

Abstract
Introduction
Analysis of histone post-translational modifications (PTMs) by mass spectrometry (MS) has become a fundamental tool for the characterization of chromatin composition and dynamics. Histone PTMs benchmark several biological states of chromatin, including regions of active enhancers, active/repressed gene promoters and damaged DNA. These complex regulatory mechanisms are often defined by combinatorial histone PTMs; for instance, active enhancers are commonly occupied by both marks H3K4me1 and H3K27ac. The traditional bottom-up MS strategy identifies and quantifies short (aa 4-20) tryptic peptides, and it is thus not suitable for the characterization of combinatorial PTMs.
Areas covered
Here, we review the advancement of the middle-down MS strategy applied to histones, which consists in the analysis of intact histone N-terminal tails (aa 50-60). Middle-down MS has reached sufficient robustness and reliability, and it is far less technically challenging than PTM quantification on intact histones (top-down). However, the very few chromatin biology studies applying middle-down MS resulting from PubMed searches indicate that it is still very scarcely exploited, potentially due to the apparent high complexity of method and analysis.
Expert Opinion/Commentary
We will discuss the state-of-the-art workflow and examples of existing studies, aiming to highlight its potential and feasibility for studies of cell biologists interested in chromatin and epigenetics.
Keywords: chromatin, cross-talk, histones, mass spectrometry, middle-down, post-translational modifications, proteomics
1. Introduction: Chromatin Biology and Histone PTM Cross-Talk
Chromatin of eukaryotes is assembled in the cell nucleus, composed of DNA and histone proteins highly organized in a complex three-dimensional structure. Beside its complexity, chromatin is also dynamic and in constant remodeling. These dynamics modulate a large variety of biological functions, mostly starting with protein recruitment in hotspots; for instance, enzymes and/or transcription factors are recruited for gene transcription, DNA repair, and condensation of DNA into chromosomes during mitosis and meiosis [1]. The nucleosome is the basic unit of chromatin, an assembly of eight histones (two copies of H3, H4, H2A and H2B) plus a histone linker (H1) wrapped around by DNA every ~200 base pairs. In some regions of the chromatin canonical histones are replaced by variants to tune specific functions [2], many of which are still unknown. Histones are heavily decorated with post-translational modifications (PTMs), especially on their tail, which is the N-terminal part of the sequence that protrudes outside the nucleosome. Some modifications benchmark regions of active transcription, e.g. H3K4me3; other gene repression, e.g. H3K9me3; others are “flags” of DNA damage, e.g. H2AS139ph (reviewed in [3]). Their aberrant regulation could have fatal effects; several histone PTMs have been found to drive or benchmark critical diseases such as cancer [4-6]. The ability of histone PTMs in recruiting chromatin readers led to the realization that the myriad of possible combinations of histone PTMs might compose a «“histone code” that considerably extends the information potential of the genetic code» in modifying and modulating chromatin activities [7]. This idea was strengthened by the discovery of histone binding proteins with multiple domains recognizing multiple PTMs, including the histone reader TAF1 (double bromodomain), the histone reader CHD1 (double chromodomain) and the histone reader/writer KDM4A (double tudor domain) [8]. Known chromatin readers are now a long list, and their classification is accurately explained by Yun et al. in a comprehensive review [9]. As well, we know currently more than 50 different histone writers, including acetyltransferases, deacetylases, methyltransferases demethylases and kinases [10], most of which are actively modifying histones upon recruitment by other proteins. The hypothesis of the “histone code” has been frequently considered fascinating but controversial, as by definition a code requires a universal readout. However, not only we cannot decipher this code yet, but it has become more and more evident that histone PTMs have interdependent mechanisms with other chromatin elements like, for instance, DNA methylation [11]. Moreover, almost every possible protein PTM has been identified on histones [10], making this hypothetical code even harder to crack. Several of these modifications are derivatives of metabolic products, mostly from carbohydrates and lipids (i.e. acyl-CoA) [12], highlighting also the link between metabolism and chromatin regulation. However, we can temporarily conclude that the number of discovered histone PTMs with unclear function is growing at a faster rate than the functional characterization of known PTMs.
A number of histone modifications have become unequivocal benchmarks of specific chromatin states, including examples of combinatorial patterns [13,14]. For instance, both H3K4me1 and H3K27ac benchmark active gene enhancers, and they are used almost as a dogma in genomics to identify those hotspots [15]. The modification H3K9me3 benchmarks heterochromatic regions, which are recognized by the chromodomain of HP1 (histone reader) and propagated/maintained by the trivalent interaction H3K9me3 - HP1 - SUV39H1/2 (histone writer); however, the binding between H3K9me3 - HP1 is lost when H3S10ph is catalyzed on the same histone [16]. In yeast, the binding of the protein spChp1 to H3K9me2/me3 is inhibited by H3K4ac [17], and the histone demethylase PHF8 binds most efficiently on the combinatorial pattern H3K4me3K9acK14ac [18]. In conclusion, even though lots has been discovered about the role of single histone PTMs [19], it has become clear that identification and quantification of single histone PTMs is not anymore an exhaustive practice to determine the role of a specific chromatin region.
2. Identification and Quantification of Co-existing Histone PTMs
While identification of novel histone modifications is now almost exclusively performed by mass spectrometry (MS), quantification of single histone PTMs is still performed by either (or both) antibody-based and MS-based techniques. Both have advantages and drawbacks, although the choice is mostly based on skills and capabilities of individual research labs. On one hand, MS has currently the highest throughput, can quantify hundreds of histone PTMs in once and it provides a significantly more accurate quantification. On the other hand, quantification based on antibodies is generally a much faster procedure, more sensitive and does not require very expensive instrumentation. Quantification of combinatorial PTMs is technically more challenging with either of the two strategies, although for this specific instance methods based on antibodies are more scarcely exploited (Fig. 1). For instance, co-existence of two PTMs 1 and 2 can be identified by immunoprecipitating histones carrying PTM 1 followed by western blotting of PTM 2 on the eluate. Yuan et al. proved that K27me3 and K36me3 are mutually exclusive on histone H3 in HeLa cells by using this approach [20]. Alternatively, immunofluorescence can be adopted to define chromatin domains; Chandra et al. showed that H3K9me3, H3K27me3 and H3K36me3 form distinct domains during embryonic development [21]. Recently, a genomics approach was proposed to quantify co-existing PTMs on single nucleosomes by performing high-throughput single-molecule imaging [22]. However, all these methods have all the typical limitations of antibody-based techniques, mostly related to the challenge of developing an efficient and unbiased antibody [23], and the fact that the analysis is currently limited to maximum 2-3 co-existing histone marks. In addition, MS is currently far more accurate as compared to antibodies in terms of relative quantification.
Figure 1. Combinatorial PTM analysis in literature.
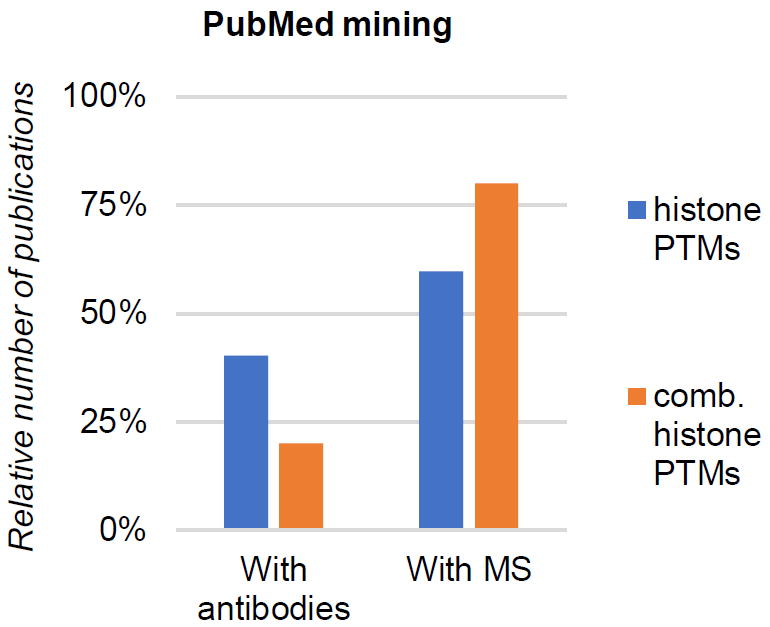
Relative count of scientific publications (source: PubMed) including histone PTM analysis using antibodies (left) and mass spectrometry (right). The graph is generated from a total of 861 studies. The publications are divided between generic “histone PTM analysis” and “combinatorial histone PTM analysis” as key words.
The high mass accuracy, speed and flexibility of MS makes it the most suitable strategy to identify and quantify combinations of PTMs [24], and it is widely used for histone analysis [25]. The traditional peptide centric analysis of histone proteins (bottom-up) is similar to the typical proteomics pipeline; however, histones are very basic proteins and thus derivatization of lysine residues is frequently applied to generate medium size Arg-C like peptides using trypsin [26]. Since 2004, the sample preparation strategy has been periodically optimized and different laboratories applied different derivatization methods, from the use of D6-acetic anhydride [27], to propionic anhydride [28] to NHS-propionate [29] to phenyl isocyanate [30]. Independently from the protocol applied, it is now clear that by performing chemical labeling of lysine residues and peptide N-termini assists more confident and accurate PTM quantification. In fact, this approach leads to the generation of peptide between 4 (e.g. KVLR, histone H4, aa 20-23) and about 20 amino acid residues (e.g. AGGKAGKDSGKAKTKAVSR, histone H2A.Z, aa 1-19); peptides as short as 4 amino acid residues are hard to resolve by reversed-phase chromatography if no derivatization is used. Bottom-up proteomics can only quantify the co-existence frequency of very close modified sites, such as H3K9-S10-K14, H3K18-K23, H3K27-K36 and H4K5-K8-K12-K16. To cope with the quantification of combinatorial PTMs other workflows have been introduced, namely middle-down and top-down MS.
Top-down MS applied to histones is the analysis of the intact histone proteins, of which pipeline has been pioneered by the group of Neil Kelleher (e.g. [31]). In the early 1990s top-down MS experiments used low-energy CID for MS/MS fragmentation [32], but this method is not ideal for histones due to the large amount of positive charges that produce incomplete fragmentation and poor sequence coverage. After the introduction of Electron Capture Dissociation (ECD) [33] and Electron Transfer Dissociation (ETD) [34] the Kelleher and Pesavento lab applied this fragmentation technique for the analysis of each canonical histone in top-down MS mode, including histone H3 [35], H2A [36], H2B [37] and H4 [38]. Regarding online separation, reversed phase chromatography is the most used [39], but it hardly resolves differently modified histone proteoforms. More recently, capillary electrophoresis was introduced as alternative separation strategy [40], although it currently remains a scarcely exploited method. Overall, top-down appears to be the ideal strategy to quantify co-existing histone PTMs, but its sensitivity is limited by the huge number of proteoforms that cannot be discriminated by HPLC-MS/MS. For instance, an average bottom-up size peptide with two lysine residues has 10-30 potential combinations of modifications (e.g. me1-ac, me2-ac, me3-me1…); of these, maybe a handful are isobaric, i.e. have identical precursor mass (e.g. me1-ac vs ac-me1). In an intact histone this issue grows exponentially, as 30 modifiable residues have potentially trillions of combinatorial PTM patterns [41]. Likely, most of them do not even exist, as histones are in the order of 10e7 in a cell. However, each intact mass corresponds to a number of potential proteoforms that we are currently unable to resolve by chromatography and discriminate within the same MS/MS spectrum.
Middle-down MS has gained interest as a “compromise” between bottom-up and top-down [42]; it is used to analyze long polypeptides that retain more information about co-existing PTMs than bottom-up, but it is not as technically challenging as top-down MS. Incidentally, middle-down MS is very suitable for histone analysis, as the intact histone N-terminal tail (~50-60 aa residues) carries most PTMs, and it can be easily cleaved off the nucleosome core using enzymes such as GluC (cleaves after glutamic acid) or AspN (cleaves before aspartic acid). As example, the human canonical histone H3 gets cleaved by GluC at the residue 50, generating the peptide aa 1-50 that can be modified in 23 different residues considering lysine, arginine, serine and threonine. This polypeptide carries most studied residues on histone H3, leaving out just H3K56, H3K79 and H3K122 as well studied modified sites. Middle-down MS also allows for discrimination of highly similar histone variants, as the few sequence variations are usually present in the N-terminal tail (Fig. 2A). In mid-2000, middle-down MS analysis of long hypermodified histone peptides was introduced mostly as proof of concept to show the potential of fragmentation methods emerging as alternative to the traditional collision based dissociation (CID or HCD); Donald Hunt, and then Joshua Coon, introduced ECD and ETD as “softer” MS/MS techniques for specific applications in proteomics. To prove their efficiency they showed examples of MS/MS spectra of long histone peptides with excellent sequence coverage [43,44], proving that for long basic sequences ECD and ETD outperforms collision based fragmentation similarly to data presented by Neil Kelleher on intact histones [31]. The technique was rapidly adopted by the group of David Allis to show combinatorial coexistences of methylation and acetylation on histone H3 N-terminal tails [42], and soon it became clear the potential of middle-down MS for histone analysis. A milestone in the throughput of middle-down MS is the work of Young et al. in 2009, where they applied to a chromatographic resin named PolyCAT A™ (PolyLC, discussed in section 3) a gradient of organic/water and pH [45]. Briefly, they applied a gradient of increasing water and decreasing pH without using any salt in the buffer that almost baseline separated histone H3 N-terminal tails with different amounts of acetyl groups and even defined distinct chromatographic peaks for tails with different number of methyl equivalents (Fig. 2B). Interestingly, this chromatographic material can be adopted also for separation of intact modified histones (top-down) [46].
Figure 2. Discrimination of histone variants and modified forms by middle-down MS; example with histone H3.
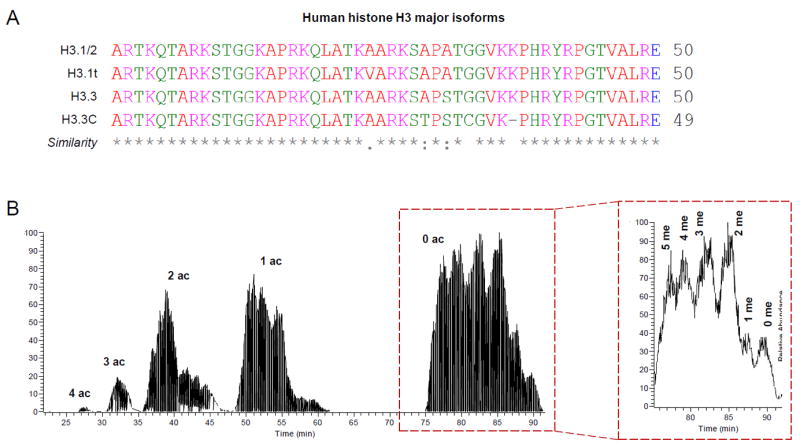
(A) Histone H3 N-terminal tails in silico digested with GluC. The major variants are listed, together with the similarity symbols (generated using Clustal Omega). Histone H3.1 cannot be discriminated from histone H3.2 based on the N-terminal sequence. (B) Middle-down LC-MS chromatogram of histone H3. The figure was generated by extracting the MS/MS fragment ion 474.31, corresponding to the fragment c4 of the histone H3 N-terminal tail. Chromatography was performed by using a nano column (75μm internal diameter) packed with WCX-HILIC. On the right, zoom of the peaks corresponding to the 0 acetylation; separation of methylated peptides can be observed.
DiMaggio et al. proposed a software developed specifically to analyze LC-MS runs of middle-down size histone peptides [47]. In 2014, Sidoli et al. improved the workflow by automating column loading and introducing a software tool, namely isoScale, to filter results of Mascot database searching (Matrix Science) and quantify intact histone tails [48]. Other software tools are potentially suitable to identify and quantify middle-down size polypeptides, including e.g. ProSightPTM [49,50], BIG Mascot [51] and spectral alignment [52] (more comprehensive list reported in the review of Sidoli et al. [25]). However, software developed for top-down MS are not ideal to quantify histone tails, as they usually do not discriminate the intensity of co-fragmented isobaric forms. In the next section, we will describe all the necessary steps to set up a typical workflow for middle-down MS of histone tails.
3. The State-of-the-art Middle-Down Workflow
There is not a single LC-MS configuration for middle-down MS of histone polypeptides, and there is no study where a comprehensive comparison of different LC-MS settings has been performed. Therefore, the setup we are about to describe has been decided at the best of our knowledge based on different publications that proposed optimizations on the method (Fig. 3A). We thus anticipate that it is potentially biased in favor of setups proposed by laboratories with more experience and/or with more sensitive instrumentation.
Figure 3. Middle-down MS workflow.
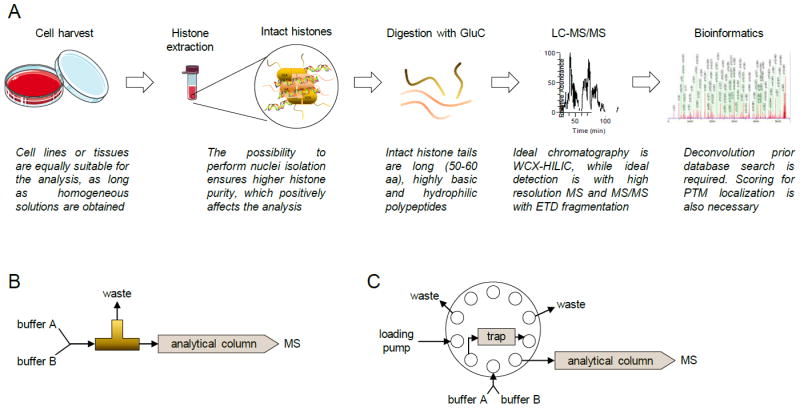
(A) Complete workflow. After histone extraction, digestion is performed with GluC. Samples are then ideally separated using WCX-HILIC chromatography coupled online with high resolution MS equipped with ETD. Identification of spectra can be performed with traditional software, but additional result filtering is recommended due to the issues of estimating a proper FDR. (B) HPLC valve configuration for single column setup and (C) for two-column setup.
Sample preparation is identical to the more widely used bottom-up MS strategy up to the point where purified histones are obtained [53]. Briefly, cell pellets are homogenized into a lysis buffer that opens the cell membranes in a hypothonic manner, maintaining cell nuclei intact. Histones are then purified from nuclei using acid extraction followed by precipitation with trichloroacetic acid (TCA). At this point the sample preparation for bottom-up, middle-down and top-down MS diverges. In middle-down MS, histones are resuspended in either ammonium bicarbonate (pH 8.0) or ammonium acetate (pH 4.0) and incubated with GluC at room temperature for six hours or overnight (detailed protocol described by Sidoli et al. at [54]). Other enzymes are suitable to generate similar size histone N-terminal tails, e.g. AspN [55].
Two chromatographic materials are suitable to separate hypermodified N-terminal tails, reversed-phase and a mixed bed weak cation exchange / hydrophilic interaction chromatography (WCX-HILIC). The WCX-HILIC (commercial name PolyCAT A, PolyLC™) has clearly shown more effective separation of modified histone tails (Fig. 2B) [45,48]. Two configurations are possible, a single column setup (Fig. 3B) or a two-column setup with reversed-phase trap column (Fig. 3C). While the first one is intuitively more straightforward to install and maintain, the second one allows solubilization of histone tails in aqueous buffers and copes with sample containing salts. Reversed-phase chromatography is also suitable for middle-down MS; it is convenient for proteomics laboratories that have exclusively C18 columns coupled online with MS and it is a more robust type of chromatography than WCX-HILIC (statement based on personal, unpublished, experience). However, C18 separation is by far less efficient than WCX-HILIC, and thus leads to less sensitive results. Liao et al. presented a derivatization strategy that drastically improves sensitivity for histone tails when separated with reversed-phase chromatography [56]. However, WCX-HILIC currently remains the most suitable chromatographic resin for the purpose.
High resolution MS and MS/MS and ETD fragmentation are currently required. As discussed by Good et al. [44] ETD outperforms collision based fragmentation methods for long basic peptides such as intact histone tails. Comprehensive fragmentation is critical, as histone tails are rich of amino acid residues that can be modified with the same PTMs. For instance, a fragment ion corresponding to the first four amino acids of histone H3 (ARTK) plus two methylations can be obtained by three potential modified forms: R2me2, K4me2, R2me1K4me1. In the absence of other fragment ions in between R and K residues those three forms cannot be distinguished, and thus the identification remains ambiguous. Histones are rich of basic residues, and thus this problem is overwhelming in case of incomplete fragmentation. High resolution MS and MS/MS are also necessary, as both precursor and fragment mass ions can be heavily charged, and determination of the charge state requires a sufficient m/z resolution to distinguish isotopic patterns. The acquisition method is usually data-dependent acquisition (DDA); however, since the number of precursor mass signals is very limited due to the large number of isobaric forms, it has been proved that by programming targeted scans sensitivity can be improved [57].
As previously mentioned, only a handful of software are suitable to identify long hypermodified polypeptides and filter out false positives. First, MS and MS/MS signals should be deconvoluted into singly charged m/z values. A few popular deconvolution algorithms for this purpose are THRASH [58], Xtract (Thermo Scientific) and MS-Deconv [59]. Peptide identification from MS/MS spectra is a more complex issue. Software developed for bottom-up proteomics are not ideal for the purpose. The estimation of the false discovery rate (FDR) using decoy peptides [60] often indicates 0% false positives, as the probability of a scrambled or reversed middle-down size sequence to match a histone sequence database by chance is close to none. LeDuc et al. recently developed the C-score, a score for characterization of intact proteins that overcomes the limits of the simple decoy search for large masses [61]. However, histone polypeptides have the additional problem of being hypermodified. There is still no scoring system sophisticated to the point of confidently assigning a probability to the localization of all potential combinatorial PTMs present in a MS/MS spectrum. On the same page, quantification is highly challenging, due to the large variety of species with the same intact mass, i.e. the intensity of the precursor mass signal corresponds to many isobaric forms. As previously mentioned, only two software tools are currently developed specifically to identify and quantify histone N-terminal tails, the algorithm of DiMaggio et al. [47] and isoScale [48]. Unfortunately, the first is currently not available in a user-friendly user interface. The second adopts a combination of spectral counting and estimation of the fragment ion relative ratio (FIRR) of isobaric forms introduced by Pesavento et al. [38]; even though it is still not an ideal solution, and it requires Mascot (Matrix Science) for the identification process, it is currently the only software tool developed on purpose to filter false positives and quantify hypermodified histone tails [48] (freely available at http://middle-down.github.io/Software/). In the next section, we will discuss how to elaborate a final list of identified and quantified combinatorial histone PTMs.
4. Analysis of Results: How to Decode Middle-Down MS Data
The most challenging part of middle-down MS is very likely data interpretation. This is probably the main reason keeping proteomics research labs from exploiting more the methodology. Middle-down MS results have more layers of information as compared to bottom-up MS, i.e. co-existence frequency of combinatorial PTMs. This type of output is overwhelming and thus it is usually deconvoluted in abundances of single PTMs (Fig. 4A). This operation simplifies the output, but it does not exploit the potential of the middle-down MS results, leading to an inevitable inertia that keeps chromatin biologists into bottom-up MS for histone PTM quantification. Some steps have been taken to better decode the myriad of information contained in such dataset; we will present a few examples of (i) sorting, (ii) transforming and (iii) visualizing these data.
Figure 4. Examples of graphical representation of combinatorial PTMs.
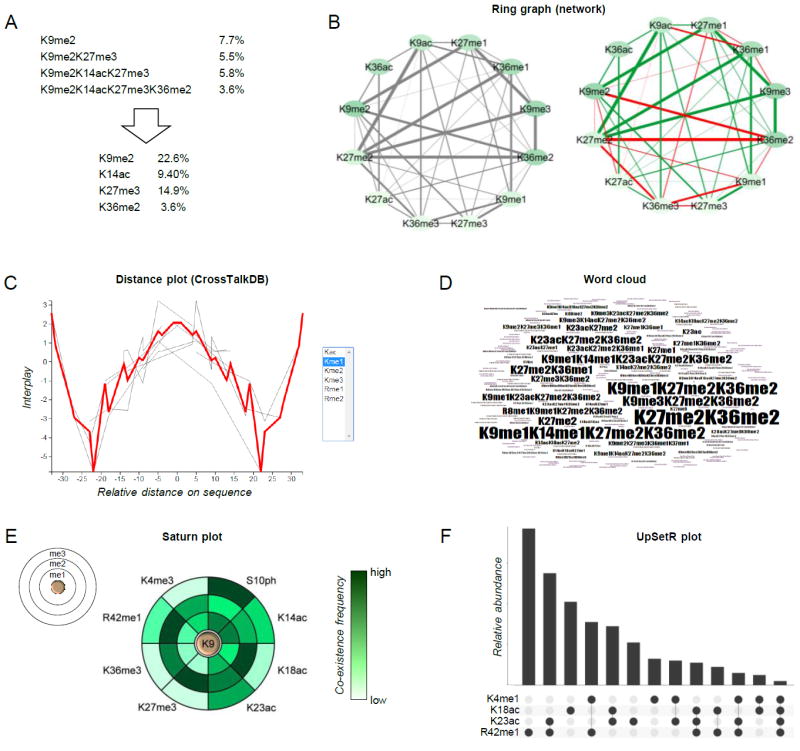
(A) Schematic showing how quantitative values of combinatorial PTMs can be deconvoluted into single mark abundance. Briefly, the total relative abundance of individual mark is the sum of the relative abundance of all modified forms containing the given mark. (B) Representation of co-existence frequency of PTMs using a ring graph layout. Line thickness indicates higher frequency; line coloring displays the interplay between two marks (green: positive, red: negative) [65]. (C) Distance plot and (D) word cloud, tools of CrossTalkDB [66]. (E) Saturn plot [70], which displays the co-existence frequencies of a modified residue together with others (on the rings). (F) UpSetR plot [71], an alternative to the Venn diagram for displaying co-existence frequencies of multiple PTMs.
Sorting middle-down MS data is usually performed using only two aspects, the abundance of a PTM pattern and the number of modified residues. Back in 2002, Zhang et al. defined a model of sequential acetylation of the tail of histone H4 using bottom-up MS data [62]. Briefly, they sorted the abundance of the quantified peptides and realized that N-terminal peptides carrying H4K16ac were always the most abundant combination. This led to the speculation that histone H4 is sequentially acetylated from the residue K16 to K5, i.e. K16ac→K12ac→K8ac→K5ac. Today, thanks to advancements in LC-MS sensitivity, we discovered that several combinatorial acetylation patterns on histone H4 lacking K16ac are also common (e.g. [63]). However, the approach was interesting, and Jung et al. used middle-down MS data to show that PTMs on histone H3 of mouse embryonic stem cells have “priorities” of deposition [64]. Specifically, H3K27 and H3K36 methylations were catalyzed first, and then other PTMs would follow on the same histone tail. This interpretation was also oversimplified, but it was a small step towards interpreting hypermodified histone tails.
In 2014, Schwämmle et al. proposed to transform the co-existence frequency of PTMs obtained from middle-down MS data to normalize their abundance bias and identify potential cross-talk [65]. Briefly, the co-existence frequency of two modifications was divided by the product of their abundance on the histone; high values implied that two PTMs are found on the same histone tail for a frequency higher than random chance of co-occurring (due to their abundance on the chromatin), while low values implied they were mutually exclusive. Cross-talk was then implied, as always co-existing marks must have a reason to do so. Data showed that selected PTMs had a consistent mutual exclusion from the same histone H3 tail, e.g. K9me3/K27me3/K36me3, while others were almost always detected together [65,66]. This was not surprising, as it is well known from other datasets such as chromatin immunoprecipitation sequencing (ChIP-seq) that K9me3/K27me3/K36me3 occupy different chromatin domains, as they have been characterized to benchmark distinct chromatin domains (K9me3: constitutive heterochromatin; K27me3: more dynamic, or facultative, heterochromatin; K36me3: enriched in actively transcribed gene bodies). As side note, ChIP-seq is currently the ideal technique to map genome-wide histone PTMs, and eventually regions of co-localization. Wang et al. performed a very extensive ChIP-seq experiment, comprising 39 different histone PTMs [67]; in their work they determined which and how often different histone modifications co-localize on human CD4+ T cells. However, ChIP-seq and middle-down MS are still complementary, as two PTMs might occupy the same chromatin domain but not be on the same histone protein, e.g. asymmetrically modified nucleosomes [68].
Graphical visualization of co-existing PTMs from a middle-down MS dataset is also a challenge. While representing the co-existence between two PTMs requires a two-dimensional graph (Fig. 4B), the co-existence between three PTMs would require a 3D graph, and so displaying the co-existence frequency of several PTMs becomes rapidly an impossible task. A few atypical plots can be used to visualize such complex dataset, but none of them can comprehensively display the full information of co-existing histone PTMs. Those examples are presented in Fig. 4B-4F. Ring graphs (Fig. 4B) have been also used to display proteoforms identified in top-down MS data [69]. Briefly, they can be used to provide visual overview of which PTMs co-exist with higher frequency (line thickness). Color coding the nodes (bubbles) can also be used to display the individual abundance of single marks. Color coding the connector lines can also be used to display a ratio between two conditions, or the interplay between two marks (green: positive, red: negative) [65]. Distance plots (Fig. 4C) show the distribution of interplays for a given modification type (in the figure, me1) along the protein sequence. In the example, mono-methylation is likely to be found together with other mono-methylations within 5 amino acid residues or at more than 30 residues in distance, while it is very unlikely to observe two mono-methyl marks distant ~20 residues. This representation can be helpful to identify binding sites of histone readers with multiple domains with a specific spatial distance. The word cloud (Fig. 4D) is currently the only representation that provides a full overview of the combinatorial PTM patterns. Larger font corresponds to more abundant modified species. However, it can display one dataset at the time. The Saturn plot (Fig. 4E) was introduced in the manuscript of Guan et al. [70] not for middle-down datasets, but it is applicable when the aim is to display the co-existence frequencies of a modified form with several others. In the example, H3K9 methylation is the reference (the three discs represent me1, me2 and me3), and the color coding represents their co-existence frequency with the PTMs listed externally. Finally, the UpSetR plot [71] is a format of Venn diagram (Fig. 4F), which becomes more intuitive and less crowded than an actual Venn diagram for visualizing more than 3-4 intersections. The dots underneath the bar plot indicate which co-existing PTMs correspond to the relative abundance displayed by the bars. Graphs of Fig. 4B, 4C and 4E are automatically generated by the database CrossTalkDB [65,66], developed on purpose to store and analyze hypermodified proteins. Collectively, a few steps have been taken towards simplifying the interpretation of middle-down MS data of hypermodified histone tails. However, none is exhaustive due to the complexity of the dataset, revealing that our limitation in understanding the full picture of histone PTM cross-talk is mostly limited by our ability to visualize it. In the next two sections we describe a number of examples of studies that applied middle-down MS for the characterization of a biological system, aiming to discuss already exploited approaches to data interpretation.
5. Expert Commentary
Middle-down MS is the result of numerous technological improvements, from improved chromatography to MS fragmentation, to software advancements, to data visualization (summarized in Fig. 5). However, to date only a handful of publications have exploited middle-down MS to understand aspects of a biological system that could not be addressed with other techniques. To our knowledge, the first publication is the work of Taverna et al. where they identified correlations between K4 methylation and other site acetylation on the histone H3 of Tetrahymena thermophila [42]. Other models have been investigated, like Toxoplasma gondii [72], Caenorhabditis elegans [73], Neurospora crassa [74] and 3D spheroids cell cultures [75]. In this last publication, the authors characterized the PTM composition of histones that underwent biological proteolytic cleavage, a.k.a. histone clipping, another phenomenon that can hardly be investigated with bottom-up MS or any other technique. Nevertheless, so much can still be done using middle-down MS. Somehow, this is the most attractive aspect of the technique; generated data can still be mined from unexplored points of view, and there is much room of application on almost every model system where histones can be efficiently extracted.
Figure 5. Timeline of important publications for the development of the middle-down MS strategy for histone analysis.

In chronological order, the seven publications listed in the figure are referenced as [7] (2001), [43] (2006), [42] (2007), [45] (2009), [80] (2012), [48] (2014) and [66] (2016).
The reluctance of scientists requiring a histone PTM analysis from using middle-down MS is understandable; very few laboratories have such system in place (meaning long delays for instrument setup), and the poor coverage of software quality evaluation raises doubts on the confidence of identification and quantification. Bottom-up MS has also intrinsic biases (discussed in [76]), mostly related to the ionization efficiency of the peptides, enzyme preference of cleavage of modified vs. unmodified sequences, peptide solubilization efficiency and also derivatization [26] when applied. Not all of them can be solved by using, e.g., internal standards. Middle-down MS is less prone to these specific biases, as unmodified and modified N-terminal tails have similar physicochemical properties due to their size. A recent publication also suggests that middle-down MS is at least as good as bottom-up MS in estimating PTM stoichiometry [77]. Therefore, in our opinion investing in middle-down MS for histone analysis is definitely worthwhile, as the system at its state-of-the-art has many more possibilities of applications with similar level of imprecision as bottom-up MS.
6. Five-Years View: Middle-Down MS to Study Chromatin Domains and PTM Functions
For the abovementioned reasons, we speculate that middle-down MS will grow in popularity and potentially replace the bottom-up MS strategy for histone analysis. However, this will happen only when we will be able to match combinatorial PTMs with unambiguous biological functions or chromatin domains. This appears to be not a short term plan, but we are convinced that functional characterization of combinatorial PTMs will be simpler if flanked by middle-down MS screening. In the very next years, middle-down MS will be most likely applied to niche studies, where very few selected PTMs are investigated. For instance, the technique is currently the most suitable to quantify the relative abundance on the chromatin of bivalent domains, i.e. chromatin regions with both active and repressive marks such as H3K4me3K27me3, or active gene enhancers (H3K4me1K27ac). Another application would be to exploit the co-existence frequency of characterized PTMs with uncharacterized ones for predicting the biological role of the latters. For instance, a study of Vandamme et al. characterized H3K23me2 in C. elegans as a new heterochromatic mark by identifying a strong co-existence frequency on histone tails carrying H3K9me3 [78]. Combining middle-down MS with ChIP has also a wide range of applications. For instance, immunoprecipitation of histone readers followed by middle-down MS could reveal whether readers are recruited by specific combinations of PTMs, discriminating transcription factors that are currently thought to be recruited by the same histone modifications. Vermeulen et al. identified readers of H3K4me3 and H3K36me3 and subsequently mapped them genome-wide using ChIP-seq [79]; including middle-down MS of the histones bound to the readers could have revealed whether other PTMs promote this binding on the chromatin. Metabolic labeling is also an interesting approach that could be combined with middle-down MS. By labeling precursors of PTMs it is possible to calculate their turnover rate; it could then be possible to identify co-existing PTMs with the same turnover and predict whether some combinations of modifications are co-catalyzed during specific biological events.
From the technical side, there is still ample room for improvement. We speculate that ion mobility will play a role at one point to separate isobaric forms that cannot be resolved by liquid chromatography. Pioneering studies have been performed using FAIMS with interesting results [80], although this technique is still limited in sensitivity. As well, bioinformatics tools will also expand. In particular, discriminating multiple co-fragmented isobaric forms is an informatics process that would drastically increase the sensitivity of the analysis. Altogether, we are confident that the supply of middle-down MS methodological improvements will improve once the demand grows, and this will happen when laboratories investigating chromatin biology will find advantageous to investigate combinatorial histone PTMs rather than single modifications. Our objective in this review was to provide an overview of the system and its capabilities.
Key Issues.
Middle-down MS for histone proteins is the analysis of intact N-terminal tails; it is a suitable compromise between bottom-up and top-down MS, as it allows for the analysis of the region where most PTMs reside
The middle-down MS workflow optimally performs using WCX-HILIC chromatography and high resolution MS equipped for ETD fragmentation; it is therefore a different setup than traditional bottom-up MS for peptide analysis
Identification and quantification of middle-down MS/MS spectra is currently not optimal with any software tool, although there are a few options available in literature
Middle-down MS of histone tails provides quantification of PTMs including their combinatorial co-frequency. This type of dataset can be overwhelming and of difficult interpretation. However, it is important in light of the potential cross-talk of PTMs on histone proteins
Combinatorial PTM data have the problem of being properly visualized; there is currently no graph that can easily display all co-existing frequencies of multiple PTMs
Middle-down MS is still scarcely exploited in chromatin biology studies, and it has thus a variety of potential applications that could unravel unexplored biological aspects
Acknowledgments
We acknowledge funding from NIH grants R01GM110174 and P01CA196539, and DOD grant W81XWH-113-1-0426. Also, we are grateful to the Leukemia and Lymphoma Society for the Dr. Robert Arceci Scholar Award.
References
Note: all cited literature is of high importance and quality. The publications highlighted below were not chosen based on their overall importance in science, but as milestones for the development of the middle-down MS strategy.
- 1.Xu D, Bai J, Duan Q, Costa M, Dai W. Covalent modifications of histones during mitosis and meiosis. Cell cycle. 2009;8(22):3688–3694. doi: 10.4161/cc.8.22.9908. [DOI] [PubMed] [Google Scholar]
- 2.Weber CM, Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev. 2014;28(7):672–682. doi: 10.1101/gad.238873.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar R, Horikoshi N, Singh M, et al. Chromatin modifications and the DNA damage response to ionizing radiation. Frontiers in oncology. 2012;2:214. doi: 10.3389/fonc.2012.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nature reviews Genetics. 2012;13(5):343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 6.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nature reviews Cancer. 2010;10(7):457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. **White paper introducing the concept of the histone code. [DOI] [PubMed] [Google Scholar]
- 8.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nature structural & molecular biology. 2007;14(11):1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yun MY, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Research. 2011;21(4):564–578. doi: 10.1038/cr.2011.42. **Accurate review describing the known histone readers and their respective domains to date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Developmental Cell. 2003;4(4):497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- 12.Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nature reviews Molecular cell biology. 2017;18(2):90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Current opinion in cell biology. 2003;15(2):172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 14.Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010;142(5):682–685. doi: 10.1016/j.cell.2010.08.011. **Very popular minireview challenging the existence of a histone code. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438(7071):1176–1180. doi: 10.1038/nature04254. **First example of phospho/methyl switch on histones defining boundaries of heterochromatin. [DOI] [PubMed] [Google Scholar]
- 17.Xhemalce B, Kouzarides T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 2010;24(7):647–652. doi: 10.1101/gad.1881710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vermeulen M, Eberl HC, Matarese F, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142(6):967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Y, Garcia BA. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harbor perspectives in biology. 2015;7(9):a025064. doi: 10.1101/cshperspect.a025064. *Poster, and popular image, showing the plethora of known histone PTMs to date. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. H3K36 Methylation Antagonizes PRC2-mediated H3K27 Methylation. Journal of Biological Chemistry. 2011;286(10):7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandra T, Kirschner K, Thuret JY, et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Molecular Cell. 2012;47(2):203–214. doi: 10.1016/j.molcel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shema E, Jones D, Shoresh N, Donohue L, Ram O, Bernstein BE. Single-molecule decoding of combinatorially modified nucleosomes. Science. 2016;352(6286):717–721. doi: 10.1126/science.aad7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egelhofer TA, Minoda A, Klugman S, et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011;18(1):91–93. doi: 10.1038/nsmb.1972. *An eye-opener overview of the quality of antibodies for histone PTM analysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walther TC, Mann M. Mass spectrometry-based proteomics in cell biology. J Cell Biol. 2010;190(4):491–500. doi: 10.1083/jcb.201004052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sidoli S, Cheng L, Jensen ON. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. Journal of proteomics. 2012;75(12):3419–3433. doi: 10.1016/j.jprot.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 26.Sidoli S, Bhanu NV, Karch KR, Wang X, Garcia BA. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. Journal of visualized experiments : JoVE. 2016;(111) doi: 10.3791/54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonaldi T, Imhof A, Regula JT. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics. 2004;4(5):1382–1396. doi: 10.1002/pmic.200300743. *Introduction of histone derivatization for facilitated peptide analysis by trypsin digestion. [DOI] [PubMed] [Google Scholar]
- 28.Garcia BA, Mollah S, Ueberheide BM, et al. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nature protocols. 2007;2(4):933–938. doi: 10.1038/nprot.2007.106. **Protocol describing histone propionylation for bottom-up MS analysis, currently the most widely adopted sample preparation strategy for the analysis of (un)modified histone peptides. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao R, Wu H, Deng H, et al. Specific and efficient N-propionylation of histones with propionic acid N-hydroxysuccinimide ester for histone marks characterization by LC-MS. Analytical chemistry. 2013;85(4):2253–2259. doi: 10.1021/ac303171h. [DOI] [PubMed] [Google Scholar]
- 30.Maile TM, Izrael-Tomasevic A, Cheung T, et al. Mass spectrometric quantification of histone post-translational modifications by a hybrid chemical labeling method. Molecular & cellular proteomics : MCP. 2015;14(4):1148–1158. doi: 10.1074/mcp.O114.046573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelleher NL. Top-down proteomics. Analytical chemistry. 2004;76(11):197A–203A. [PubMed] [Google Scholar]
- 32.Loo JA, Edmonds CG, Smith RD. Primary sequence information from intact proteins by electrospray ionization tandem mass spectrometry. Science. 1990;248(4952):201–204. doi: 10.1126/science.2326633. [DOI] [PubMed] [Google Scholar]
- 33.Zubarev RA, K NL, McLafferty FW. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 34.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(26):9528–9533. doi: 10.1073/pnas.0402700101. **Introduction of ETD for proteomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas CE, Kelleher NL, Mizzen CA. Mass spectrometric characterization of human histone H3: a bird’s eye view. Journal of proteome research. 2006;5(2):240–247. doi: 10.1021/pr050266a. [DOI] [PubMed] [Google Scholar]
- 36.Boyne MT, 2nd, Pesavento JJ, Mizzen CA, Kelleher NL. Precise characterization of human histones in the H2A gene family by top down mass spectrometry. Journal of proteome research. 2006;5(2):248–253. doi: 10.1021/pr050269n. [DOI] [PubMed] [Google Scholar]
- 37.Siuti N, Roth MJ, Mizzen CA, Kelleher NL, Pesavento JJ. Gene-specific characterization of human histone H2B by electron capture dissociation. Journal of proteome research. 2006;5(2):233–239. doi: 10.1021/pr050268v. [DOI] [PubMed] [Google Scholar]
- 38.Pesavento JJ, Mizzen CA, Kelleher NL. Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Analytical chemistry. 2006;78(13):4271–4280. doi: 10.1021/ac0600050. *Introduction of fragment ion relative ratio (FIRR) for the differential quantification of isobarically modified histones. [DOI] [PubMed] [Google Scholar]
- 39.Banks GC, Deterding LJ, Tomer KB, Archer TK. Hormone-mediated dephosphorylation of specific histone H1 isoforms. The Journal of biological chemistry. 2001;276(39):36467–36473. doi: 10.1074/jbc.M104641200. [DOI] [PubMed] [Google Scholar]
- 40.Sarg B, Faserl K, Kremser L, Halfinger B, Sebastiano R, Lindner HH. Comparing and combining capillary electrophoresis electrospray ionization mass spectrometry and nano-liquid chromatography electrospray ionization mass spectrometry for the characterization of post-translationally modified histones. Molecular & cellular proteomics : MCP. 2013;12(9):2640–2656. doi: 10.1074/mcp.M112.024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Onder O, Sidoli S, Carroll M, Garcia BA. Progress in epigenetic histone modification analysis by mass spectrometry for clinical investigations. Expert review of proteomics. 2015;12(5):499–517. doi: 10.1586/14789450.2015.1084231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taverna SD, Ueberheide BM, Liu Y, et al. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(7):2086–2091. doi: 10.1073/pnas.0610993104. **First application of middle-down MS to address a biological question. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mikesh LM, Ueberheide B, Chi A, et al. The utility of ETD mass spectrometry in proteomic analysis. Biochimica et biophysica acta. 2006;1764(12):1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance characteristics of electron transfer dissociation mass spectrometry. Molecular & cellular proteomics : MCP. 2007;6(11):1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. High throughput characterization of combinatorial histone codes. Molecular & cellular proteomics : MCP. 2009;8(10):2266–2284. doi: 10.1074/mcp.M900238-MCP200. **First high-throughput nanoLC-MS/MS setup for the quantification of histone N-terminal tails. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tian Z, Tolic N, Zhao R, et al. Enhanced top-down characterization of histone post-translational modifications. Genome Biol. 2012;13(10):R86. doi: 10.1186/gb-2012-13-10-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DiMaggio PA, Jr, Young NL, Baliban RC, Garcia BA, Floudas CA. A mixed integer linear optimization framework for the identification and quantification of targeted post-translational modifications of highly modified proteins using multiplexed electron transfer dissociation tandem mass spectrometry. Mol Cell Proteomics. 2009;8(11):2527–2543. doi: 10.1074/mcp.M900144-MCP200. *First software attempting the challenging process of quantifying differentially modified histone tails, in particular isobaric forms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sidoli S, Schwammle V, Ruminowicz C, et al. Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics. 2014;14(19):2200–2211. doi: 10.1002/pmic.201400084. [DOI] [PubMed] [Google Scholar]
- 49.LeDuc RD, Taylor GK, Kim YB, et al. ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004;32(Web Server issue):W340–345. doi: 10.1093/nar/gkh447. *Free version and precursor of the software ProSightPC, the first commercial tool for MS/MS identification of intact proteins via top-down MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zamdborg L, LeDuc RD, Glowacz KJ, et al. ProSight PTM 2.0: improved protein identification and characterization for top down mass spectrometry. Nucleic acids research. 2007;35(Web Server issue):W701–706. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karabacak NM, Li L, Tiwari A, et al. Sensitive and specific identification of wild type and variant proteins from 8 to 669 kDa using top-down mass spectrometry. Mol Cell Proteomics. 2009;8(4):846–856. doi: 10.1074/mcp.M800099-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frank AM, Pesavento JJ, Mizzen CA, Kelleher NL, Pevzner PA. Interpreting top-down mass spectra using spectral alignment. Analytical Chemistry. 2008;80(7):2499–2505. doi: 10.1021/ac702324u. [DOI] [PubMed] [Google Scholar]
- 53.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nature protocols. 2007;2(6):1445–1457. doi: 10.1038/nprot.2007.202. **The most adopted protocol for the purification of histones from cells. [DOI] [PubMed] [Google Scholar]
- 54.Sidoli S, Garcia BA. Characterization of Individual Histone Posttranslational Modifications and Their Combinatorial Patterns by Mass Spectrometry-Based Proteomics Strategies. Methods in molecular biology. 2017;1528:121–148. doi: 10.1007/978-1-4939-6630-1_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalli A, Sweredoski MJ, Hess S. Data-dependent middle-down nano-liquid chromatography-electron capture dissociation-tandem mass spectrometry: an application for the analysis of unfractionated histones. Analytical chemistry. 2013;85(7):3501–3507. doi: 10.1021/ac303103b. [DOI] [PubMed] [Google Scholar]
- 56.Liao R, Zheng D, Nie A, et al. Sensitive and Precise Characterization of Combinatorial Histone Modifications by Selective Derivatization Coupled with RPLC-EThcD-MS/MS. Journal of proteome research. 2017 doi: 10.1021/acs.jproteome.6b00788. [DOI] [PubMed] [Google Scholar]
- 57.Sweredoski MJ, Moradian A, Raedle M, Franco C, Hess S. High resolution parallel reaction monitoring with electron transfer dissociation for middle-down proteomics. Analytical chemistry. 2015;87(16):8360–8366. doi: 10.1021/acs.analchem.5b01542. [DOI] [PubMed] [Google Scholar]
- 58.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J Am Soc Mass Spectrom. 2000;11(4):320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 59.Liu X, Inbar Y, Dorrestein PC, et al. Deconvolution and database search of complex tandem mass spectra of intact proteins: a combinatorial approach. Mol Cell Proteomics. 2010;9(12):2772–2782. doi: 10.1074/mcp.M110.002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aggarwal S, Yadav AK. False Discovery Rate Estimation in Proteomics. Methods in molecular biology. 2016;1362:119–128. doi: 10.1007/978-1-4939-3106-4_7. [DOI] [PubMed] [Google Scholar]
- 61.LeDuc RD, Fellers RT, Early BP, Greer JB, Thomas PM, Kelleher NL. The C-score: a Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. Journal of proteome research. 2014;13(7):3231–3240. doi: 10.1021/pr401277r. *Introduction of the C-score, a computational scoring system to define confident identification of intact proteoforms by top-down MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang K, Williams KE, Huang L, et al. Histone acetylation and deacetylation: identification of acetylation and methylation sites of HeLa histone H4 by mass spectrometry. Molecular & cellular proteomics : MCP. 2002;1(7):500–508. doi: 10.1074/mcp.m200031-mcp200. [DOI] [PubMed] [Google Scholar]
- 63.Gonzales-Cope M, Sidoli S, Bhanu NV, Won KJ, Garcia BA. Histone H4 acetylation and the epigenetic reader Brd4 are critical regulators of pluripotency in embryonic stem cells. BMC genomics. 2016;17:95. doi: 10.1186/s12864-016-2414-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jung HR, Sidoli S, Haldbo S, et al. Precision mapping of coexisting modifications in histone H3 tails from embryonic stem cells by ETD-MS/MS. Analytical chemistry. 2013;85(17):8232–8239. doi: 10.1021/ac401299w. [DOI] [PubMed] [Google Scholar]
- 65.Schwammle V, Aspalter CM, Sidoli S, Jensen ON. Large scale analysis of co-existing post-translational modifications in histone tails reveals global fine structure of cross-talk. Molecular & cellular proteomics : MCP. 2014;13(7):1855–1865. doi: 10.1074/mcp.O113.036335. *Introduction of the interplay score to predict cross-talk between histone PTMs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwammle V, Sidoli S, Ruminowicz C, et al. Systems Level Analysis of Histone H3 Post-translational Modifications (PTMs) Reveals Features of PTM Crosstalk in Chromatin Regulation. Molecular & cellular proteomics : MCP. 2016;15(8):2715–2729. doi: 10.1074/mcp.M115.054460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nature genetics. 2008;40(7):897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Voigt P, LeRoy G, Drury WJ, 3rd, et al. Asymmetrically modified nucleosomes. Cell. 2012;151(1):181–193. doi: 10.1016/j.cell.2012.09.002. **Discovery confirming the suspect that the two copies of the same histone variant in the same nucleosome can be differentially modified. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shortreed MR, Frey BL, Scalf M, Knoener RA, Cesnik AJ, Smith LM. Elucidating Proteoform Families from Proteoform Intact-Mass and Lysine-Count Measurements. Journal of proteome research. 2016;15(4):1213–1221. doi: 10.1021/acs.jproteome.5b01090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guan X, Rastogi N, Parthun MR, Freitas MA. Discovery of histone modification crosstalk networks by stable isotope labeling of amino acids in cell culture mass spectrometry (SILAC MS) Molecular & cellular proteomics : MCP. 2013;12(8):2048–2059. doi: 10.1074/mcp.M112.026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H. UpSet: Visualization of Intersecting Sets. IEEE transactions on visualization and computer graphics. 2014;20(12):1983–1992. doi: 10.1109/TVCG.2014.2346248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nardelli SC, Che FY, Silmon de Monerri NC, et al. The histone code of Toxoplasma gondii comprises conserved and unique posttranslational modifications. mBio. 2013;4(6):e00922–00913. doi: 10.1128/mBio.00922-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sidoli S, Vandamme J, Salcini AE, Jensen ON. Dynamic changes of histone H3 marks during Caenorhabditis elegans lifecycle revealed by middle-down proteomics. Proteomics. 2016;16(3):459–464. doi: 10.1002/pmic.201500285. [DOI] [PubMed] [Google Scholar]
- 74.Jamieson K, Wiles ET, McNaught KJ, et al. Loss of HP1 causes depletion of H3K27me3 from facultative heterochromatin and gain of H3K27me2 at constitutive heterochromatin. Genome research. 2016;26(1):97–107. doi: 10.1101/gr.194555.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tvardovskiy A, Wrzesinski K, Sidoli S, Fey SJ, Rogowska-Wrzesinska A, Jensen ON. Top-down and Middle-down Protein Analysis Reveals that Intact and Clipped Human Histones Differ in Post-translational Modification Patterns. Molecular & cellular proteomics : MCP. 2015;14(12):3142–3153. doi: 10.1074/mcp.M115.048975. *Application of the middle-down MS method to study the abundance of histone clipping and PTMs present on clipped histone tails. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zheng Y, Huang X, Kelleher NL. Epiproteomics: quantitative analysis of histone marks and codes by mass spectrometry. Current opinion in chemical biology. 2016;33:142–150. doi: 10.1016/j.cbpa.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sidoli S, Lin S, Karch KR, Garcia BA. Bottom-up and middle-down proteomics have comparable accuracies in defining histone post-translational modification relative abundance and stoichiometry. Analytical chemistry. 2015;87(6):3129–3133. doi: 10.1021/acs.analchem.5b00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vandamme J, Sidoli S, Mariani L, et al. H3K23me2 is a new heterochromatic mark in Caenorhabditis elegans. Nucleic acids research. 2015;43(20):9694–9710. doi: 10.1093/nar/gkv1063. **Application of the middle-down MS method to identify the function of an unknown histone PTM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vermeulen M, Eberl HC, Matarese F, et al. Quantitative Interaction Proteomics and Genome-wide Profiling of Epigenetic Histone Marks and Their Readers. Cell. 2010;142(6):967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 80.Shvartsburg AA, Zheng Y, Smith RD, Kelleher NL. Separation of variant methylated histone tails by differential ion mobility. Analytical chemistry. 2012;84(15):6317–6320. doi: 10.1021/ac301541r. [DOI] [PMC free article] [PubMed] [Google Scholar]