

SUMMARY
During antibacterial autophagy, ubiquitination of intracellular bacteria recruits proteins that mediate bacterial delivery to the lysosome for degradation. Smurf1 is an E3 ubiquitin-ligase whose role in selective bacterial autophagy is unknown. We show that Smurf1 facilitates selective autophagy of the human pathogen Mycobacterium tuberculosis (Mtb). Smurf1−/− macrophages are defective in recruiting polyubiquitin, the proteasome, the ubiquitin-binding autophagy adaptor NBR1, the autophagy protein LC3, and the lysosomal marker LAMP1 to Mtb-associated structures, and are more permissive for Mtb growth. This function of Smurf1 requires both its ubiquitin-ligase and C2 phospholipid-binding domains, and involves K48-rather than K63-linked ubiquitination. Chronically infected Smurf1−/− mice have increased bacterial load, increased lung inflammation, and accelerated mortality. SMURF1 controls Mtb replication in human macrophages and associates with bacteria in lungs of patients with pulmonary tuberculosis. Thus, Smurf1 is required for selective autophagy of Mtb and host defense against tuberculosis infection.
Keywords: M. tuberculosis, selective autophagy, Smurf1, K48 ubiquitin
eTOC Blurb
Franco et al. describe a role for the ubiquitin ligase Smurf1 in selective autophagy of intracellular bacteria including M. tuberculosis (Mtb) and L. monocytogenes. Smurf1 mediates K48-ubiquitination of Mtb and recruitment of the proteasome and autophagy machinery components. Smurf1 helps restrict Mtb replication in macrophages and in mice.
INTRODUCTION
Autophagy is a conserved catabolic process that recycles intracellular components to provide nutrients during starvation and maintain organelle and protein quality control (Levine and Kroemer, 2008). In addition to cellular homeostasis, autophagy functions in the elimination of intracellular pathogens such as viruses, parasites and bacteria (Levine et al., 2011). Autophagic elimination of intracellular bacteria is largely dependent on targeting bacteria (or cellular membranes associated with the bacteria) with ubiquitin chains. Ubiquitin-binding autophagy adaptors, such as p62, NDP52, NBR1 and optineurin (OPTN) are then recruited to the ubiquitin-associated bacteria and bind to the autophagosomal membrane-associated protein, LC3, resulting in the delivery of intracellular bacteria to lysosomes for degradation (Gomes and Dikic, 2014).
Mycobacterium tuberculosis (Mtb) is a human pathogen responsible for several million deaths annually. Autophagic elimination of mycobacteria plays an important role in host defense in vitro and IFN-γ or starvation-induced autophagy in macrophages accelerates maturation of mycobacteria-containing phagosomes and increases lysosome-mediated bacterial killing (Castillo et al., 2012; Gutierrez et al., 2004; Manzanillo et al., 2013; Watson et al., 2012). However, recent studies have challenged the role of autophagy in host defense against Mtb in vivo. Mice with granulocyte- and macrophage-specific deletion of Atg5, a protein required for autophagosome formation, have markedly enhanced susceptibility to tuberculosis (Castillo et al., 2012; Watson et al., 2012), but the heightened susceptibility of these mice was recently shown to be due to a hyperinflammatory syndrome (Kimmey et al., 2015). Furthermore, loss of other core autophagy genes in the mouse germline (Ulk1, Ulk2, Atg4b) or via conditional deletion in granulocytes and macrophages (Atg14l, Atg12, Atg16l, Atg7, Atg3) does not affect susceptibility to acute tuberculosis infection (Kimmey et al., 2015). Thus, it is unclear what roles bacterial autophagy play in the pathogenesis of tuberculosis infection.
Ubiquitination represents an important step in antibacterial autophagy. Parkin, an E3 ubiquitin ligase, not only mediates autophagic elimination of damaged mitochondria (Winklhofer, 2014) but also catalyzes the binding of K63- but not K48-ubiquitin chains to Mtb-associated structures and promotes autophagy-mediated host resistance to tuberculosis and listeriosis (Manzanillo et al., 2013). DRAM1 (DNA damage-regulated autophagy modulator 1) recruits p62 to M. marinum-containing vesicles and promotes resistance to infection in zebrafish (van der Vaart et al., 2014). UBQLN1, a member of a protein family that contains a ubiquitin-like domain, a ubiquitin-associated domain, and STI1 motifs, recruits ubiquitin, p62 and LC3 to Mtb-containing vacuoles and restricts bacterial replication (Sakowski et al., 2015). It is likely that other as-of-yet-determined factors also contribute to ubiquitin-dependent autophagic elimination of Mtb.
Our previous genome-wide siRNA screen identified 96 genes that were essential for selective autophagy of viruses and damaged mitochondria (Orvedahl et al., 2011). Murine embryonic fibroblasts (MEFs) deficient for one of these genes, Smurf1 (SMAD-specific E3 ubiquitin-ligase protein 1) were defective in targeting Sindbis and herpes simplex viruses to autophagosomes, but demonstrated a normal autophagy response to starvation. Additionally, Smurf1-deficient MEFs infected with Sindbis virus accumulated viral nucleocapsids, suggesting a possible role for Smurf1 in immune defense by targeting viruses to the autophagy (Orvedahl et al., 2011).
Smurf1 is an E3 ubiquitin ligase that catalyzes the ubiquitination of substrates for subsequent proteasomal degradation. Smurf1 targets for degradation several cytoplasmic proteins, including Smad1/5, RhoA, TGF-βR, MEKK2, Prickle1, JunB, STAT1 and MyD88, thereby regulating embryonic development, cell polarity, adhesion, bone homeostasis and immune responses (Cao and Zhang, 2013). Because Smurf1 is essential in virophagy and mitophagy and also ubiquitinates several targets, we hypothesized that it may be required for Mtb ubiquitination and selective bacterial autophagy.
Our results indicate an essential role for Smurf1 in targeting Mtb for autophagic degradation in macrophages via a mechanism involving K48-linked ubiquitination.
RESULTS
Smurf1 Functions in Selective Autophagy of M. tuberculosis and the Control of M. tuberculosis Replication
To evaluate whether Smurf1 is involved in bacterial autophagy, we infected bone marrow-derived macrophages (BMDMs) from wild-type or Smurf1−/− mice (Yamashita et al., 2005) crossed to mice that express GFP-LC3 (a fluorescent marker of autophagomes) (Mizushima et al., 2004) with mCherry-expressing Mtb (Collins et al., 2015) and evaluated the colocalization of mCherry-Mtb with GFP-LC3. Compared to BMDMs from wild-type littermate controls, Smurf1−/− BMDMs were defective in GFP-LC3 recruitment to Mtb-associated structures (Figure 1A and 1B). (We use the term Mtb-associated structures as it is unknown whether the GFP-LC3 is in direct association with Mtb or host organelles that contain Mtb). Similar results were observed upon staining endogenous LC3 in wild-type and Smurf1−/− BMDMs derived from mice without the GFP-LC3 transgene (Figure S1). Thus, there is a partial defect LC3 recruitment to Mtb-associated structures in murine Smurf1−/− BMDMs.
Figure 1. Smurf1 Functions in Selective Autophagy of M. tuberculosis and the Control of M. tuberculosis Replication.
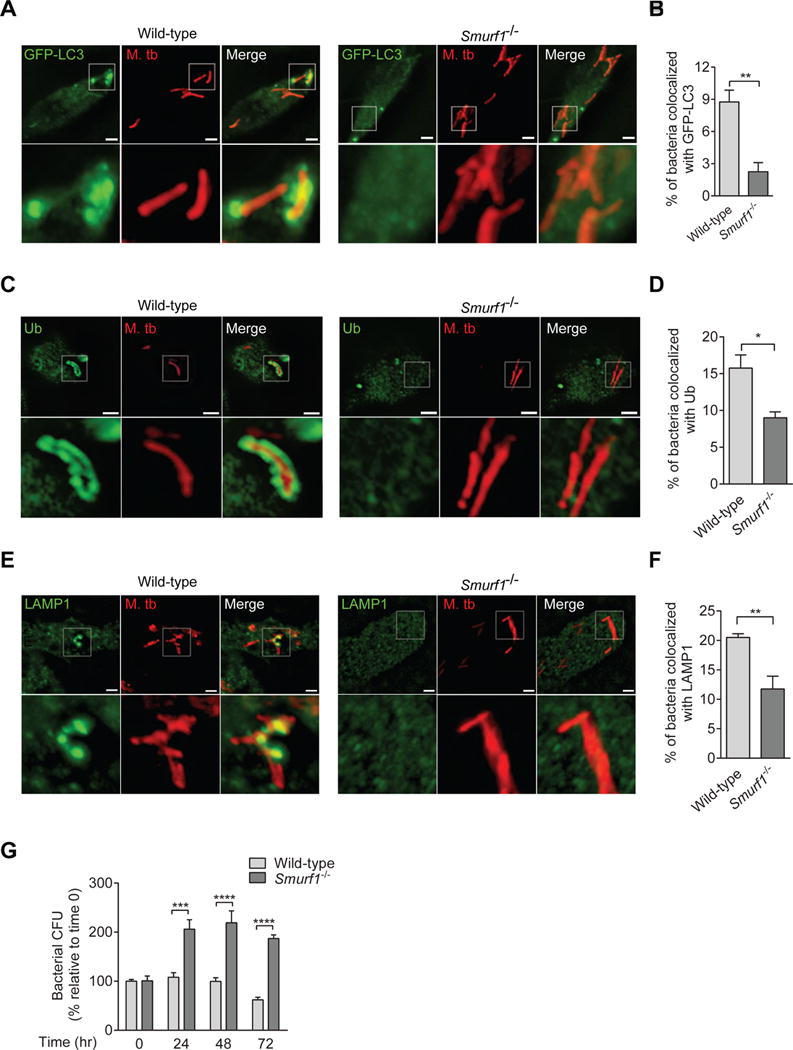
(A–F) Photomicrographs (A, C, E) and quantitation (B, D, F) of the colocalization of mCherry-Mtb and GFP-LC3 (A, B), polyubiquitin (C, D) or LAMP1 (E, F) 15 hr after infection of BMDMs from wild-type or Smurf1−/− mice. Insets show representative mycobacteria that would be considered colocalized with respective markers in wild-type BMDMs or not colocalized in Smurf1−/− BMDMs. Scale bars, 3 μm. See also Figure S1.
(G) Mycobacterial growth in wild-type or Smurf1−/− BMDMs infected with Mtb. Infected cells were lysed at the indicated time-points and mycobacterial growth was determined by counting colony-forming-units (CFUs). Bars are mean ± SEM of quadruplicate samples; each sample was normalized to day 0. Similar results were observed in three independent experiments. ***P<0.01; ****P<0.0001 for indicated comparison; two-way ANOVA.
For B, D and F, bars are mean ± SEM of quadruplicate samples (100 bacteria evaluated per sample per genotype) from a representative experiment. Similar results were observed in at least three independent experiments. *P<0.05, **P<0.01, for indicated comparison; t-test.
Next, we evaluated whether Smurf1 is involved in the recruitment of polyubiquitin, a signal necessary for mycobacterial autophagy (Watson et al., 2012), to Mtb. Compared to wild-type BMDMs, Smurf1−/− BMDMs showed a significant reduction in polyubiquitin/Mtb colocalization (Figure 1C and 1D). To investigate if the defective recruitment of polyubiquitin and LC3 to Mtb-associated structures in Smurf1−/− BMDMs impairs subsequent Mtb association with lysosomes, we evaluated Mtb colocalization with the lysosomal marker LAMP1. Smurf1−/− BMDMs were defective in trafficking Mtb to LAMP1-positive organelles (Figures 1E and 1F). The defect in LC3 recruitment, polyubiquitination, and LAMP1 colocalization in Smurf1−/− BMDMS was associated with a significant increase in Mtb bacterial replication at 24, 48 and 72 hr after infection (Figure 1G). Thus, Mtb delivery to lysosomes mediated by Smurf1 restricts Mtb replication in murine macrophages.
Tat-Beclin 1 Peptide Enhances Autophagic Targeting and Control of M. tuberculosis Replication in a Smurf1-Dependent Manner
To investigate whether Smurf1 mediates the antimycobacterial effects of autophagy-inducing agents, we evaluated the effects of the autophagy-inducing peptide, Tat-Beclin 1, on Mtb/GFP-LC3 colocalization and Mtb replication in wild-type and Smurf1−/− BMDMs. We used a cell-penetrating peptide, Tat-Beclin 1, derived from a short sequence of the autophagy protein Beclin 1 that induces autophagy in vitro and in vivo, and inhibits the replication of several viruses (West Nile, chikungunya, HIV) and the intracellular bacterium, Listeria monocytogenes (Shoji-Kawata et al., 2013). We used a modified, more active version of the original Tat-Beclin 1, called Tat-Beclin 1-L11, which consists of 11 rather than 18 amino acids of Beclin 1 sequence (Pietrocola et al., 2016). As a control, we used a mutant peptide with a Phe to Ser mutation at position 2 of the Beclin 1 sequence (hereafter called control peptide) that lacks autophagy-inducing activity.
Tat-Beclin 1-L11 increased Mtb/GFP-LC3 colocalization in wild-type, but not Smurf1−/− BMDMs (Figure 2A). This was not due to impaired Tat-Beclin 1-L11-induced general autophagy, as Tat-Beclin 1-L11 increased GFP-LC3 puncta numbers, (Figure 2B and 2C), increased conversion of LC3-I to the autophagosome-associated lipidated form, LC3-II, and decreased levels of the autophagy substrate, p62, (Figure 2D) similarly in wild-type and Smurf1−/− BMDMs. In all conditions, the numbers of GFP-LC3 puncta increased further in the presence of the lysosomal inhibitor, bafilomycin A1 (Baf A1), indicating a complete autophagic response (i.e. enhanced autophagic flux). In addition, Tat-Beclin 1-L11 decreased Mtb replication in wild-type but not Smurf1−/− BMDMs (Figure 2E). Taken together, our data suggest that Smurf1 plays a role in the antimycobacterial effects of Tat-Beclin 1-L11 by selectively targeting Mtb for autophagolysosomal degradation.
Figure 2. Tat-Beclin 1-L11 Peptide Enhances M. tuberculosis/GFP-LC3 Colocalization and Decreases M. tuberculosis Replication in a Smurf1-Dependent Manner.
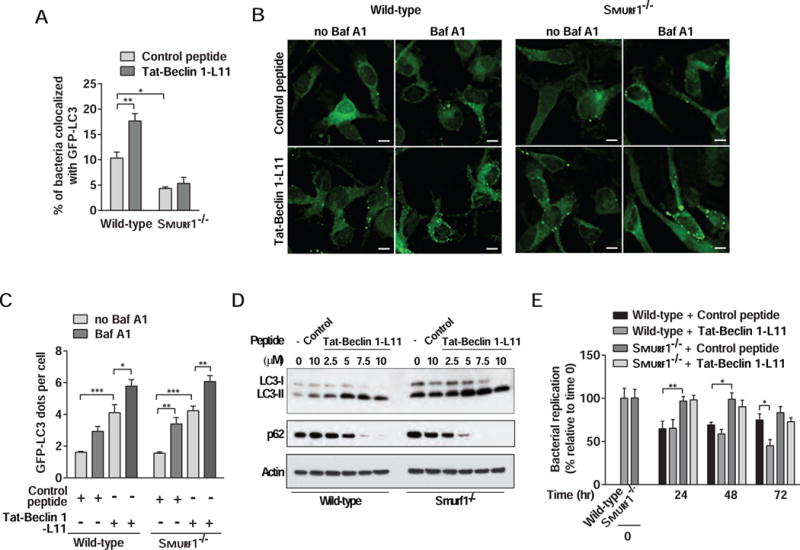
(A) Quantitation of the colocalization of mCherry-Mtb with GFP-LC3 in wild-type or Smurf1−/− BMDMs infected with Mtb and treated with inactive control (Tat-Beclin 1 F270S) or autophagy-inducing active Tat-Beclin 1-L11 peptide for 15 hr. Bars are mean ± SEM of quadruplicate samples (100 bacteria evaluated per sample) from a representative experiment.
(B and C) Representative images (B) and quantitation (C) of the GFP-LC3 dots in GFP-LC3 wild-type or Smurf1−/− BMDMs treated with Tat-Beclin 1 F270S or Tat-Beclin 1-L11 peptide for 6 hr. Bafilomycin A1 (Baf A1) (100 nM) was added during the last 3 hr of treatment. Scale bars, 5 μm. Bars are mean ± SEM of triplicate samples (100 cells evaluated per sample).
(D) Western blot analyses to detect autophagy induction (LC3-I to LC3-II conversion and p62 degradation) in BMDMs from wild-type or Smurf1−/− mice treated with Tat-Beclin 1-L11 peptide or Tat-Beclin 1 F270S for 2 hr.
(E) Mtb growth in wild-type or Smurf1−/− BMDMs treated with Tat-Beclin 1 F270S or autophagy-Tat-Beclin 1-L11 peptide. Infected cells were lysed at the indicated time-points and Mtb growth was determined by counting CFUs. Bars are mean ± SEM of quadruplicate samples; each sample was normalized to day 0.
For A, C and E, similar results were observed in at least three independent experiments. *P<0.05, **P<0.01, ***P<0.001; for indicated comparison; two-way ANOVA.
The C2 Domain and Ubiquitin Ligase Activity of Smurf1 are Required for M. tuberculosis Autophagy
Smurf1 is composed of an N-terminal C2 domain involved in membrane binding, two central WW domains involved in protein-protein interactions, and a C-terminal HECT (homologous to the E6-AP carboxyl terminus) ubiquitin-ligase domain (Figure 3A) (Cao and Zhang, 2013). To determine which domains of Smurf1 are required for bacterial autophagy, we expressed in Smurf1−/− murine BMDMs Flag-tagged versions of human wild-type SMURF1 or SMURF1 mutants, including SMURF1 C699A which has a loss-of ubiquitin-ligase function mutation in the HECT domain (Cheng et al., 2011; Yuan et al., 2012) or SMURF1ΔC2, which is deleted of amino acids 1–119 and is defective in membrane localization but not ubiquitin ligase activity (Suzuki et al., 2002).
Figure 3. Smurf1-Dependent Selective Autophagy of M. tuberculosis Requires the Smurf1 C2 domain and its Ubiquitin Ligase Activity.
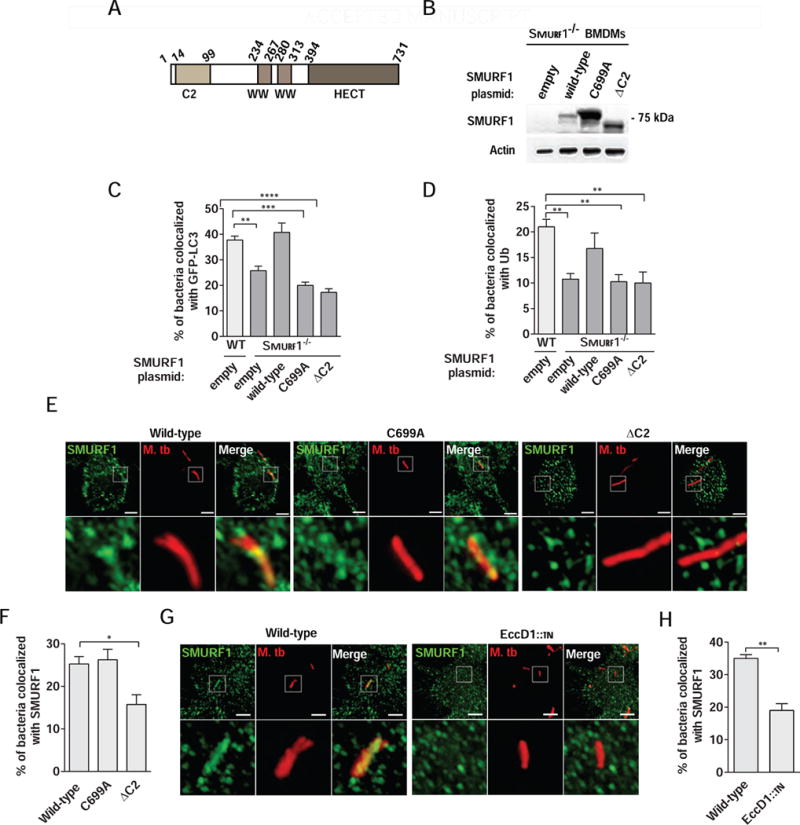
(A) Schematic representation of human SMURF1.
(B) Western blot of lysates from Smurf1−/− BMDMs transduced with lentivirus encoding Flag epitope-tagged wild-type SMURF1, SMURF1 C699A, SMURF1ΔC2, or empty plasmid detected with an anti-FLAG antibody.
(C and D) Rescue of mycobacterial colocalization with GFP-LC3 (C) or polyubiquitin (D) by wild-type but not mutant forms of SMURF1 in Smurf1−/− BMDMs. Wild-type BMDMs expressing GFP-LC3 (C) were transduced with empty lentivirus and Smurf1−/− BMDMs expressing GFP-LC3 (C) were transduced as described in (B), and infected with mCherry-expressing Mtb for 15 hr. For polyubiquitin staining (D), wild-type and Smurf1−/− BMDMs lacking GFP-LC3 were transduced and infected as above prior to immunostaining with anti-polyubiquitin antibody.
(E and F) Photomicrographs (E) and quantitation (F) of the colocalization of mCherry-Mtb and SMURF1 15 hr after infection of RAW 264.7 transduced with lentivirus encoding Flag epitope-tagged wild-type SMURF1, SMURF1 C699A or SMURF1ΔC2. Insets (lower panels) in (E) show representative mycobacteria that would be considered colocalized in wild-type or SMURF1 C699A expressing cells, or not colocalized in SMURF1ΔC2 expressing cells. Scale bars, 3 μm.
(G and H) Photomicrographs (G) and quantitation (H) of the colocalization of mCherry-Mtb wild-type or EccD1∷tn and SMURF1 15 hr after infection of RAW 264.7 cells. Insets (lower panels) in (G) show representative wild-type Mtb which would be considered colocalized with SMURF1 and representative Mtb EccD1∷tn which would not be considered colocalized with SMURF1. Scale bars, 3 μm. **P<0.01 for indicated comparison; t-test.
For C, D, F, and H, bars are mean + SEM for quadruplicate samples (100 bacteria analyzed per sample) from a representative experiment. Similar results were observed in at least three independent experiments. *P<0.05; **P< 0.01; ***P<0.001; ****P<0.0001; one-way ANOVA.
We compared the effects of wild-type SMURF1, SMURF1 C699A, and SMURF1ΔC2 on polyubiquitin/Mtb and GFP-LC3/Mtb colocalization. SMURF1 undergoes autoubiquitination and rapid degradation by the proteasome, and the SMURF1 C699A mutation, which abolishes its E3 ligase-activity, thereby stabilizes the protein (Wang et al., 2003). Thus, as expected, SMURF1 C699A expression levels in BMDMs were increased as compared to wild-type SMURF1 (Figure 3B). Lentivirus transduction increased GFP-LC3/Mtb and polyubiquitin/Mtb colocalization in all groups compared to non-transduced cells, presumably due to innate sensing of lentivirus infection by cGAS (Gao et al., 2013), with subsequent cGAS-mediated autophagy induction (Liang et al., 2014). As expected, Smurf1−/− BMDMs transduced with empty vector were defective in GFP-LC3/Mtb and polyubiquitin/Mtb colocalization compared to wild-type BMDMs (Figure 3C and 3D). Expression of wild-type SMURF1 restored GFP-LC3/Mtb and polyubiquitin/Mtb colocalization to levels similar to those observed in wild-type BMDMs transduced with control vector. However, neither SMURF1 C699A nor SMURF1ΔC2 was able to rescue GFP-LC3/Mtb (Figure 3C) or polyubiquitin/Mtb colocalization (Figure 3D). Thus, Smurf1 mediates selective autophagy of Mtb through a mechanism dependent on both its C2 domain and ubiquitin ligase activity.
Because the Smurf1 C2 domain is required to mediate Smurf1 interaction with membrane phospholipids (Lu et al., 2011; Suzuki et al., 2002), we hypothesized that the C2 domain targets Smurf1 to Mtb-associated structures. We expressed Flag-tagged wild-type SMURF1 or SMURF1 C699A or SMURF1ΔC2 mutants in RAW 264.7 murine macrophage cells and evaluated colocalization with Mtb. Both wild-type and the SMURF1 C699A mutant colocalized similarly with Mtb-associated structures, while colocalization of SMURF1ΔC2 was reduced (Figure 3E and 3F), suggesting that the Smurf1 C2 domain is required for proper localization of Smurf1 to Mtb-associated structures.
The Mtb ESX-1 protein secretion system was previously shown to activate selective autophagy and recruit autophagy factors via membrane damage and phagosomal permeabilization (Watson et al., 2012). To determine if recruitment of Smurf1 to Mtb-associated structures is dependent on ESX-1 activity, we infected RAW 264.7 cells expressing wild-type SMURF1 with either wild-type Mtb or Mtb EccD1∷tn, an ESX-1 mutant strain (Stanley et al., 2003). Recruitment of Smurf1 to Mtb-associated structures was reduced when cells were infected with Mtb EccD1∷tn (Figure 3G and 3H). These data suggest that phagosomal permeabilization is required to recruit Smurf1 to Mtb-associated structures and via the Smurf1 C2 domain.
Smurf1 Mediates K48-Linked Polyubiquitination of M. tuberculosis and L. monocytogenes
Previous reports have shown that Mtb is surrounded by both K63- and K48-linked polyubiquitin chains within macrophages and that Parkin mediates K63-, but not K48-linked ubiquitination (Manzanillo et al., 2013). As Smurf1 induces K48-linked ubiquitination of STAT1 for subsequent degradation (Yuan et al., 2012), we evaluated if Smurf1 mediates K48-linked ubiquitination of Mtb-associated structures. We infected wild-type and Smurf1−/− BMDMs with Mtb and quantified K63- and K48-linked ubiquitin colocalization with Mtb-associated structures. Colocalization of Mtb with K63-linked ubiquitin was similar between wild-type and Smurf1−/− BMDMs (Figure 4A and 4B). In contrast, there was a marked decrease in K48-linked ubiquitin/Mtb-associated structure colocalization in Smurf1−/− BMDMs (Figure 4A and 4B). This defect in K48-linked ubiquitin colocalization with Mtb in Smurf1−/− BMDMs was fully rescued by transduction with a lentivirus vector expressing wild-type but not the C699A or ΔC2 SMURF1 mutants (Figure 4C). Thus, Smurf1 mediates K48-linked ubiquitination of Mtb through a mechanism reliant on both its ubiquitin ligase activity and its C2 domain.
Figure 4. Smurf1 Mediates K48-Linked, But Not K63-Linked, Polyubiquitination of M. tuberculosis and L. monocytogenes.
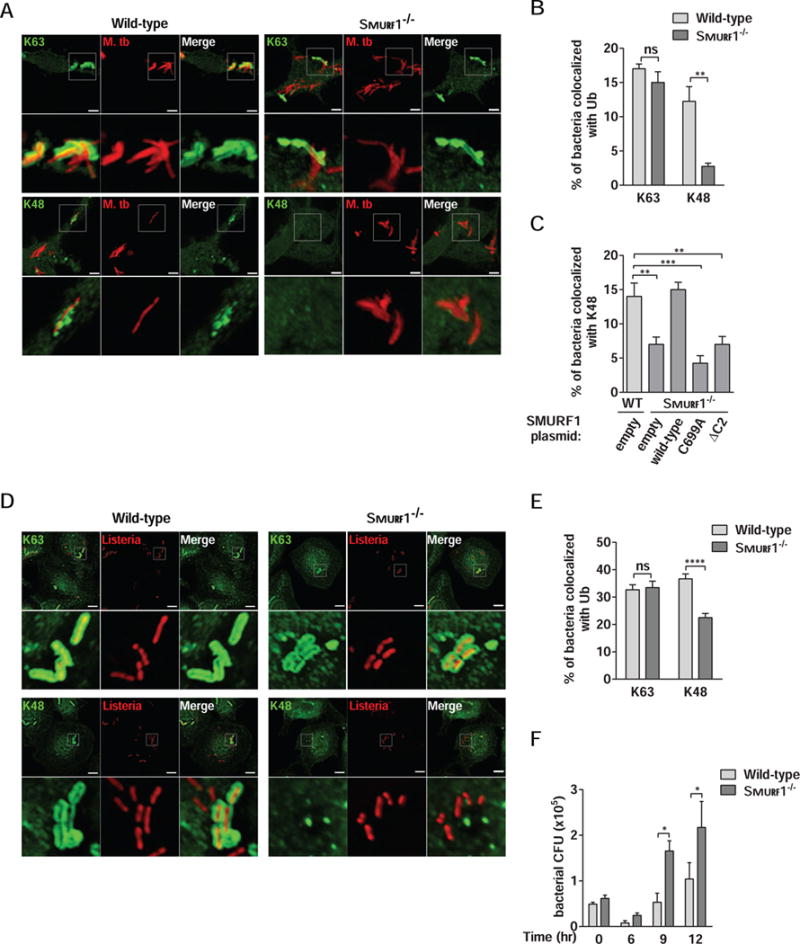
(A and B) Photomicrographs (A) and quantitation (B) of the colocalization of mCherry-Mtb and K63 or K48 polyubiquitin in wild-type or Smurf1−/− BMDMs 15 hr after infection. Scale bars, 3 μm. Bars are mean ± SEM for quadruplicate samples (at least 100 bacteria analyzed per sample) from a representative experiment. **P<0.01; ns: non-significant; t-test.
(C) Rescue of mycobacterial colocalization with K48-linked polyubiquitin by wild-type but not mutant forms of SMURF1 in Smurf1−/− BMDMs. Wild-type BMDMs were transduced with empty lentivirus and Smurf1−/− BMDMs were transduced as described in Figure 3B, and infected with mCherry-Mtb for 15 hr. Bars are mean ± SEM for quadruplicate samples (at least 100 bacteria analyzed per sample) from a representative experiment. **P<0.01; ***P<0.001; one-way ANOVA.
(D and E) Photomicrographs (D) and quantitation (E) of the colocalization of RFP-expressing L. monocytogenes ΔActA mutant and K63 or K48 polyubiquitin in wild-type or Smurf1−/− BMDMs 2 hr after infection. Scale bars, 5 μm. Bars are mean ± SEM for quadruplicate samples (at least 100 bacteria analyzed per sample) from a representative experiment. ****P<0.0001; ns: non-significant; t-test.
(F) L. monocytogenes growth in wild-type or Smurf1−/− BMDMs. Infected cells were lysed at the indicated time-points and L. monocytogenes growth was determined by counting CFU. Bars are mean ± SEM for quadruplicate samples. *P<0.05 for indicated comparison; two-way ANOVA. For B, C, E, and F, similar results were observed in at least three independent experiments.
See also Figure S2.
To further visualize K48 ubiquitin in Mtb-infected cells, we performed immune electron microscopy to detect K48 ubiquitin. We observed three major patterns of K48 immunogold staining. Some mycobacteria were labeled with K48 immunogold either on the bacterial or host organellar membrane (Figure S2A). (We note that we were unable to define fixation conditions that permitted both detection of K48 immunogold labeling and optimal membrane preservation; thus, we were not able to discern whether K48 was in direct association with mycobacteria or host membranes). For other mycobacteria, K48-ubiquitin aggregates were observed in close proximity to mycobacteria, either in well-circumscribed spherical structures near the mycobacteria (Figure S2B, left panels) or as a more diffuse pattern of strong K48 labeling in the vicinity of mycobacteria (Figure S2B, right panels). The K48-ubiquitin aggregates appeared morphologically similar to K63-ubiquitin aggregates observed in the cytoplasm near Salmonella-containing vacuoles in HeLa cells (Mesquita et al. 2012) which were postulated to serve as recruitment sites for p62 and LC3.
We next evaluated whether Smurf1 also mediates K48-linked ubiquitination of L. monocytogenes, an intracellular bacteria that resides in the cytoplasm. To avoid rapid cell-to-cell spread of L. monocytogenes, we used a mutant strain lacking the ActA protein that is targeted to autophagy via a mechanism involving protein ubiquitination (Perrin et al., 2004) and recruitment of autophagy adaptors (Lam et al., 2013). Similar to our observations with Mtb, Smurf1−/− BMDMs had a significant reduction in polyubiquitin/L. monocytogenes colocalization (Figure S2C and S2D). Moreover, there was no difference in the percentage of L. monocytogenes that stained positive for K63-linked ubiquitin in Smurf1−/− and wild-type BMDMs, but there was a significant decrease in K48-linked ubiquitin staining of L. monocytogenes in Smurf1−/− BMDMs (Figures 4D and 4E). We also observed a significant increase in L. monocytogenes replication in Smurf1−/− BMDMs (Figure 4F). Thus, Smurf1 mediates K48-linked, but not K63-linked, ubiquitin association with both Mtb and L. monocytogenes (or host membranes surrounding these bacteria).
Smurf1 Combines With Parkin to Mediate Distinct Steps in Ubiquitin-Dependent Selective Autophagy of M. tuberculosis
Since Smurf1 (this study) and Parkin (Manzanillo et al., 2013) mediate K48 and K63 ubiquitination of Mtb-associated structures, respectively, we evaluated whether the two ligases act coordinately to mediate polyubiquitination, selective autophagy and control of Mtb replication in macrophages. We compared the colocalization of Mtb-associated structures with polyubiquitin, K48-ubiquitin, K63-ubiquitin, or GFP-LC3, as well as mycobacterial growth in Smurf1−/− or Parkin−/− single knockout or Smurf1−/−/Parkin−/− double knockout (DKO) BMDMs. Both Smurf1−/− or Parkin−/− BMDMs had similarly reduced polyubiquitination of Mtb-associated structures, whereas DKO BMDMs displayed the greatest reduction in polyubiquitin/Mtb colocalization (Figure 5A). As expected, Smurf1 and Parkin specifically mediated K48- and K63-ubiquitination of Mtb-associated structures, respectively, and colocalization of both markers in DKO BMDM was reduced to the same extent as in the individual knockouts (Figure 5B and 5C). DKO BMDMs had the greatest reduction in GFP-LC3 colocalization with Mtb-associated structures (Figure 5D). This reduction was significant when compared to Smurf1−/− knockout BMDMs and had a trend that did not reach statistical significance when compared to Parkin−/− BMDMs. Both Smurf1−/− and Parkin−/− single knockout BMDMs were more permissive for Mtb replication at 48 hr after infection, and the absence of both ubiquitin ligases in DKO BMDMs led to higher Mtb replication as compared to in single knockout BMDMs (Figure 5E). Thus, Smurf1 and Parkin work together to mediate ubiquitin-dependent selective autophagy of Mtb and the activity of both ubiquitin ligases is required for optimal control of Mtb replication within macrophages.
Figure 5. Smurf1 Cooperates With Parkin to Trigger Ubiquitin-Dependent Selective Autophagy of M. tuberculosis.
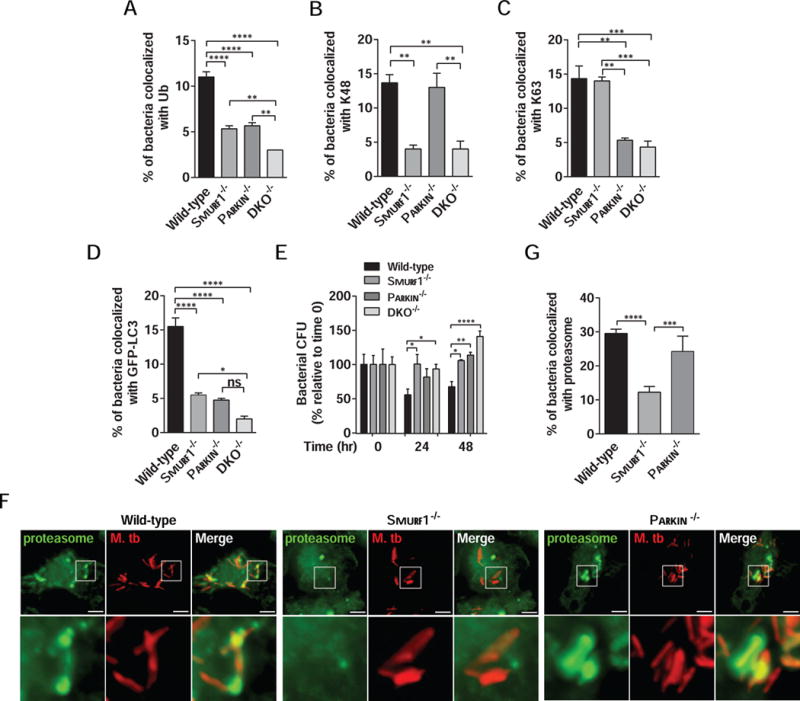
(A–D) Quantitation of the colocalization of mCherry-Mtb and polyubiquitin (A), K48 (B), K63 (C) or GFP-LC3 (D) in wild-type, Smurf1−/−, Parkin−/− or Smurf1−/−Parkin−/− double knockout (DKO−/−) BMDMs 15 hr after infection. Bars are mean ± SEM for quadruplicate samples (at least 100 bacteria analyzed per sample) from a representative experiment. *P<0.05; **P< 0.01; ***P<0.001; ****P<0.0001 for indicated comparison; ns: non-significant; one-way ANOVA.
(E) Mtb growth in wild-type, Smurf1−/−, Parkin−/− or DKO−/− BMDMs. Infected cells were lysed at the indicated time-points and Mtb growth was determined by counting CFU. Bars are mean ± SEM of quadruplicate samples; each sample was normalized to day 0. *P<0.05; **P< 0.01; ***P<0.001; ****P<0.0001 for indicated comparison; two-way ANOVA.
(F and G) Photomicrographs (F) and quantitation (G) of the colocalization of mCherry-Mtb and the proteasome 15 hr after infection of BMDMs from wild-type, Smurf1−/− or Parkin−/− mice. Insets (lower panels) in (F) show representative mycobacteria that would be considered colocalized with the proteasome in wild-type or Parkin−/− BMDMs or not colocalized with the proteasome in Smurf1−/− BMDMs. In (G), bars are mean ± SEM of quadruplicate samples (100 bacteria evaluated per sample per genotype) from a representative experiment. Scale bars, 5 μm. ***P<0.001; ****P<0.0001 for indicated comparison; one-way ANOVA. For For A–E and F, similar results were observed in at least three independent experiments.
See also Figure S3.
The autophagy receptors p62 and NBR1, which recognize ubiquitinated targets, are selectively recruited to Mtb-associated structures in a Parkin-dependent manner (Manzanillo et al., 2013). We therefore evaluated whether Smurf1 is required for recruitment of the autophagy receptors, p62 and NBR1. Similar levels of p62/Mtb colocalization were observed in wild-type and Smurf1−/− BMDMs (Figure S3A and S3B). In contrast, NBR1 recruitment to Mtb-associated structures was significantly reduced in Smurf1−/− BMDMs (Figure S3C and S3D). These results suggest that different types of ubiquitin linkages have distinct roles in the differential recruitment of certain autophagy receptors in selective bacterial autophagy.
In addition to p62 and NBR1 recruitment, ubiquitin functions in the recruitment of the proteasome to ubiquitin-coated Salmonella in the cytoplasm of murine macrophages (Perrin et al., 2004). Because K48-ubiquitination directs proteins to the proteasome (Grice and Nathan, 2016), we hypothesized that Smurf1, but not Parkin, might function in recruitment of the proteasome to Mtb-associated structures. Indeed, there was a significant reduction in colocalization of the proteasomal 20S β2i subunit with Mtb-associated structures in Smurf1−/− BMDMs compared to wild-type BMDMs (Figure 5F and 5G). However, there was no defect in proteasomal colocalization with Mtb in Parkin−/− BMDMs. Together, our data suggest that Smurf1 and Parkin have both shared and distinct activities in selective autophagy. Smurf1 has a unique role, not shared by Parkin, in K48-mediated ubiquitination and recruitment of the proteasome to sites of Mtb bacilli in murine macrophages.
Smurf1 Is Required For Anti-Mycobacterial Host Defense in vivo
Given the role of Smurf1 in the control of Mtb autophagic targeting and replication in BMDMs, we investigated whether Smurf1 is required for host immune defense during Mtb infection in vivo. We used an established murine model of tuberculosis (Collins et al., 2015) in which mice are infected with a low-dose inoculum via aerosol (~200 CFUs). We measured equal numbers of bacteria in the lungs of wild-type and Smurf1−/− mice on day zero of infection (Figure 6A). During the acute phase of infection (21 and 42 days after inoculation), wild-type and Smurf1−/− mice had equal numbers of Mtb CFUs in lungs, spleen (Figure S4A and S4B) and liver (data not shown). In contrast, the bacterial burden in the lungs and spleens of Smurf1−/− mice during the chronic stage of infection (90 or 120 days after inoculation) was higher (Figure 6B and 6C). Importantly, Smurf1−/− mice succumbed to disease earlier than their wild-type counterparts (Figure 6D). At 120 days after infection, when Mtb CFUs were significantly higher in the lungs of Smurf1−/− mice compared to wild-type littermate controls, there was a significant increase in the area of lung inflammation (Figure 6E and 6F). Upon histopathological examination by a pathologist blinded to genotype, there was an increase in mononuclear, but not polymorphonuclear, inflammatory cells as well as bronchiolitis in the lungs of Smurf1−/− mice (Table S1). At 120 days but not at earlier time points (42 and 90 days), there was an increase in lung concentrations of the pro-inflammatory cytokine, IL-17, but not in other cytokines such as IFN-γ, IL-1α, IL-1β and IL-6 (Figure S4). Thus, Smurf1 deficiency during the chronic phase of Mtb infection results in increased bacterial load in lung and spleen, increased pulmonary inflammation, and accelerated mortality.
Figure 6. Smurf1 Controls Chronic M. tuberculosis Infection In Vivo.
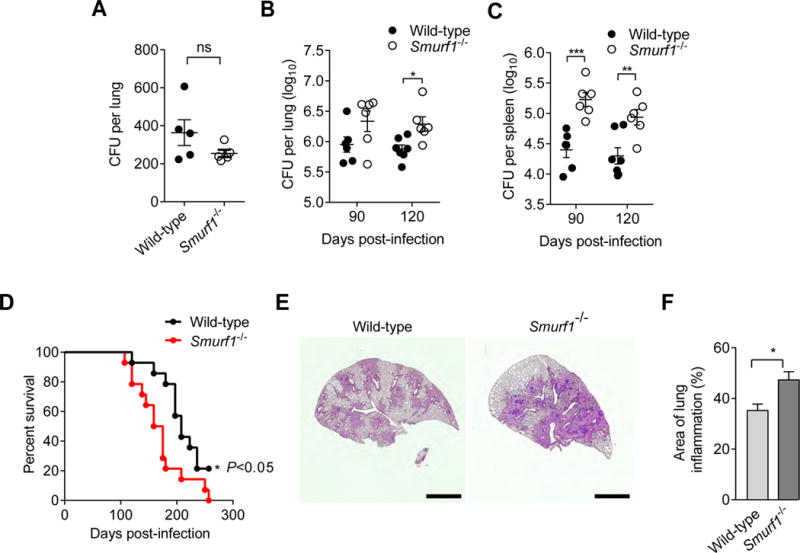
(A) Bacterial CFUs in lungs of wild-type or Smurf1−/− mice at day 0 of aerosol infection with Mtb.
(B and C) Bacterial CFUs in lungs (B) and spleens (C) of wild-type or Smurf1−/− mice at indicated time point after infection with Mtb. Bars are mean ± SEM of 6–7 animals/group. *P<0.05; **P<0.01; ***P<0.001; two-way ANOVA.
(D) Kaplan-Meier curve of survival of Mtb-infected mice (n=14 animals/genotype). *P<0.05; Log-rank test.
(E) Representative photomicrographs of H&E-stained lungs 120 days post-infection with Mtb. Scale bars, 2 mm.
(F) Quantitation of inflammatory areas in lungs of Mtb-infected mice at 120 days post-infection. Measurement was performed with ImageJ software. Results are mean ± SEM for 6–7 animals/group. *P<0.05; Mann-Whitney U-test.
See also Figure S4 and Table S1.
SMURF1 Controls M. tuberculosis Growth in Human Macrophages
We evaluated the effects of SMURF1 shRNA knockdown (Figure 7A) on Mtb replication in primary monocyte-derived human macrophages. Knockdown of SMURF1 with two independent shRNAs resulted in increased mycobacterial growth (Figure 7B). Similar results were also observed in primary monocyte-derived human macrophages derived from a second independent anonymous donor (Figure S5A). These findings suggest that SMURF1 may also function as a selective autophagy factor in the control of tuberculosis in humans.
Figure 7. SMURF1 Controls Mycobacterial Growth in Human Macrophages, is Expressed in Granulomas, and Colocalizes with M. tuberculosis in Lungs of Patients with Active Pulmonary Tuberculosis.
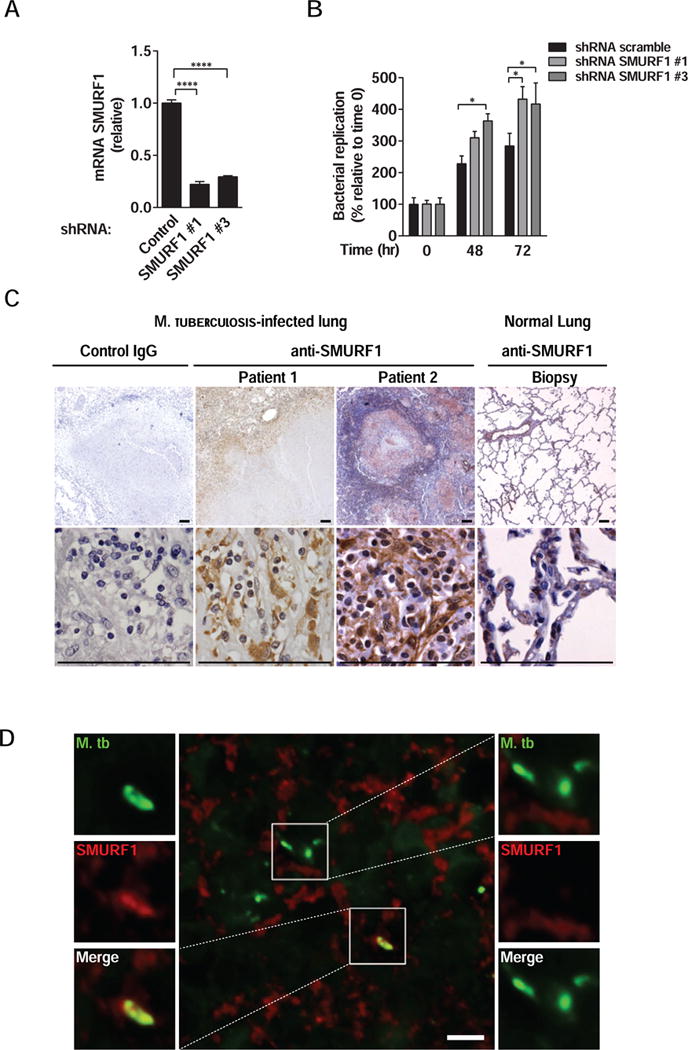
(A) Smurf1 knockdown in primary human monocyte-derived macrophages transduced with controls or two different SMURF1 shRNA constructs. mRNA levels of SMURF1 were analyzed by quantitative RT-PCR. Bars represent mean ± SEM (normalized to control scrambled shRNA). Similar results were observed in three independent experiments. ****P<0.0001; t-test.
(B) Mtb growth in primary human monocyte-derived macrophages transduced with control scrambled or SMURF1shRNA. Infected cells were lysed at the indicated time-points and Mtb growth was determined by counting CFUs. Bars are mean ± SEM of quadruplicate samples; each sample was normalized to day 0. Similar results were observed in two independent experiments. See Figure S5A for results from one additional donor. *P<0.05; **P<0.01 for indicated comparison; two-way ANOVA.
(C) Immunohistochemical staining for SMURF1 in lung biopsies of two human patients with active pulmonary tuberculosis or from a normal lung. Scale bars, 100 μm.
(D) Immunofluorescence staining of a lung biopsy of a human patient with active pulmonary tuberculosis using anti-Mtb and anti-SMURF1 antibodies. Left panel, image of Mtb that colocalizes with SMURF1. See Figure S5 for additional examples. Right panel, image of Mtb that do not colocalize with SMURF1. Scale bar, 5 μm. See also Figure S5.
SMURF1 is Expressed in Granulomas and Colocalizes with Mtb in Lungs of Patients With Active Pulmonary Tuberculosis
We examined SMURF1 expression in lung biopsy samples from normal lungs and those with active pulmonary tuberculosis. SMURF1 was highly expressed in cells with morphological characteristics of macrophages located within and surrounding granuloma lesions in human lungs (Figure 7C). In biopsies from normal human lung, SMURF1 was expressed in bronchial epithelial cells, vascular endothelial cells, and at lower levels, in pneumocytes (Figure 7C). Moreover, immunofluorescence staining revealed that human SMURF1 colocalized with Mtb in a patient lung biopsy sample (Figure 7D and Figure S5B). Quantification of images encompassing the entire section revealed that 24 out of 59 total bacteria colocalized with SMURF1. Thus, SMURF1 may play a role in the autophagic targeting of Mtb in patients with pulmonary tuberculosis.
DISCUSSION
Here we demonstrate that Smurf1 functions in selective autophagy of Mtb via a mechanism that involves K48-linked ubiquitination and recruitment of LC3 to intracellular mycobacteria and/or mycobacteria-containing organelles. Moreover, Smurf1 is required for Mtb-associated structures to be directed to the lysosome for degradation, for control of acute replication in murine macrophages, for host defense against chronic tuberculosis in mice and for control of Mtb growth in human macrophages. In addition, Smurf1 also mediates K48-linked ubiquitination of another intracellular pathogenic bacterium, L. monocytogenes, and restricts growth of Listeria in macrophages. Taken together, our findings suggest that Smurf1 may function in K48-ubiquitination and lysosomal targeting of different intracellular bacterial pathogens in primary macrophages. Furthermore, our findings suggest a role for Smurf1/SMURF1 in host defense against M. tuberculosis infection in mice and humans.
Previous studies have shown that general autophagy activation in mycobacteria-infected macrophages accelerates the maturation of mycobacteria-containing phagosomes and increases lysosome-mediated killing of tuberculous bacilli (Gutierrez et al., 2004). In our study, we found that Smurf1 partially mediates antimycobacterial activity in naive macrophages, as well as the killing of Mtb in macrophages treated with an autophagy-inducing peptide, Tat-Beclin 1. Although Tat-Beclin 1 enhanced autophagic flux similarly in wild-type and Smurf1−/− BMDMs, Tat-Beclin 1 failed to increase LC3 targeting to Mtb bacilli and failed to reduce Mtb replication in Smurf1−/− BMDMs. Thus, Smurf1 may function in naïve macrophages as well as in macrophages treated with autophagy inducers to enhance Mtb degradation.
The ubiquitination of intracellular Mtb-containing organelles is an important early step in anti-mycobacterial autophagy (Watson et al., 2012). Ubiquitin-coated mycobacterial-containing organelles recruit ubiquitin-binding autophagy adaptors such as p62 and NBR1, which bind to LC3 and mediate the delivery of bacteria to lysosomes (Levine et al., 2011; Manzanillo et al., 2013; Watson et al., 2012). Whereas Parkin mediates K63- but not K48-ubiquitination of Mtb-associated structures (Manzanillo et al., 2013), our data demonstrate a role for Smurf1 in K48-ubiquitination of Mtb-associated structures. The presence of a minor amount of residual K48-ubiquitination of Mtb-associated structures in Smurf1−/− BMDMs suggests that another as-of-yet unidentified ubiquitin-ligase may also act independently of Smurf1 to mediate K48-ubiquitination.
Our data indicate that Parkin-mediated K63 and Smurf1-mediated K48 ubiquitination have distinct roles in bacterial selective autophagy and function in the differential recruitment of downstream factors. For example, the recruitment of the selective autophagy factor, p62, to Mtb-associated structures is independent of Smurf1 (this study), whereas it has been previously shown to require Parkin (Manzanillo et al., 2013). Moreover, we found that Smurf1, but not Parkin, is required for recruitment of proteasomes to Mtb-associated structures. Thus, taken together with prior studies (Manzanillo et al., 2013), our results suggest that efficient ubiquitin-mediated anti-M. tuberculosis selective autophagy may require the simultaneous action of both Smurf1 and Parkin which act via K48-ubiquitination or K63-ubiquitination, respectively, to coordinate delivery of intracellular Mtb-associated structures or bacterial antigens to the proteasome or lysosome.
The precise role of K48-linked, ubiquitin-dependent proteasomal recruitment to Mtb-associated structures in selective bacterial autophagy and anti-tuberculous host defense is unknown. However, of note, the role of Smurf1 in promoting K48-ubiquitination is not restricted to Mtb, as we found that Smurf1 can also mediate K48-ubiquitination and growth restriction of L. monocytogenes in murine macrophages. As ubiquitin is targeted to a number of other intracellular pathogens such as Legionella pneumophila (Ivanov and Roy, 2009), Francisella tularensis (Chong et al., 2012), Toxoplasma gondii (Haldar et al., 2015) and Shigella flexneri (Baxt and Goldberg, 2014), our findings raise the question of whether Smurf1 and K48-linked ubiquitination also function in cell autonomous immunity to these, and potentially other, intracellular organisms.
Most proteins ubiquitinated by Smurf1 bind to its WW domains through their PY motifs (Suzuki et al., 2002; Yuan et al., 2012) and this WW-PY interaction is considered the canonical Smurf1-substrate interaction. However, non-canonical interactions between Smurf1 and its substrates have also been reported; Smurf1-dependent ubiquitination and degradation of RhoA and Axin is dependent on their direct interaction with Smurf1 C2 domain (Fei et al., 2014; Tian et al., 2011). Here we found that both Smurf1 HECT E3 ligase and C2 membrane phospholipid-binding domains are required for bacterial autophagy. The HECT domain is likely essential to ligate K48-ubiquitin to Smurf1 target(s) and our results demonstrate that the C2 domain facilitates the localization of Smurf1 to Mtb-associated structures. However, we do not rule out the possibility that the Smurf1 C2 domain may also be necessary to promote the binding of Smurf1 to its substrate in a non-canonical interaction. The identity of the protein(s) targeted for ubiquitination by Smurf1 remains to be explored.
Consistent with the role of Smurf1 in selective bacterial autophagy in vitro, we found that Smurf1−/− mice had increased Mtb replication during chronic stages of infection and succumbed earlier to lethal disease, similar to another selective autophagy factor, Parkin (Manzanillo et al., 2013). During acute Mtb infection in vivo, multiple immune mechanisms act to control bacterial growth (O’Garra et al., 2013). Even though Smurf1−/− macrophages have a defect in selective bacterial autophagy and bacterial growth during acute infection, Smurf1−/− mice likely control Mtb replication in vivo in the early stages of infection via other anti-mycobacterial mechanisms. However, long-term absence of Smurf1 and defects in bacterial selective autophagy may lead to cumulative effects on intracellular bacterial replication as well as other possible indirect effects on immunity, resulting in a higher susceptibility to infection.
As we found an increase in the area of inflammatory lesions, increased mononuclear cell infiltration and increased bronchitis/bronchiolitis in lungs of Mtb-infected Smurf1−/− mice, we do not exclude the possibility that Smurf1−/− mice could be more susceptible to tuberculosis due to a hyperinflammatory response as reported previously with Atg5fl/fl;LysM-Cre+ mice (Castillo et al., 2012; Kimmey et al., 2015; Watson et al., 2012). However, the phenotype we observed is quite different from that reported in the Atg5fl/fl;LysM-Cre+ mice, which succumb early to lethal disease. Moreover, the only cytokine abnormality we detected was an increase in IL-17 at late stages, when bacterial replication was increased in lungs and spleen, and this increased IL-17 was not associated with an increase in neutrophilic inflammation. Thus, the precise significance of elevated IL-17 remains unknown, and it is unclear whether it is an indirect consequence of enhanced bacterial growth and/or other effects of Smurf1-deficiency on immunity, such as its negative effects on IFN-γ signaling (Yuan et al., 2012) which regulates the production of IL-17 by CD4 T lymphocytes (Mangan et al., 2006).
Mtb infection in C57Bl/6 mice with either germline (Ulk1, Ulk2, Atg4b) or conditional deletion in macrophages (Atg14l, Atg12, Atg16l, Atg7, Atg3) of core macroautophagy genes did not result in increased mouse mortality or lung CFUs within the first 80 days of infection (Kimmey et al., 2015). Because C57Bl/6 mice are among the most resistant mouse strains to Mtb (Medina and North, 1998), and Smurf1−/− mice with a C57Bl/6 genetic background were more susceptible to Mtb beyond 90 days after infection, we propose that selective autophagy may be important for long-term, but not short-term, control of Mtb infection. During chronic infection, Smurf1-dependent selective autophagy may either directly control bacterial replication (via a slow cumulative effect over time), enhance antigen presentation by dendritic cells (Jagannath et al., 2009) (as K48 polyubiquitination plays an important role in MHC class I antigen processing (Fiebiger et al., 2015)), facilitate the recognition of pathogenic molecules by innate immune receptors (Lee et al., 2007), and/or contribute to antituberculous host defense via other effects on innate or adaptive immunity. Furthermore, as the E3 ubiquitin ligase activity of Smurf1 and Parkin are also important for ubiquitination of signaling molecules and bacteria-containing structures, their roles during infection may extend beyond initiating canonical macroautophagy or phagolysosomal maturation.
In conclusion, our data demonstrate that Smurf1 represents a key component in anti-M. tuberculosis autophagic targeting in macrophages and in anti-tuberculous host defense in vivo. These findings have implications for understanding the roles of different types of ubiquitin ligases in innate immunity against intracellular pathogens.
EXPERIMENTAL PROCEDURES
Mice
Transgenic mice expressing GFP-LC3 were obtained from N. Mizushima (Mizushima et al., 2004) and rederived in a C57BL/6J background. Smurf1−/− mice (Yamashita et al., 2005) (already backcrossed to C57BL/6J mice for >10 generations) were bred with C57BL/6J mice (The Jackson Laboratory) to obtain Smurf1+/− heterozygotes. Smurf1−/− mice and littermate controls obtained from heterozygote crosses were used for all experiments. Parkin−/− mice were obtained from Jackson Laboratory. Smurf1−/−/Parkin−/− double knockout mice were obtained by crossing Smurf1−/− mice with Parkin−/− mice. For experiments quantitating autophagy in bone marrow-derived macrophages (BMDMs), mice were crossed with GFF-LC3 transgenic mice. All mice were housed under specific pathogen-free conditions. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas (UT) Southwestern Medical Center.
Cells and Cell Culture
BMDMs were generated from mice by culturing bone marrow cells from mouse femurs and tibia as described (Collins et al., 2015). The RAW 264.7 murine macrophage cell line was cultured in DMEM supplemented with 10% FBS. Primary human macrophages were obtained from buffy coats from anonymous donors provided by a local blood bank.
Bacterial Strains
For Mtb experiments, Mtb Erdman was used as the wild-type strain throughout. The EccD1 mutant and mCherry-expressing strain were described previously (Collins et al., 2015). For listeria experiments, L. monocytogenes 10403s ΔActA stably expressing RFP under the control of actA promoter (ΔActA-PACTA-RFP) was used (gift of D. Portnoy). Bacteria were cultured as described in detail in the Supplemental Experimental Procedures.
Macrophage and Animal Infections
BMDMs and primary human monocyte-derived macrophages (obtained from anonymous donors at a local blood bank) were infected with Mtb and/or L. monocytogenes, and Mtb low-dose aerosol infection (Collins et al. 2015) was performed in wild-type or Smurf1−/− mice as described in detail in the Supplemental Experimental Procedures.
Immunofluorescence Studies
BMDMs were fixed and stained as described in the Supplemental Experimental Proceudres using the following primary antibodies anti-LC3 (Sigma L7543); anti-LAMP1 (Santa Cruz SC-8098); anti-polyubiquitin (Enzo FK1), humanized anti-K63 and anti-K48 (Genentech), anti-proteasome 20S β2i subunit (Enzo BML-PW8150), anti-Flag M1 (Sigma), anti-p62 (Abnova H00008878-M01), anti-NBR1 (Abcam ab55474). Immunofluorescence imaging was performed using a Zeiss AxioImager M2 microscope equipped with a Photometrics CoolSnap HQ2 camera and deconvolution microscopy using AutoDeBlur (Bitplane). Imaris (7.4.0 version; Bitplane) was used for image analysis.
Immunohistochemistry and Immunofluorescence of Human Lung Tissue
Archived lung biopsies from patients at University of Texas Southwestern Medical Center were subjected to immunohistochemical analyses to detect SMURF1 expression and immunofluorescence analyses to detect SMURF1 and Mtb as described in Supplemental Experimental Procedures. This study was approved by the Institutional Review Board of the University of Texas Southwestern Medical Center.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 6.01. Two-tailed unpaired Student’s t test was used for single comparisons; Analysis of Variance (ANOVA) was used for experiments with multiple comparisons; Log-rank (Mantel-Cox) test was used for survival studies.
Supplementary Material
Highlights.
-
-
The ubiquitin ligase Smurf1 functions in selective autophagy of M. tuberculosis (Mtb)
-
-
Smurf1 recruits K48 ubiquitin, the proteasome, LC3 and LAMP1 to Mtb-associated structures
-
-
Smurf1 controls Mtb replication in murine and human macrophages, and in mice
Acknowledgments
We thank BEI Resources, Genentech, N. Mizushima, and D. Portnoy for providing critical reagents; Zhongju Zou and Lori Nguyen for technical assistance; K. Luby-Phelps and A. Darehshouri for helpful discussions and assistance with immunogold EM; and H. Smith for assistance with manuscript preparation. EM studies were carried out in the UT Southwestern Live Cell and Electron Microscopy Facility. This work was supported by NIH grants RO1 AI099439 and R21 AI111023 (M.U.S.), RO1 DK097485 (R.J.X), T32 AI528436 (C.R.S) and U19 AI109725 (R.J.X, M.U.S., B.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes detailed experimental procedures, five figures, and one table and can be found with this article online at___
AUTHOR CONTRIBUTIONS
L.H.F, R.J.X, M.U.S. and B.L designed research; L.H.F., V.R.N. and C.R.S performed research; J.R.T. performed blinded pathologic analysis; L.H.F, M.U.S. and B.L. analyzed data; and L.H.F, M.U.S. and B.L. wrote the paper.
References
- Baxt LA, Goldberg MB. Host and bacterial proteins that repress recruitment of LC3 to Shigella early during infection. PLoS One. 2014;9:e94653. doi: 10.1371/journal.pone.0094653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Zhang L. A Smurf1 tale: function and regulation of an ubiquitin ligase in multiple cellular networks. Cell Mol Life Sci. 2013;70:2305–2317. doi: 10.1007/s00018-012-1170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA. 2012;109:E3168–3176. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PL, Lu H, Shelly M, Gao H, Poo MM. Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron. 2011;69:231–243. doi: 10.1016/j.neuron.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Chong A, Wehrly TD, Child R, Hansen B, Hwang S, Virgin HW, Celli J. Cytosolic clearance of replication-deficient mutants reveals Francisella tularensis interactions with the autophagic pathway. Autophagy. 2012;8:1342–1356. doi: 10.4161/auto.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, Scharn CR, Stamm CE, Levine B, Chen ZJ, et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe. 2015;17:820–828. doi: 10.1016/j.chom.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei C, He X, Xie S, Miao H, Zhou Z, Li L. Smurf1-mediated axin ubiquitination requires Smurf1 C2 domain and is cell cycle-dependent. J Biol Chem. 2014;289:14170–14177. doi: 10.1074/jbc.M113.536714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebiger BM, Pfister H, Behrends U, Mautner J. Polyubiquitination of lysine-48 is an essential but indirect signal for MHC class I antigen processing. Eur J Immunol. 2015;45:716–727. doi: 10.1002/eji.201444830. [DOI] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell. 2014;54:224–233. doi: 10.1016/j.molcel.2014.03.009. [DOI] [PubMed] [Google Scholar]
- Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci. 2016;73:3497–3506. doi: 10.1007/s00018-016-2255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Haldar AK, Foltz C, Finethy R, Piro AS, Feeley EM, Pilla-Moffett DM, Komatsu M, Frickel EM, Coers J. Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc Natl Acad Sci USA. 2015;112:E5628–5637. doi: 10.1073/pnas.1515966112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov SS, Roy CR. Modulation of ubiquitin dynamics and suppression of DALIS formation by the Legionella pneumophila Dot/Icm system. Cell Microbiol. 2009;11:261–278. doi: 10.1111/j.1462-5822.2008.01251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15:267–276. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–569. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy. 2013;9:985–995. doi: 10.4161/auto.24406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, Shi M, Leslie BJ, Hopfner KP, Ha T, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15:228–238. doi: 10.1016/j.chom.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Li P, Zhang M, Xing G, Li X, Zhou W, Bartlam M, Zhang L, Rao Z, He F. Pivotal role of the C2 domain of the Smurf1 ubiquitin ligase in substrate selection. J Biol Chem. 2011;286:16861–16870. doi: 10.1074/jbc.M110.211979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina E, North RJ. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology. 1998;93:270–274. doi: 10.1046/j.1365-2567.1998.00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesquita FS, Thomas M, Sachse M, Santos AJ, Figueira R, Holden DW. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 2012;8:e1002743. doi: 10.1371/journal.ppat.1002743. 22719249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol. 2013;31:475–527. doi: 10.1146/annurev-immunol-032712-095939. [DOI] [PubMed] [Google Scholar]
- Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol. 2004;14:806–811. doi: 10.1016/j.cub.2004.04.033. [DOI] [PubMed] [Google Scholar]
- Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, Levesque S, Castoldi F, Jacquelot N, Yamazaki T, et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell. 2016;30:147–160. doi: 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, Maurer K, Cadwell K, Philips JA. Ubiquilin 1 Promotes IFN-gamma-Induced Xenophagy of Mycobacterium tuberculosis. PLoS Pathog. 2015;11:e1005076. doi: 10.1371/journal.ppat.1005076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki C, Murakami G, Fukuchi M, Shimanuki T, Shikauchi Y, Imamura T, Miyazono K. Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane. J Biol Chem. 2002;277:39919–39925. doi: 10.1074/jbc.M201901200. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Raghavan S, Hwang WW, Cox JS. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci USA. 2003;100:13001–13006. doi: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Bai C, Lin Q, Lin H, Liu M, Ding F, Wang HR. Binding of RhoA by the C2 domain of E3 ligase Smurf1 is essential for Smurf1-regulated RhoA ubiquitination and cell protrusive activity. FEBS Lett. 2011;585:2199–2204. doi: 10.1016/j.febslet.2011.06.016. [DOI] [PubMed] [Google Scholar]
- van der Vaart M, Korbee CJ, Lamers GE, Tengeler AC, Hosseini R, Haks MC, Ottenhoff TH, Spaink HP, Meijer AH. The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLP-MYD88 to authophagic defense. Cell Host Microbe. 2014;15:753–767. doi: 10.1016/j.chom.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science. 2003;302:1775–1779. doi: 10.1126/science.1090772. [DOI] [PubMed] [Google Scholar]
- Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer KF. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol. 2014;24:332–341. doi: 10.1016/j.tcb.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, Zhang YE. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell. 2005;121:101–113. doi: 10.1016/j.cell.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, Qi J, Zhao X, Gao C. Smurf1 protein negatively regulates interferon-gamma signaling through promoting STAT1 protein ubiquitination and degradation. J Biol Chem. 2012;287:17006–17015. doi: 10.1074/jbc.M112.341198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.