developed to obtain the purified ABC transporter in active form, it may be possible to obtain structural information with inhibitor bound to facilitate development of transport inhibitors for therapeutic use.
Supplementary Material
Acknowledgments
Funding Sources
This work was funded by NIH grants U19AI109764 and P01AI08324, and NSF GRFP grant DGE1144152 (ARH).
We thank: the staff at ICCB-Longwood Screening Facility for help; the CETR DTR Core for toxicity testing; and Michael Cameron, Drug Metabolism and Pharmacokinetics Core, Scripps Research Institute, for the microsomal stability and solubility assays.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Supporting Information is available free of charge on the ACS Publications website. Methods, SI figures and tables (PDF).
No competing financial interests are declared.
References
- 1.(a) Wright GD. ACS Infect Dis. 2015;1:80. doi: 10.1021/id500052s. [DOI] [PubMed] [Google Scholar]; (b) Chambers HF, Deleo FR. Nat Rev Microbiol. 2009;7:629. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Okano A, Nakayama A, Schammel AW, Boger DL. J Am Chem Soc. 2014;136:13522. doi: 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bouley R, Kumarasiri M, Peng Z, Otero LH, Song W, Suckow MA, Schroeder VA, Wolter WR, Lastochkin E, Antunes NT, Pi H, Vakulenko S, Hermoso JA, Chang M, Mobashery S. J Am Chem Soc. 2015;137:1738. doi: 10.1021/jacs.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee W, Schaefer K, Qiao Y, Srisuknimit V, Steinmetz H, Müller R, Kahne D, Walker S. J Am Chem Soc. 2016;138:100. doi: 10.1021/jacs.5b11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Weidenmaier C, Kokai-Kun JF, Kristian SA, Chanturiya T, Kalbacher H, Gross M, Nicholson G, Neumeister B, Mond JJ, Peschel A. Nat Med. 2004;10:243. doi: 10.1038/nm991. [DOI] [PubMed] [Google Scholar]; (b) Swoboda JG, Meredith TC, Campbell J, Brown S, Suzuki T, Bollenbach T, Malhowski AJ, Kishony R, Gilmore MS, Walker S. ACS Chem Biol. 2009;4:875. doi: 10.1021/cb900151k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Campbell J, Singh AK, Santa Maria JP, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S. ACS Chem Biol. 2011;6:106. doi: 10.1021/cb100269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilkens S. F1000Prime Rep. 2015;7:14. doi: 10.12703/P7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) D'Elia MA, Pereira MP, Chung YS, Zhao W, Chau A, Kenney TJ, Sulavik MC, Black TA, Brown ED. J Bacteriol. 2006;188:4183. doi: 10.1128/JB.00197-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) D'Elia MA, Henderson JA, Beveridge TJ, Heinrichs DE, Brown ED. J Bacteriol. 2009;191:4030. doi: 10.1128/JB.00611-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiao Y, Lebar MD, Schirner K, Schaefer K, Tsukamoto H, Kahne D, Walker S. J Am Chem Soc. 2014;136:14678. doi: 10.1021/ja508147s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Lee K, Campbell J, Swoboda JG, Cuny GD, Walker S. Bioorg Med Chem Lett. 2010;20:1767. doi: 10.1016/j.bmcl.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo N, She X, Kales S, Liang L, Liu J, Wu J, Santa Maria J, Su J, Pan J, Hailey J, Mcguinness D, Tan CM, Flattery A, Walker S, Black T, Roemer T. Chem Biol. 2013;20:272. doi: 10.1016/j.chembiol.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng TJ, Wu YT, Yang ST, Lo KH, Chen SK, Chen YH, Huang WI, Yuan CH, Guo CW, Huang LY, Chen KT, Shih HW, Cheng YS, Cheng WC, Wong CH. Bioorg Med Chem. 2010;18:8512. doi: 10.1016/j.bmc.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 9.(a) Pasquina L, Santa Maria JP, McKay Wood B, Moussa SH, Matano LM, Santiago M, Martin SE, Lee W, Meredith TC, Walker S. Nat Chem Biol. 2016;12:40. doi: 10.1038/nchembio.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schaefer K, Matano LM, Qiao Y, Kahne D, Walker S. Nat Chem Biol. 2017;13:396. doi: 10.1038/nchembio.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Matano LM, Morris HG, Wood BM, Meredith TC, Walker S. Bioorg Med Chem. 2016;24:6307. doi: 10.1016/j.bmc.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schirner K, Stone LK, Walker S. ACS Chem Biol. 2011;6:407. doi: 10.1021/cb100390w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Science. 2009;323:1718. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jin MS, Oldham ML, Zhang Q, Chen J. Nature. 2012;490:566. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Loo TW, Clarke DM. Biochem Pharmacol. 2014;92:558. doi: 10.1016/j.bcp.2014.10.006. [DOI] [PubMed] [Google Scholar]; (d) Loo TW, Clarke DM. J Biol Chem. 2015;290:29389. doi: 10.1074/jbc.M115.695171. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sherman DJ, Okuda S, Denny WA, Kahne D. Bioorg Med Chem. 2013;21:4846. doi: 10.1016/j.bmc.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Nat Rev Drug Discov. 2006;5:219. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]; (b) Crowley E, McDevitt CA, Callaghan R. Methods Mol Biol. 2010;596:405. doi: 10.1007/978-1-60761-416-6_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falasca M, Linton KJ. Expert Opin Investig Drugs. 2012;21:657. doi: 10.1517/13543784.2012.679339. [DOI] [PubMed] [Google Scholar]
- 14.Moody JE, Millen L, Binns D, Hunt JF, Thomas PJ. J Biol Chem. 2002;277:21111. doi: 10.1074/jbc.C200228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Borths EL, Poolman B, Hvorup RN, Locher KP, Rees DC. Biochemistry. 2005;44:16301. doi: 10.1021/bi0513103. [DOI] [PubMed] [Google Scholar]; (b) Perez C, Gerber S, Boilevin J, Bucher M, Darbre T, Aebi M, Reymond JL, Locher KP. Nature. 2015;524:433. doi: 10.1038/nature14953. [DOI] [PubMed] [Google Scholar]
- 16.Lee JY, Kinch LN, Borek DM, Wang J, Urbatsch IL, Xie XS, Grishin NV, Cohen JC, Otwinowski Z, Hobbs HH, Rosenbaum DM. Nature. 2016;533:561. doi: 10.1038/nature17666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Dawson RJ, Locher KP. Nature. 2006;443:180. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]; (b) Ward A, Reyes CL, Yu J, Roth CB, Chang G. Proc Natl Acad Sci U S A. 2007;104:19005. doi: 10.1073/pnas.0709388104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.