

Abstract
We describe an unusual net 2+2 cycloaddition reaction between boron alkylidenes and unactivated alkenes. This reaction provides a new method for construction of carbocyclic ring systems bearing versatile organoboronic esters. In addition to surveying the scope of this reaction, we provide details about the mechanistic underpinnings of this process, and examine application to the synthesis of the natural product aphanamal.
Keywords: Cyclization, Boron, Total Synthesis, Alkenes, Reaction mechanisms
COMMUNICATION
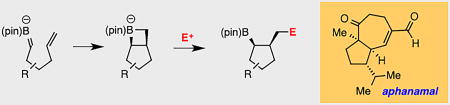
Two plus two, or not two plus two, that is the question: Boron alkylidenes are generated by alkoxide induced deborylation of geminal bis(boronates). These alkene analogs are shown to undergo a net 2+2 cycloaddition reaction with tethered alkenes and the resulting boronate complex can be trapped with electrophiles to generate substituted carbocyclic products. Mechanistic studies and application to synthesis are described.
Geminal bis(boronates) are readily available reagents[1] that engage in an array of functional group transformations under catalytic[1b,g,i,j, 2] and non-catalytic[1e, 3] conditions. In recent studies, it was found that metal-alkoxide-induced deborylative alkylation (eq. 1, Scheme 1) of these compounds can be brought about under mild conditions, even for relatively hindered substrate pairings.[3b] Mechanistic investigations suggested that these reactions proceed through the intermediacy of a boron-stabilized carbanion (i.e. 3), a compound that is generated upon reaction of the bis(boronate) 1 with the metal alkoxide. Spectroscopic study of the boron-stabilized carbanion revealed significant π-bonding between the carbanionic carbon and the three-coordinate boron center indicating that the anion can alternately be represented as boron alkylidene structure 4, a compound related to other[4] boron alkylidenes. To probe the reactivity of the boron alkylidene, we examined deborylative alkyation of bis(boronate) 5, a compound bearing a tethered alkene. In the presence of an alkyl halide electrophile, alkoxideinduced deborylation of 5 furnished alkylation product 6 without involvement of the tethered olefin. In this manuscript, we report that an alternate reactivity mode operates when the electrophile is omitted from deborylation of alkene-containing substrates such as 5: in this circumstance, a deborylative cyclization reaction is observed that is consistent with a net 2+2 cycloaddition between the tethered, unactivated alkene and the boron alkylidene giving 7 (eq. 3).[5] After 14 hours, addition of an electrophile furnishes 8. Overall, this unusual mode of reactivity results in net non-catalyzed carboboration[6] of an unactivated alkene, a process that has some resemblance to hexenyllithium and related cyclization reactions[7], but as far as we are aware, does not find precedent in reactions of stabilized carbanion equivalents. Importantly, this reaction provides a compound with a new carbocyclic ring system and has the capacity to establish three new stereocenters from simple precursor building blocks. Herein, we describe mechanistic and synthetic features of this process and examine the utility of this reaction in the construction of the terpene-derived natural product aphanamal.
Scheme 1.

Studies of the 2+2 reaction in eq. 3 began with the observation that treatment of geminal bis(boronate) 5 with sodium tert-butoxide furnished a small amount (13% yield) of cyclopentane-containing product 9 (Figure 1). Notably, while the cyclization product was formed in low yield, it was furnished as a 13:1 ratio of diastereomers. After optimization (see Supporting Information, Table S1), it was determined that KOt-Bu gave improved yield of the reaction product. It was also found that while the reaction efficiency was higher in toluene solvent at room temperature, when THF was employed as the reaction solvent, an efficient reaction and a higher level of diastereoselectivity was generally observed. Subsequently, we surveyed other substrates and electrophiles. As depicted in Figure 1, when deborylative cyclization of 5 was followed by addition of either D2O, NBS, or I2, electrophilic trap[8] of the reaction intermediate occurred and delivered 10, 11, and 12, respectively. It merits mention that relative to the example with protic work-up, the diastereoselectivity in formation of compounds 10–13 is considerably enhanced (>20:1 dr vs. 4:1 dr). The enhanced diastereoselection in production of 10–13 relative to 9 appears to result from protonolysis of the minor trans diastereomer prior to addition of electrophile such that electrophilic trap of the remaining cis isomer furnishes isomerically-enriched product.[9] As noted in Figure 1, reactions of other substrates show that a tethered styrene can also engage in the reaction (product 15) and that with substitution on the alkene, a third stereocenter can be generated with useful levels of stereocontrol (product 16). Also of note, prospective internally-coordinating Lewis bases such as benzyl ethers (product 14) and silyl ethers (18) are tolerated. Similarly, substrates bearing substituents at the alkylidene center ranging from large aliphatic groups (products 17, 19) to a simple proton (20) provide cyclic products in reasonable yields and selectivity. Lastly, a tethered diene engages in the reaction and furnishes an internal alkene upon protic work-up (22→23).
Figure 1.

Boron alkylidene-alkene 2+2 cycloaddition followed by reaction with an electrophile. After deborylative cyclization, the following electrophiles were employed: H2O (
 ), D2O (
), D2O (
 ), N-bromosuccinimide (
), N-bromosuccinimide (
 , I2 (
, I2 (
 ), benzyl bromide (
), benzyl bromide (
 ).
).
We propose that the deborylative cyclization proceeds through the intermediacy of a 5-borata-[3.2.0]-bicycloheptane "ate" complex 7 (Scheme 1). Structure 7 explains the following observations: (1) a carbanion equivalent (boron "ate" complex) is formed and is stable enough that it persists for the duration of the first step (up to 14 h) such that it may be trapped with an electrophile; (2) only one stereoisomer of cyclization intermediate (syn) appears to be stable and subject to trap with the electrophile, while the other is protonated prior to addition of trapping reagent. To gain evidence for the intermediacy of complexes such 7, the experiments in Figure 2A were conducted. In the first, 13C-labeled compound 24 was subjected to KOtBu in THF-d8 and the reaction followed by proton-decoupled 13C NMR spectroscopy.[10] Over the course of the reaction, the 13C resonance for the starting material (20.1 ppm; broad due to the boron quadrupole) was replaced by a broad resonance at 44.0 ppm (assigned to 25).[11] Upon quench of the reaction mixture with H2O, the signal at 44.0 ppm was replaced with a broad resonance at 38.5 ppm. In a second experiment, reaction of 13C-labeled 26 (δ = 114.0 ppm; sharp singlet) furnished a new structure that exhibits a broad resonance at 26.9 ppm (assigned to 27). Addition of H2O to putative bicycle 27 furnishes a compound with a labeled methyl group resonating at δ = 18.0 ppm. The similar chemical shift and linewidth of the labeled carbons in 25 and 27 is suggestive of a related environment for both carbon atoms, a feature most readily accommodated by the intermediacy of the boracyclic compound.
Figure 2.

Mechanistic investigation of the boron alkylidene/alkene cycloaddition reaction. (a) Isotope labeling studies support the intermediacy of a four-membered boretane. (b) Stereochemical and radical clock experiments suggest that radical intermediates may not be involved. (c) Use of a labeled alkene suggests the 2+2 reaction is not a stereospecific process. (d) The lack of stereospecificity in 2+2 reactions enables stereoconvergent reactions.
To learn about the properties of the boron alkylidene/α-boryl carbanion, the experiments in Figure 2B were conducted. First, it was determined that deborylative alkylation of a non-racemic secondary alkyl bromide (29) occurs stereospecifically, furnishing 30 with inversion of configuration at the electrophilic carbon. Thus, in the context of an alkyl halide substitution reaction, the nature of the α-boryl carbanion is more consistent with a two-electron nucleophile as opposed to a single electron donor that undergoes alkylation by single-electron transfer to the alkyl halide, followed by fragmentation and radical combination.[12] In a second experiment, it was shown that a cyclopropyl group attached to the boron-stabilized carbanion center does not undergo fragmentation during the course of deborylative cyclization (31→32).[13] While these experiments suggest that the boron-stabilized carbanion does not have a proclivity to react by single-electron-transfer pathways, the concerted carbometallation pathway by which hexenyllithium[7a] undergoes cyclization does not appear to operate: cyclization/iodination of 33 (Figure 2C) leads to a 1:1 mixture of deuterium-labeled diastereomers 34 (epimeric at the iodination site). Assuming iodination of the ate complex is stereospecific[8a], this observation suggests that the cycloaddition is a non-concerted step-wise process, proceeding through an intermediate capable of bond rotation prior to closure to the boretane. We considered that a thermally-accessible open shell structure[14] such as 35 – formed by a process analogous to that proposed for dimerization of strained olefins[15] and for alleneyne intramolecular 2+2 reactions[16] – and closed-shell system 36 (see inset, Figure 2C) were most plausible intermediates; however, both raise questions. The radical-based pathway is not supported by experiments in Figure 2B, while addition of a stabilized carbanion to an unactivated alkene appears to have little precedent.[17] Nonetheless, the capacity for stereochemical scrambling at the reactive olefin site extends to other systems: cyclization/alkylation of cis and trans substrates 37 and 21 (Figure 2D) occurs with stereoconvergence – the intermediate "ate" complex is presumed to undergo stereoinvertive SE2 alkylation[8] via 39 – furnishing 38 in excellent yield and stereoselection.
Key steps of the reaction mechanism were probed using density functional theory (DFT). As depicted in Figure 3, minimization of the α-boryl carbanion gives the structure GSSM bearing a planar boron atom and a shortened B–C bond (1.46 Å) indicative of π-bonding. Cyclization of the boron alkylidene gives a non-stablized carbanion (INTcis) by way of transition state TS1CIS (calculated barrier = 20.7 kcal/mol) wherein the terminal carbon of the alkene undergoes pyramidalization as it accommodates a pair of electrons. An important feature of INTcis is that the carbanion center does not yet exhibit bonding with the three-coordinate boron atom and therefore may be subject to bond rotation and stereoinversion, features required for stereoconvergence with styrene derivatives 18 and 39, as well as the stereochemical scrambling observed with deuterium-labeled terminal alkene substrate 35. As a final step in the reaction sequence, internal electrophilic trap of the carbanion in INTcis by the adjacent boron atom occurs with stereoinversion at carbon and furnishes a stable four-membered boracyclic intermediate. Importantly, for the simplified model system used in these calculations, a similar reaction pathway is accessible for production of the trans product and the barrier is comparable to that observed for the syn compound (ΔGrel TS1TRANS = +19.8 kcal/mol, INTTRANS = +9.4 kcal/mol, TS2TRANS = +15.1 kcal/mol); however, the trans boracycle is much less stable (PDTTRANS = +1.2 kcal/mol) and this feature may explain its inability to persist until the end of the reaction period. Overall, the mechanism that appears most consistent with experimental and computational observations is a step-wise 2+2 cycloaddition proceeding through the intermediacy of a carbanionic intermediate.
Figure 3.

DFT Analysis of the boron alkylidene/alkene cycloadddition reaction. Reaction to give the syn stereoisomer of product proceeds through an intermediate carbanion. Values are relative ΔG in kcal/mol at 298 K; calculations performed by density functional theory (DFT; M06-2×/6–31+G* with PCM solvent model (THF).
Considering the prevalence of cyclopentane-containing natural products (>49,000 according to the Reaxys database), it was of interest to study the cycloaddition in construction of natural product targets.[18] In this context, we addressed the natural product aphanamal, a bicyclic terpene isolated from the folk-medicine plant chromolaena laevigata.[19,20] As depicted in Figure 4, when geminal bis(boronate) 40 was subjected to deborylative 2+2 cycloaddition followed by trap with Eschenmoser salt[21], alkyl amine derivative 41 was isolated in good yield and with good levels of stereoinduction. Importantly, the tertiary boronate could be readily converted to methyl ketone 42 by a modified[22] Evans-Zweifel olefination. Subsequent, oxidation of the amine and Hoffmann elimination[23] furnished terminal alkene 43 in good yield. Finally, alkylation of the methyl ketone with methallyl iodide, followed by ring-closing olefin metathesis[24], and allylic oxidation[25] furnished the natural product.
Figure 1.

Stereoselective synthesis of aphanamal.
In conclusion, the addition of a carbanion to an unactivated alkene is an uncommon event in organic chemistry. While the overall transformation of the boron alkylidene bears some similarity to transition metal alkylidene-based processes (i.e. in olefin metathesis)[26], the stepwise mechanism that operates with boron appears to be distinct from the concerted nature of transition metal-based 2+2 reactions and the energetic profile is much different, offering an opportunity to intercept the metallacyclobutane in alternate processes. This feature is likely to find use in construction of complex target structures from simple starting materials.
Supplementary Material
Acknowledgments
We thank Professors William F. Bailey (University of Connecticut) and Lawrence T. Scott (Boston College and University of Nevada, Reno) for helpful discussion, and we thank Dr. Bo Li (Boston College) for x-ray crystallography. This research was supported in part by the U.S. National Institutes of Health, Institute of General Medical Sciences (GM 59417).
References
- 1.a) Matteson DS, Moody RJ. Organometallics. 1982;1:20–29. [Google Scholar]; b) Lee JCH, McDonald R, Hall DG. Nature Chem. 2011;3:894–899. doi: 10.1038/nchem.1150. [DOI] [PubMed] [Google Scholar]; c) Ito H, Kubota K. Org. Lett. 2012;14:890–893. doi: 10.1021/ol203413w. [DOI] [PubMed] [Google Scholar]; d) Li H, Wang L, Zhang Y, Wang J. Angew. Chem. Int. Ed. 2012;51:2943–2946. doi: 10.1002/anie.201108139. [DOI] [PubMed] [Google Scholar]; e) Wommack AJ, Kingsbury JS. Tetrahedron Lett. 2014;55:3163–3166. [Google Scholar]; f) Li H, Shangguan X, Zhang Z, Huang S, Zhang Y, Wang J. Org. Lett. 2014;16:448–451. doi: 10.1021/ol403338s. [DOI] [PubMed] [Google Scholar]; g) Feng X, Jeon H, Yun J. Angew. Chem. Int. Ed. 2013;52:3989–3992. doi: 10.1002/anie.201208610. [DOI] [PubMed] [Google Scholar]; h) Lee S, Li D, Yun J. Chem. Asian J. 2014;9:2440–2443. doi: 10.1002/asia.201402458. [DOI] [PubMed] [Google Scholar]; i) Cho SH, Hartwig JF. Chem. Sci. 2014;5:694–698. [Google Scholar]; j) Zhang Z-Q, Yang C-T, Liang L-J, Xiao B, Lu X, Liu J-H, Sun Y-Y, Marder TB, Fu Y. Org. Lett. 2014;16:6342–6345. doi: 10.1021/ol503111h. [DOI] [PubMed] [Google Scholar]; k) Cuenca AB, Cid J, García-López D, Carbó JJ, Fernández E. Org. Biomol. Chem. 2015;13:9659–9664. doi: 10.1039/c5ob01523e. [DOI] [PubMed] [Google Scholar]; l) Palmer WN, Obligacion JV, Pappas I, Chirik PJ. J. Am. Chem. Soc. 2016;138:766–769. doi: 10.1021/jacs.5b12249. [DOI] [PubMed] [Google Scholar]; m) Zuo Z, Huang Z. Org. Chem. Front. 2016;3:434–438. [Google Scholar]; n) Zhao H, Tong M, Wang H, Xu S. Org. Biomol. Chem. 2017;15:3418–3422. doi: 10.1039/c7ob00654c. [DOI] [PubMed] [Google Scholar]; o) Wang L, Zhang T, Sun W, He Z, Xia C, Lan Y, Liu C. J. Am. Chem. Soc. 2017;139:5257–5264. doi: 10.1021/jacs.7b02518. [DOI] [PubMed] [Google Scholar]
- 2.a) Endo K, Ohkubo T, Hirokami M, Shibata T. J. Am. Chem. Soc. 2010;132:11033–11035. doi: 10.1021/ja105176v. [DOI] [PubMed] [Google Scholar]; b) Endo K, Ohkubo T, Shibata T. Org. Lett. 2011;13:3368–3371. doi: 10.1021/ol201115k. [DOI] [PubMed] [Google Scholar]; c) Endo K, Ishioka T, Ohkubo T, Shibata T. J. Org. Chem. 2012;77:7223–7231. doi: 10.1021/jo3015165. [DOI] [PubMed] [Google Scholar]; d) Endo K, Ohkubo T, Ishioka T, Shibata T. J. Org. Chem. 2012;77:4826–4831. doi: 10.1021/jo3004293. [DOI] [PubMed] [Google Scholar]; e) Lee JCH, Sun H-Y, Hall DG. J. Org. Chem. 2015;80:7134–7143. doi: 10.1021/acs.joc.5b00991. [DOI] [PubMed] [Google Scholar]; f) Sun H-Y, Kubota K, Hall DG. Chem. Eur. J. 2015;21:19186–19194. doi: 10.1002/chem.201406680. [DOI] [PubMed] [Google Scholar]; g) Sun C, Potter B, Morken JP. J. Am. Chem. Soc. 2014;136:6534–6537. doi: 10.1021/ja500029w. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Potter B, Szymaniak AA, Edelstein EK, Morken JP. J. Am. Chem. Soc. 2014;136:17918–17921. doi: 10.1021/ja510266x. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Xu S, Shangguan X, Li H, Zhang Y, Wang J. J. Org. Chem. 2015;80:7779–7784. doi: 10.1021/acs.joc.5b01100. [DOI] [PubMed] [Google Scholar]; j) Joannou MV, Moyer BS, Meek SJ. J. Am. Chem. Soc. 2015;137:6176–6179. doi: 10.1021/jacs.5b03477. [DOI] [PubMed] [Google Scholar]; k) Joannou MV, Moyer BS, Goldfogel MJ, Meek SJ. Angew. Chem. Int. Ed. 2015;54:14141–14145. doi: 10.1002/anie.201507171. [DOI] [PubMed] [Google Scholar]; l) Murray SA, Green JC, Tailor SB, Meek SJ. Angew. Chem. Int. Ed. 2016;55:9065–9069. doi: 10.1002/anie.201603465. [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Shi Y, Hoveyda AH. Angew. Chem. Int. Ed. 2016;55:3455–3458. doi: 10.1002/anie.201600309. [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Kim J, Park S, Park J, Cho SH. Angew. Chem. Int. Ed. 2016;55:1498–1501. doi: 10.1002/anie.201509840. [DOI] [PubMed] [Google Scholar]; o) Park J, Lee Y, Kim J, Cho SH. Org. Lett. 2016;18:1210–1213. doi: 10.1021/acs.orglett.6b00376. [DOI] [PubMed] [Google Scholar]; p) Kim J, Park S, Park J, Cho SH. Angew. Chem. int. Ed. 2016;55:1498–1501. doi: 10.1002/anie.201509840. [DOI] [PubMed] [Google Scholar]; q) Cui L-C, Zhang Z-Q, Lu X, Xiao B, Fu Y. RSC Adv. 2016;6:51932–51935. [Google Scholar]; r) Ebrahim-Alkhalil A, Zhang Z-Q, Gong T-J, Su W, Lu X-Y, Xiao B, Fu Y. Chem. Commun. 2016;52:4891–4893. doi: 10.1039/c5cc09817c. [DOI] [PubMed] [Google Scholar]; s) Li F, Zhang Z-Q, Lu X, Xiao B, Fu Y. Chem. Commun. 2017;53:3551–3554. doi: 10.1039/c7cc00129k. [DOI] [PubMed] [Google Scholar]; t) Zhang Z-Q, Zhang B, Lu X, Liu J-H, Lu X-Y, Xiao B, Fu Y. Org. Lett. 2016;18:952–955. doi: 10.1021/acs.orglett.5b03692. [DOI] [PubMed] [Google Scholar]; u) Zhan M, Li R-Z, Mou Z-D, Cao C-G, Liu J, Chen Y-W, Niu D. ACS Catal. 2016;6:3381–3386. [Google Scholar]
- 3.a) Endo K, Hirokami M, Shibata T. J. Org. Chem. 2010;75:3469–3472. doi: 10.1021/jo1003407. [DOI] [PubMed] [Google Scholar]; b) Hong K, Liu X, Morken JP. J. Am. Chem. Soc. 2014;136:10581–10584. doi: 10.1021/ja505455z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Coombs JR, Zhang L, Morken JP. Org. Lett. 2015;17:1708–1711. doi: 10.1021/acs.orglett.5b00480. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jo W, Kim J, Choi S, Cho SH. Angew. Chem. Int. Ed. 2016;55:9690–9694. doi: 10.1002/anie.201603329. [DOI] [PubMed] [Google Scholar]; e) Blair DJ, Tanini D, Bateman JM, Scott HK, Myers EL, Aggarwal VK. Chem. Sci. 2017:2898–2903. doi: 10.1039/c6sc05338f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Olmstead MM, Power PP, Weese KJ, Doedens RJ. J. Am. Chem. Soc. 1987;109:2541–2542. [Google Scholar]; b) Pelter A, Singaram B, Warren L, Wilson JW. Tetrahedron. 1993;49:2965–2978. [Google Scholar]; c) Kawashima T, Yamashita N, Okazaki R. J. Am. Chem. Soc. 1995;117:6142–6143. [Google Scholar]; d) Cooke MP. J. Org. Chem. 1994;59:2930–2031. [Google Scholar]
- 5.For important precedents in B-element addition across π-systems, see: Hirner JJ, Faizi DJ, Blum SA. J. Am. Chem. Soc. 2014;136:4740–4745. doi: 10.1021/ja500463p.Chong E, Blum SA. J. Am. Chem. Soc. 2015;137:10144–10147. doi: 10.1021/jacs.5b06678.Suginome M, Shirakura M, Yamamoto A. J. Am. Chem. Soc. 2006;128:14438–14439. doi: 10.1021/ja064970j.Semba K, Nakao Y. J. Am. Chem. Soc. 2014;136:7567–7570. doi: 10.1021/ja5029556.Smith KB, Logan KM, You W, Brown MK. Chem. Eur. J. 2014;20:12032–12036. doi: 10.1002/chem.201404310.
- 6.For recent uncatalyzed alkene carboboration, see: Sanzone JR, R J, Hu C, Woerpel KA. J. Am. Chem. Soc. doi: 10.1021/jacs.7b03986. Just Accepted.
- 7.a) Bailey WF, Gavaskar KV. Tetrahedron. 1994;50:5957–5970. [Google Scholar]; b) Richey HG, Rees TC. Tetrahedron Lett. 1966;36:4297–4301. [Google Scholar]; c) St Denis J, Dolzine T, Oliver JP. J. Am. Chem. Soc. 1972;94:8260–8261. [Google Scholar]
- 8.For electrophilic SE2 trap of alkyl boronate complexes, see: Larouche-Gauthier R, Elford TG, Aggarwal VK. J. Am. Chem. Soc. 2011;133:16794–16797. doi: 10.1021/ja2077813.Sanford C, Rasappan R, Aggarwal VK. J. Am. Chem. Soc. 2015;137:10100–10103. doi: 10.1021/jacs.5b05848.Mohiti M, Rampalakos C, Feeney K, Leonori D, Aggarwal VK. Chem. Sci. 2014;5:602–607.
- 9.Although we have yet to determine the source of the hydrogen atom that traps the minor diastereomer (use of d8-THF and d9-KOtBu do not give deuterated products), protonolysis compound epi-9 is always generated in the reaction regardless of electrophile employed.
- 10.Analysis of reactions by 11B NMR are complicated by boron-containing products of the deborylation step.
- 11.For comprehensive overview of NMR spectroscopy of organoboron compounds, see: Wrackmeyer B. Ann. Rep. NMR Spectrosc. 1988;20:61–203.
- 12.a) Ashby EC. Accts. Chem. Res. 1988;21:414–421. [Google Scholar]; b) Rossi RA, Pierini AB, Peñéñory AB. Chem Rev. 2003;103:71–167. doi: 10.1021/cr960134o. [DOI] [PubMed] [Google Scholar]
- 13.For an overveiw of cyclopropylcarbinyl radical reactions, see: Nonhebel D. Chem. Soc. Rev. 1993:347–359.
- 14.Perrotta RF, Winter AH, Coldren WH, Falvey DE. J. Am. Chem. Soc. 2011;133:15553–15558. doi: 10.1021/ja204711a. [DOI] [PubMed] [Google Scholar]
- 15.Lukin K, Eaton PE. J. Am .Chem. Soc. 1995;117:7652–7656. and references cited therein. [Google Scholar]
- 16.Siebert MR, Osbourn JM, Brummond KM, Tantillo DJ. J. Am. Chem. Soc. 2010;132:11952–11966. doi: 10.1021/ja102848z. [DOI] [PubMed] [Google Scholar]
- 17.In contrast to the intramolecular reaction of non-stabilized carbanions (ref. 7), we are unaware of non-catalyzed addition of stabilized anions (enolates, α–metallonitriles, etc.) to unactivated alkenes.
- 18.Kurteva VB, Afonso CAM. Chem. Rev. 2009;109:6809–6857. doi: 10.1021/cr900169j. [DOI] [PubMed] [Google Scholar]
- 19.Misra LN, Jakupovic J, Bohlmann F, Schmeda-Hirschmann G. Tetrahedron. 1985;41:5353–5356. [Google Scholar]
- 20.Previous syntheses of aphanamal/aphanamol, see: Mehta G, Krishnamurthy N, Karra SR. J. Chem. Soc. Chem. Commun. 1989:1299–1300.Hansson T, Wickberg B. J. Org. Chem. 1992;57:5370–5376.Harmata M, Carter KW. Tetrahedron Lett. 1997;38:7985–7988.Wender PA, Zhang L. Org. Lett. 2000;2:2323–2326. doi: 10.1021/ol006085q.Ferrara SJ, Burton JW. Chem. Eur. J. 2016;22:11597–11600. doi: 10.1002/chem.201602669.
- 21.Eschenmoser A, Schreiber J, Maag H, Hashimoto N. Angew. Chem. Int. Ed. 1971;10:330–331. [Google Scholar]
- 22.Aggarwal VK, Sonawane RP, Jheengut V, Rabalakos C, Larouche-Gauthier R, Scott HK. Angew. Chem. Int. Ed. 2011;50:3760–3763. doi: 10.1002/anie.201008067. [DOI] [PubMed] [Google Scholar]
- 23.von Hofmann AW. Annalen der Chemie und Pharmacie. 1851;78:253–286. [Google Scholar]
- 24.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168–8179. [Google Scholar]
- 25.Hayashi K, Nakamura H, Mitsuhashi H. Chem. Pharm. Bull. 1973;21:2806–2807. [Google Scholar]
- 26.Grubbs RH, Wenzel AG, editors. Handbook of Olefin Metathesis. 2. Vol. 1. Wiley-VCH; Weinhem: 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.