

Abstract
Rhomboid proteases constitute a family of intramembrane serine proteases ubiquitous in all forms of life. They differ in many aspects from their soluble counterparts. We applied molecular dynamics (MD) computational approach to address several challenging issues regarding their catalytic mechanism: How does the exosite of GlpG rhomboid protease control the kinetics efficiency of substrate hydrolysis? What is the mechanism of inhibition by the non‐competitive peptidyl aldehyde inhibitors bound to the GlpG rhomboid active site (AS)? What is the underlying mechanism that explains the hypothesis that GlpG rhomboid protease is not adopted for the hydrolysis of short peptides that do not contain a transmembrane domain (TMD)? Two fundamental features of rhomboid catalysis, the enzyme recognition and discrimination of substrates by TMD interactions in the exosite, and the concerted mechanism of non‐covalent pre‐catalytic complex to covalent tetrahedral complex (TC) conversion, provide answers to these mechanistic questions.
Keywords: rhomboid, serine proteases, membrane enzymes, exosite, effector
Introduction
Rhomboid proteases, identified in 2001 as a novel family of intramembrane serine proteses,1, 2 are ubiquitous in all kingdoms of life from bacteria to mammals. They play a regulatory role in various cell pathways by cleaving amide bonds of transmembrane proteins.3, 4, 5, 6, 7 The best studied rhomboid protease, E. coli GlpG,8, 9, 10, 11 utilizes a His‐Ser catalytic dyad, rather than the classical Asp‐His‐Ser catalytic triad of water‐soluble serine proteases. Kinetic analysis of hydrolysis of a series of intramembrane protein substrates by GlpG demonstrated that the low catalytic efficiency, kcat/KM, is the consequence of low kcat, whereas Km is similar to the corresponding values of peptide hydrolysis by the soluble serine proteases.12 Molecular dynamics combined with quantum mechanical simulations of the transformation of the Michaelis complex (MC) to tetrahedral complex (TC) revealed that the general‐base catalysis in GlpG rhomboid protease is a concerted process of proton transfer and nucleophilic attack.13 Conversely, the mechanism of general base catalysis in chymotrypsin, with Asp‐His‐Ser catalytic triad, could be described as a stepwise process, with an initial proton transfer from Ser to His in the MC, and subsequent nucleophilic attack of the Ser alkoxide on the substrate.
Recent studies showed that peptidyl aldehydes are non‐competitive inhibitors of intramembrane substrate hydrolysis by GlpG rhomboid,14 in contrast to soluble serine proteases, in which peptidyl aldehydes are competitive reversible covalent inhibitors.15, 16 It was therefore suggested that peptidyl aldehydes do not compete with intramembrane protein substrates for the same binding site when the latter form Michaelis complex with GlpG.14 This result supports the previous suggestion that the first binding contact of an intramembrane protein substrate with GlpG is in the exosite.12, 17, 18 The unwinding of the substrate helical transmembrane domain (TMD) in the rhomboid exosite is essential for rhomboid intramembrane proteolysis.12, 14, 17, 18 All currently known substrates of rhomboid proteases are intramembrane proteins.19, 20, 21
Several challenging open questions regarding the catalytic mechanism of rhomboid proteases include: How does the exosite of GlpG rhomboid protease control the kinetics efficiency of substrate hydrolysis? What is the mechanism of GlpG inhibition by the non‐competitive peptidyl aldehyde inhibitors bound in the active site? If indeed rhomboid proteases are not adopted for the hydrolysis of short peptides without TMD, what could be the origin of the phenomenon? We applied molecular modeling to address these issues.
Results and Discussion
Construction and MD simulation of GlpG non‐covalent complexes with peptide substrates
The crystal structure of E. Coli GlpG complexed with the peptidyl aldehyde inhibitor Ac‐VRMA‐CHO (5f5b.pdb)14 was used as a structural template for the construction by molecular modeling of the GlpG non‐covalent complexes with peptide substrates. Applying the visual interface and energy minimization algorithms of YASARA software,22 we constructed such complexes of GlpG with two fragments of Gurken protein substrate (UniProt P42287): 11‐pep, a peptide substrate of 11 amino acids ‐ RKVRMA∼HIVFS (corresponding to residues 241–251), and 33‐pep, a peptide substrate of 33 amino acids corresponding to residues 241–273 ‐ RKVRMA∼HIVFSFPVLLMLSSLYVLFAAVFMLRN (“∼” indicates the location of the scissile bond). The N‐termini of both substrates are located in the enzyme active site, starting from Arg241 at P6 to Ala246 at the P1 position. The initial 3D structure of the 33‐pep substrate was generated by homology modelling.23, 24, 25 Sequential steps of molecular modeling, including molecular dynamics simulation by AMBER14 software suite,26, 27 were used for the generation of membrane immersed structures of GlpG non‐covalent complexes with the two substrates (Fig. 1). The Cα RMSD graphs calculated on the MD production simulation phase of non‐covalent complexes formed by 11‐pep (750 ns, 75,000 frames) and 33‐pep (450 ns, 45,000 frames) are presented in Figure 2. The 11‐pep substrate is located in the enzyme active site [Fig. 1(C)]. The N‐terminal fragment of the 33‐pep substrate also occupies the GlpG active site. The backbones of the N‐terminal fragments of both substrates are well superposed [Fig. 1(D)]. In contrast, their C‐ends are turned in opposite directions: 11‐pep is bent up to the periplasm whereas 33‐pep turns down, occupying the GlpG exosite with its C‐end in the cytoplasm [Fig. 1(D)]. For the following analysis of the results, we used frames from the end portion of the production MD trajectories: 350 – 750 ns for 11‐pep, and 200 – 450 ns for 33‐pep. Calculated per residue RMSD values on the backbone atoms (Cα, N, C) of 11‐pep revealed that the observed large RMSD Cα fluctuations (Fig. 2) originate from the labile N‐ and C‐terminal residues of 11‐pep, P6 and P5′, respectively (Fig. 3). The 11‐pep does not include a fragment extended and aligned in the GlpG exosite like 33‐pep does (Fig. 1). This extension considerably restricts the conformational lability of 33‐pep S251 (P5′), in contrast to the very motile S251 (P5′) at the C‐end of 11‐pep [Fig. 3(A)].
Figure 1.
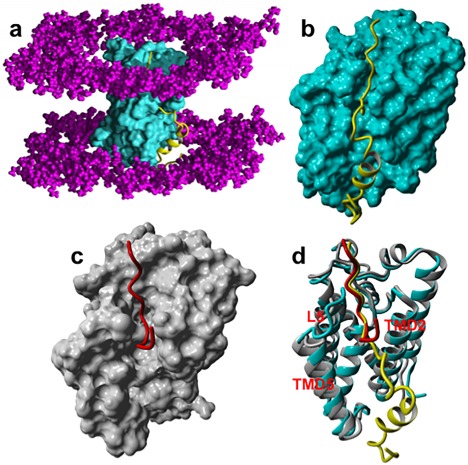
Structures of conformational cluster centroids of the pre‐catalytic non‐covalent complex with 33‐pep and 11‐pep substrates. They were identified by the condition of concerted non‐covalent complex to TC transformation, that is, the Ser201 Oγ – His254 Nɛ and Ser201 Oγ – Ala246 C interatomic distances are ≤ 3 Å, simultaneously. (A): The non‐covalent complex of GlpG with 33‐pep. The molecular surface of GlpG is cyan. The backbone of 33‐pep in ribbon style is yellow. The polar heads of the lipid bilayer are colored in magenta. (B): The non‐covalent complex with 33‐pep. (C): The non‐covalent complex of GlpG (grey) with 11‐pep (red). (D): Superposition of the non‐covalent complexes formed with 33‐pep and 11‐pep on the Cα, N, C backbone atoms of S201 and H254 catalytic residues of GlpG (RMSD = 0.18 Å). Enzyme backbone is colored cyan and grey in the pre‐catalytic complex with 33‐pep (yellow) and with 11‐pep (red), respectively.
Figure 2.
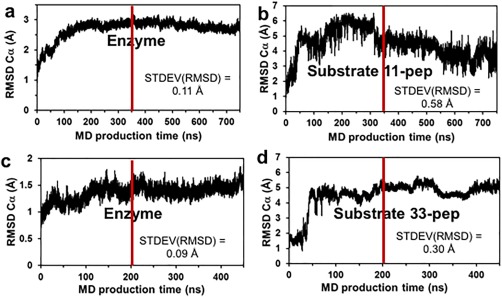
The RMSD of Cα atoms separately calculated for enzyme and substrate of the pre‐catalytic non‐covalent complex on production MD trajectories. The frames of MD trajectories to the right from the red line were used for the conformational cluster analysis and calculations of STDEV (RMSD). (A‐B): 11‐pep, 750 ns total and the last 400 ns for cluster analysis. (C‐D): 33‐pep, 450 ns total and the last 250 ns for cluster analysis.
Figure 3.
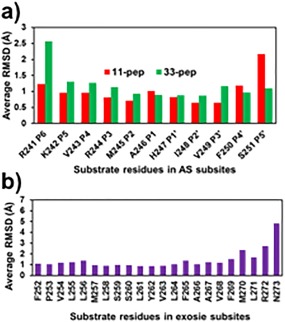
Average per residue RMSD values of substrate backbone atoms (Cα, N, and C). They were calculated by AMBER14 CPPTRAJ algorithm on production trajectory of MD simulated pre‐catalytic non‐covalent complexes formed by GlpG with the substrates. Reference to cluster centroids of conformations relevant for the non‐covalent complex to TC transformation. (A): AS residues of 11‐pep at 350–750 ns, 40,000 frames, and of 33‐pep at 200–450 ns, 25,000 frames. (B): Exosite residues of 33‐pep at 200–450 ns, 25,000 frames.
The observed large conformational flexibility of the P6 and P5′ in the non‐covalent complex of 11‐pep correlates with the structural features of the active site substrate specificity of proteases, which is usually limited to the substrate P4‐P4′ subsites interval.28, 29, 30 Moreover, our computational result about P6 conformational flexibility is in agreement with the experimental observation of the crystal structure of GlpG complexes with peptide aldehydes.14 The irregular character of RMSD Cα of 33‐pep [Fig. 2(D)], while not as pronounced as of 11‐pep, mostly originates in the contribution from large fluctuations of its very end residue in the C terminus [Fig. 3(B)], which is exposed in the cytoplasm [Fig. 1(A)]. The latter is not surprising, since 33‐pep is a truncated inner fragment of Gurken, a large intramembrane protein substrate, extended from the cell periplasm to the cytoplasm.
Analysis of probability density distribution of non‐covalent complex conformers
As mentioned above, the general base catalyzed MC to TC conversion in GlpG rhomboid protease involves a synchronous process of proton transfer from S201 to H254 and nucleophilic attack of S201 on the A246 carbonyl group of the substrate.13 A geometrical condition for the synchronous process realization is that the distance values of both atomic pairs must be ≤ 3 Å simultaneously in the non‐covalent pre‐catalytic complexes formed by both 11‐pep and 33‐pep substrates with GlpG. We analyzed the probability density distribution of conformers of GlpG non‐covalent complexes with 11‐pep and 33‐pep as a function of Ser201 Oγ – His254 Nɛ and Ser201 Oγ – Ala246 C interatomic distance fluctuations on the MD production trajectories. For this analysis, we collected frames every 100 ps from the end fragments on the production MD trajectories: 350 – 750 ns for 11‐pep (40,000 frames), and 200 – 450 ns for 33‐pep (25,000 frames). The results revealed striking difference between the two substrates. The total number of frames satisfying the condition of catalytically competent conformers is 5574 for 33‐pep and 169 for 11‐pep. Thus, the portion of catalytically competent conformers with the 33‐pep substrate amounts to 22.3% of all its conformations, whereas it is only a tiny 0.42% with 11‐pep (Fig. 4). In other words, the Ser201 Oγ – His254 Nɛ and Ser201 Oγ – Ala246 CO interatomic distances are spread much more evenly in the conformational space of GlpG/11‐pep than in the GlpG/33‐pep complex. The phenomenon originates in the binding alignment of the 33‐pep fragment in the GlpG exosite, which significantly limits the conformational lability of its fragment aligned in the enzyme AS. Thus, the selective binding of the substrate TMD fragment in the GlpG exosite, like in 33‐pep, is a critical factor favoring the catalytic processing by rhomboid protease.
Figure 4.
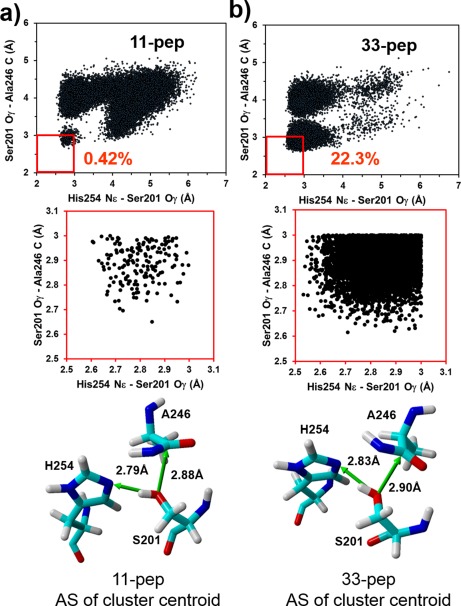
Probability density distribution of the pre‐catalytic non‐covalent complex conformers as a function of simultaneously varied interatomic distances of Ser201 Oγ – His254 Nɛ and Ser201 Oγ – Ala246 C, generated on MD production trajectories. The catalytically competent conformations of the non‐covalent complexes are localized in the red square. (A): non‐covalent complex of 11‐pep. (B): non‐covalent complex of 33‐pep.
GlpG exosite influence on the active‐site (as) substrate selectivity
We have identified catalytically productive conformational cluster centroids of GlpG non‐covalent complexes with both 33‐pep and 11‐pep substrates (Figs. 1 and 4). There is close matching between the backbone atoms of P4 – P2′ residues of both substrates [Figs. 1(d) and 5]. Nevertheless, the side chains that were aligned in the AS acquired significantly different conformations (Fig. 5), also reflected in the calculated per‐residue interaction energies with GlpG residues for 11‐pep and 33‐pep cluster centroids (Supporting Information Table SI) and the per‐residue RMSD differences (Supporting Information Table SII). The conformational difference originates from the TMD of 33‐pep aligned in the GlpG rhomboid exosite, which restricts the conformational lability of the substrate fragment aligned in the GlpG active site and also affects the enzyme conformation. It is well known that a bound ligand may cause conformational changes in the host protein, most prominently affecting the side chains of the binding site residues and their water exposure level.31 We analyzed the conformational lability of 11‐pep and 33‐pep side residues and per‐residue Van der Waals and Coulomb interaction energies with GlpG in their non‐covalent complexes (Supporting Information Table SI, Figs. 3 and 6). The most effective contribution to the substrate recognition of 33‐pep by GlpG active site is limited to the P4 – P2′ fragment, where the strongest interactions are with Arg (P3) and Ala (P1) (Supporting Information Table SI). This result is in agreement with a recent mutagenesis study of the efficiency and selectivity in the processing of transmembrane peptide substrates by E. coli GlpG, demonstrating the strongest influence of the same two substrate subsites.21 Our result also correlates well with the experimental observations that GlpG AS does not contain specific substrate‐interacting subsites beyond P4.11, 14 The 33‐pep TMD bound in the GlpG exosite leads to significant shift of its P3′ – P5′ backbone relative to 11‐pep [Figs. 1(D) and 5]. V249 (P3′) and F250 (P4′) of 33‐pep form strong binding interactions with F153 and W236 GlpG residues (Fig. 6 and Supporting Information Table SI), the exosite gatekeepers, where the former is located on TMD2 and the latter on TMD5. Two 33‐pep residues, Y262 and F265, provide the strongest binding interactions in the GlpG exosite (Fig. 6, Supporting Information Table SI). The partially unwound backbone of 33‐pep TMD, with a bent conformation, manifests negligible binding interactions (< |0.1| kcal/mol) with some residues of the GlpG exosite in the non‐covalent pre‐catalytic complex (Fig. 6, Supporting Information Table SI). Our molecular modeling results fit the literature suggestions of the unwinding of the substrate helical transmembrane domain (TMD) in the rhomboid exosite.12, 14, 17, 18
Figure 5.
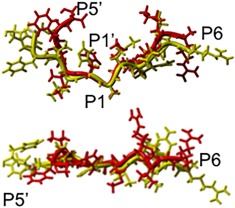
Two projections on the overlapping of P6 – P5' residues of 11‐pep (red) and 33‐pep (yellow) substrates. Superposition on S201 and H254 backbone atoms of GlpG in the pre‐catalytic non‐covalent complex.
Figure 6.
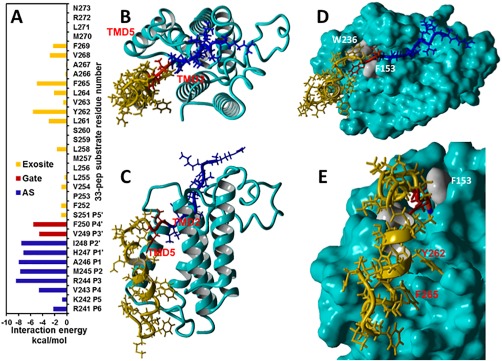
Analysis of 33‐pep substrate residues selectivity to the AS and exosite of GlpG (cyan) in the MD generated pre‐catalytic non‐covalent complex conformational cluster centroid competent for the concerted non‐covalent complex to TC conversion. (A): Interaction energies of 33‐pep residues with the AS (blue), gating residues (red) and exosite (orange) of GlpG. (B, C): The non‐covalent complex of 33‐pep (in ribbon style) in top and frontal view. The 33‐pep substrate is color‐coded as in A. (D): Top view of the 33‐pep alignment in the AS and exosite of GlpG. The gating residues F153 and W236 are colored grey. (E): Residues Y262 and F265 of 33‐pep are aligned in separate binding cavities on the molecular surface of the exosite of GlpG. Y262 and F265 have the strongest binding interactions with GlpG exosite (panel A and Supporting Information Table SI).
The combined influence of concerted TC formation and exosite interactions on the kinetic efficiency of substrate processing
The 33‐pep provides insight at the molecular level into the experimentally observed phenomenon of low kcat of rhomboid proteases in hydrolysis of intramembrane proteins.12 The experimental kcat is a macroscopic parameter of the reaction system, interpreted in statistical physics as an average value on all conformational ensemble. According to our analysis, in about 80% of the conformers of 33‐pep non‐covalent complex with GlpG one or both Ser201 Oγ – His254 Nɛ and Ser201 Oγ – Ala246 C interatomic distances are too long. Therefore, activation barriers should be high in most conformers, making the catalytic processing of 33‐pep substrate difficult, if not impossible at all. The observed conformational factor is yet another explanation, in addition to the previously identified low general base catalytic efficiency of H254 in rhomboid protease.12, 13 The calculated fraction of catalytically productive non‐covalent complex conformers of 11‐pep substrate is 53 times smaller than for 33‐pep, supporting the hypothesis that GlpG is not adopted for the hydrolysis of peptide substrates without TMD effectively aligned in the exosite. The identified crucial role of the TMD bound to the rhomboid protease exosite in substrate processing is confirmed by the experimental observation that the cleavage rate of a substrate decreases significantly upon truncating the TMD domain by more than five amino acids from the substrate C terminus.21
The substrate gating mechanism and non‐competitive character of peptidyl aldehyde inhibitors
The kinetics of rhomboid protease inhibition by covalent reversible peptide aldehydes, bound in the enzyme active site, is non‐competitive.14 The derived conclusion was that in the kinetically observed Michaelis complex the substrate is bound in the enzyme exosite with its TMD, but it does not occupy the enzyme active site. It provided a direct experimental evidence for the hypothesis of two successive, functionally separated, enzyme‐substrate non‐covalent states: a Michaelis complex and a pre‐catalytic complex with the substrate's TMD bound to the intramembrane‐located exosite while its cleavage site is aligned in the enzyme active site.12, 14, 17, 18 Our MD simulations enable atomic level comparative analysis of the geometries of the pre‐catalytic complex of GlpG with the transmembrane 33‐pep substrate and crystal structures of complexes formed by GlpG with peptidyl aldehyde inhibitors. The widely discussed role of the F153 (TMD2) and W236 (TMD5) residues in gating of intramembrane protein substrates is still controversial. The “lateral gate model” suggests that rhomboid TMD5 and L5 are simultaneously moving, with TMD5 acting as a lateral substrate gate opening for the substrate entrance.9 An alternative “L5 cap model” proposes that the flexible L5 loop is the only mobile element in the structure providing the substrate access to the active site.32 The distance between F153 and W236 Cα atoms, 9.6 Å in TC from WT GlpG (5f5b.pdb) and 11.7 Å from Y205F mutant (5f5j.pdb)14 is shorter than the 12.2 Å distance in the MD simulated pre‐catalytic complex formed with 33‐pep in this work. Our observation that the gate opening between TMD2 and TMD5 causes conformational change of the F153 and W236 side chains is in agreement with the 13 Å distance reported in the molecular modeling of the Michaelis complex of rhomboid protease with a substrate spanning the P4 – P3′ subsites.11 We superimposed the Cα, N, C backbone atoms of the S201 and H254 catalytic residues in the MD generated catalytically productive conformational cluster centroid of the pre‐catalytic complex formed by the 33‐pep substrate with TC crystal structures (WT GlpG, 5f5b.pdb and TMD2‐TMD5 gate‐open Y205F mutant, 5f5j.pdb) formed by the non‐competitive reversible covalent peptidyl aldehyde inhibitor Ac‐VRMA‐CHO (Figs. 7 and 8). The 33‐pep substrate residues H247 (P1′), V249 (P3′), and F250 (P4′) have the strongest binding interaction energies with F153 of GlpG, and V249 (P3′), F250 (P4′) interact also with W236 in the pre‐catalytic complex (Supporting Information Table SI). In contrast, alignment of 33‐pep in the GlpG exosite in both examined TC's formed by the peptidyl aldehyde is blocked by bumping into the side chains of F153 and W236 (Figs. 7 and 8). It is worth noting that in the apo form of GlpG the positions of the side chains of F153 and W236 are also too close for the penetration of the cleavable fragment of the transmembrane peptide substrate into the enzyme active site (see for instance the crystal structures 2xov.pdb and 2o7l.pdb). Our computational results favor the “lateral gate model”, where in the first step rhomboid protease discriminates substrates from non‐substrates by recognition in the exosite located between TMD2 and TMD5. The exosite in GlpG is spatially separated from the enzyme active site. The protein substrate bound to the exosite forms a non‐covalent Michaelis complex, which is thermodynamically stabile with a lifetime sufficient for conformational reorganization in the next step. The latter includes gate opening between GlpG F153 and W236 and unwinding of the substrate transmembrane helix for proper alignment of the substrate scissile fragment in the enzyme active site. Consequently, the protein substrate is non‐covalently bound to rhomboid in two distinct areas – the active site and the exosite, forming a conformationally pre‐organized enzyme‐substrate non‐covalent pre‐catalytic complex, the starting point for the following catalytic processing.
Figure 7.
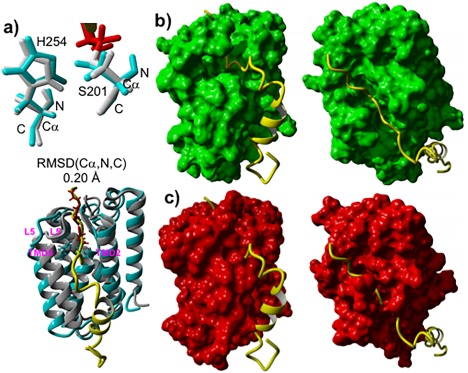
Comparison of 3D structures of GlpG complexes: TC (5f5b.pdb) formed with Ac‐VRMA‐CHO peptidyl aldehyde covalent inhibitor versus MD generated pre‐catalytic non‐covalent complex conformational cluster centroid relevant to the peptide catalytic hydrolysis formed with 33‐pep substrate. (A): Superposition on the Cα, N, C backbone atoms of S201 and H254 catalytic residues of GlpG in TC (grey) with GlpG in the pre‐catalytic non‐covalent complex (cyan), inhibitor colored in red. (B): Frontal and top view of the alignment of 33‐pep (yellow) in the non‐covalent complex molecular surface of GlpG (green). (C): Frontal and top view of the alignment of 33‐pep (yellow) in the TC crystal structure molecular surface of GlpG (red).
Figure 8.
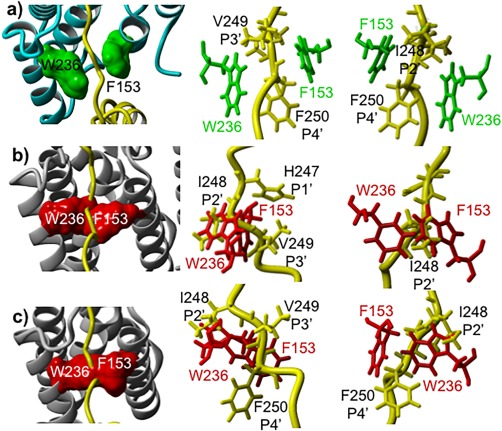
Comparison of 3D structures of the gating of 33‐pep substrate with its TMD fragment between GlpG F153 and W236 residues located on TMD2 and TMD5. MD generated catalytically productive conformational cluster centroid of the pre‐catalytic non‐covalent complex formed with 33‐pep substrate (yellow) versus TC's formed by the non‐competitive covalent peptidyl aldehyde inhibitor Ac‐VRMA‐CHO. (A): Different projections on the MD generated non‐covalent complex of GlpG (cyan) with 33‐pep substrate. F153 and W236 residues and their molecular surfaces are colored in green. (B): Different projections on the crystal structure of TC (5f5j.pdb) formed by Y205F mutant GlpG (grey) with superposed (on the backbone atoms of S201 and H254 catalytic residues of GlpG) 33‐pep substrate. F153 and W236 residues and their molecular surfaces are colored in red. (C): Different projections on the crystal structure of TC (5f5b.pdb) formed by wt GlpG (grey) with superposed 33‐pep substrate. F153 and W236 residues and their molecular surfaces are colored in red.
The calculated significant per‐residue interaction energies between R244 (P3) and M245 (P2) residues of Ac‐VRMA‐CHO inhibitor and GlpG active site residues in both TC's (Supporting Information Table SIII) confirmed the suggestion of the important role of these recognition subsites in the inhibitor binding to the enzyme AS.14 Moreover, P2 and P3 residues of the peptidyl aldehyde have stronger binding interactions with the enzyme AS in both TC's than analogous residues of the 11‐pep and 33‐pep substrates in the pre‐catalytic complexes (see for comparison Supporting Information Tables SI and SIII). It is worth noting, in addition to the oxyanion hole interactions with S201 NH backbone, H150 and N154 side chains14 (see also Supporting Information Table SIII), the strong electrostatic stabilizing interaction between the oxyanion of TC and the positively charged protonated catalytic H254. The strongest covalent binding interaction of GlpG catalytic S201 to the aldehyde group (Supporting Information Table SIII) is the dominant factor of the thermodynamic stability of TC formed by the reversible covalent inhibitor, in contrast to the short‐living species of TC formed by the amide group of a peptide substrate. The latter phenomenon originates in the intrinsic nature of serine proteases. Its theoretical explanation, as well as the nature of binding of the warheads of reversible covalent inhibitors in the enzyme active site and their application to rational inhibitor design, has been recently reviewed.33
Conclusions
In this work, we analyzed how the binding of a TMD fragment of a transmembrane peptide substrate in the exosite of GlpG rhomboid protease affects the efficiency of catalysis. We applied various methods of molecular modeling, including all atom molecular dynamics simulation in membrane of non‐covalent complexes formed by GlpG with 11‐pep and 33‐pep substrates. Statistical analysis of the MD generated conformations of the 33‐pep substrate pre‐catalytic complex demonstrated that concerted mechanism of substrate processing by GlpG rhomboid protease can be realized only for 22% of all conformers. This is yet another explanation to the poor efficiency of rhomboid protease, in addition to the experimentally identified low kcat 12 and theoretically derived weak general base catalytic efficiency of H254.13 The much lower percentage of only 0.4% for catalytically competent conformations of the non‐covalent complex with the 11‐pep substrate, which does not have a TMD fragment, correlates well with experimental observations,21 and thus supports the hypothesis of rhomboid protease inability to process substrates without TMD fragments. Our computational results favor the two‐step “lateral gate model” of the pre‐catalytic complex formation between rhomboid protease and transmembrane protein substrate. In this model, the first step is Michaelis complex formation, where substrate is bound to the enzyme exosite located between TMD2 and TMD5, spatially separated from the enzyme active site. This special role of the enzyme exosite in primary substrate recognition is the origin of the non‐competitive inhibition of rhomboid protease by peptide aldehydes. The next step is formation of the conformationally pre‐organized enzyme‐substrate non‐covalent pre‐catalytic complex, where the protein substrate is non‐covalently bound to rhomboid in both the active site and exosite. Our results on the model 33‐pep transmembrane substrate confirmed the hypothesis that gate opening between GlpG F153 and W236 is a prerequisite for the penetration and alignment of the substrate unwound scissile fragment in the enzyme active site. We conclude that the concerted mechanism of a transmembrane substrate pre‐catalytic complex to TC conversion and the exosite recognition and discrimination of substrates by their TMD fragment are two intrinsically connected key features of the rhomboid protease catalytic nature.
Methods
Construction and MD simulation of non‐covalent complex of GlpG rhomboid protease with 11‐pep substrate
The initial 3D structure of GlpG non‐covalent complex with the 11‐pep peptide substrate was constructed applying graphical interface and sequential geometry optimization protocols by YASARA Structure molecular modeling software.22 We used the 5f5b.pdb file, containing E. coli GlpG rhomboid protease covalent complex with Ac‐VRMA‐CHO peptidyl aldehyde inhibitor, as a structural template. The 11‐pep consists of 11 amino acid residues spanning P6‐P5′ binding subsites (the 241–251 amino acid sequence fragment of Gurken protein, P42287 registration code in UniProt databank). The step‐by‐step protocol includes:
Standard protonation states were used for all ionizable residues: Asp, Glu, Arg, Lys. All His residues where neutral, containing one proton on Nδ except for His141 and His150 with one proton on Nɛ. The covalent bond between the catalytic Ser201 and the carbonyl carbon of the inhibitor warhead was broken. The aldehyde warhead of the inhibitor was deleted and substituted for His247 (P1′) amino acid residue by addition to Ala246 (P1) at the peptide C‐terminal.
Vacuum minimization of the new non‐covalent complex was applied with the FF14SB force field, PME electrostatics, and 10.5 Å cutoff. The backbones of GlpG rhomboid and the peptide substrate were frozen, except for His247 residue of the substrate. The side chains of GlpG were free for optimization, except for those crucial for catalysis: Ser201, His254, His150, and Asn154, in order to maintain the required geometry of H‐bonds between His254 and Ser201 and between the substrate carbonyl group and the oxyanion hole.
In the following several steps, the substrate was extended in its C‐end by the sequential addition of Ile248 (P2′), Val249 (P3′), Phe250 (P4′), and Ser251 (P5′), and the optimization algorithm presented above. The N‐terminal of the substrate was then also extended by Lys242 (P5) and Arg241 (P6). Thus, this protocol provided the 11‐pep peptide substrate extended from P6 to P5′ residues. At every following step of the growing substrate, the previously added residue was frozen for the optimization. It is worth mentioning that every sequential addition at the C‐end of the substrate was controlled and corrected by the condition that the new carboxylate should be directed toward the gate between TM2 and TM5.
When construction of the non‐covalent complex was completed, full unrestricted geometry optimization was applied, and the resulting 3D structure was saved.
The generated non‐covalent complex was used as input structure for the Charmm‐Gui software34, 35 that immersed the complex into a membrane bilayer solubilized in water, with physiological concentration of Na+ and Cl‐ ions (0.15 M, including 3 Cl‐ counter ions to neutralize the protein charge). The simulated membrane contains 200 molecules of 1‐palmytoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphatidylethanolamine (POPE), which constitutes ∼80% of the E. coli membrane.36 The Charmm‐Gui output structure of the non‐covalent complex in the membrane system, containing 59,581 atoms, was used as an input for multistep processing by AMBER 14 software,26, 27 which included minimization, heating up to 310 K, equilibration, and 750 ns production molecular dynamics (MD) simulation consisting of fifteen 50 ns successive steps. We use SHAKE constraints to fix hydrogen atom bond lengths, allowing to run the MD with 2 fs time steps. Every sequential MD step was restarted from the previous one, applying a random seeds generator. The FF14SB37 (for protein and substrates), and Lipid14 (for lipids)38 force‐fields were used. The production dynamics was conducted with Langevin thermostat and NPT ensemble under pressure of 1 atm and temperature of 310 K. The details of the AMBER14 simulation protocol are analogous to that used for 33‐pep and are presented below in the METHODS corresponding chapter.
Generation of non‐covalent complex formed by E. Coli GlpG rhomboid protease with 33‐pep substrate
Homology modeling
In order to simulate a non‐covalent complex with a peptide substrate containing a TMD and aligned in both the GlpG exosite and active site, we used 33‐pep substrate, a peptide fragment of 33 residues of Gurken amino acid sequence, a transmembrane substrate of rhomboid proteases. Gurken does not have a 3D structure in the PDB. Therefore, we used the online homology modeling by I‐TASSER software23, 24, 25 to construct a Gurken fragment – a peptide containing 65 residues (231‐ 295). The 3D structure of the best Model 1 (C‐score = −1.94) is presented in Supporting Information Figure S1(a). C‐score is a confidence score for estimating the quality of predicted models by I‐TASSER, calculated based on the significance of threading template alignments and the convergence parameters of the structure ensemble simulations. C‐score is typically in the range of [‐5, 2], where a C‐score of higher value signifies a model with a high confidence. The best structural alignment of the Model 1, identified by I‐TASSER on the 2rp4.pdb molecule D template, has TM‐score = 0.766 and RMSD = 1.78 Å. TM‐score is a scale for measuring the structural similarity between two structures. A TM‐score > 0.5 indicates a model of correct topology.
Construction and MD relaxation of the GlpG non‐covalent pre‐catalytic complex with 33‐pep by YASARA
We used the structure of the non‐covalent complex with 11‐pep as a starting point for the construction of the non‐covalent complex with 33‐pep substrate. The three backbone atoms (Ca, N, C) of 11‐pep S251 (P5′) residue were superposed with the corresponding S251 three backbone atoms of the 251–273 helical fragment of the homology generated peptide. Applying visual modeling by YASARA, the 251–273 peptide helical fragment was manually oriented in a relevant position toward TMD2 and TMD5 of GlpG. The structure of the constructed 33‐pep pre‐catalytic complex was corrected by sequential steps of gas‐phase constrained minimization with AMBER14 (FF14SB) force field37 implemented in YASARA [Supporting Information Fig. S1(b)]. Finally, we applied the YASARA algorithm, which combines sequential automated steps by the script (that we adopted for the specificity of our system), including protein immersion in membrane (250 POPE lipids), solvation in explicit water molecules, addition of Na+ and Cl‐ in physiological concentration (including neutralizing Cl‐ counter ions), and finally MD simulation of the system in a periodic cell at 310 K and constant pressure of 1 bar (see reference 22 for the algorithm details) [Supporting Information Fig. S1(c)]. After 250 ps of MD relaxation, the full system from the final frame in the MD trajectory was optimized and the non‐covalent complex of GlpG with 33‐pep was separated from the system and used for the next modeling stage.
Generation of GlpG with 33‐pep non‐covalent complex in membrane by Charmm‐Gui
The isolated from membrane non‐covalent complex of 33‐pep, generated in the previous stage by YASARA MD, was passed to the online Charmm‐Gui package34, 35 for the preparation of the full system of the protein immersed in a membrane bilayer (300 POPE lipid molecules) and solubilized in water with the 0.15M NaCl, including neutralizing counter ions [Supporting Information Fig. S1(d)].
Simulation of non‐covalent complex of GlpG with 33‐pep substrate by AMBER14
The above generated non‐covalent complex of 33‐pep in membrane, containing 87,610 atoms in total (including 300 POPE lipids, 15,506 TIP3P water molecules and salt ions), was converted into the AMBER file format (see AMBER14 manual for details) and then subjected to a multi‐stage simulation procedure in a periodic cell (the AMBER14 simulation protocol is analogous to that used for 11‐pep). The FF14SB (for protein, substrates and solvent molecules), and Lipid14 (for lipids) force‐fields were used.
Minimization
The first stage was a four steps energy minimization (each with 5000 steps of steepest descent minimization followed by maximum 35,000 steps of conjugate gradient minimization) at constant volume with 50 kcal/mol*Å2 force constant for sequential restraints of different system fragments: (1) enzyme + substrate and lipids; (2) lipids and water solvent; (3) water solvent only; and (4) unrestrained minimization of the full system.
Heating
After the initial minimization, in order to avoid lipid structure explosion, the system was slowly heated in six simulation steps from 0 K to the production temperature of 310 K in 50 K steps (the last one of 60 K). The MD simulation of every heating step was of 70 ps. The Langevin thermostat was used to control the temperature using a collision frequency of 1.0 ps−1. The SHAKE constraints were used to fix hydrogen atom bond lengths allowing to run with a 2 fs time step. Since in low temperature the calculation of pressure is inaccurate, the response of the barostat can distort the system, so MD simulation of every heating step was conducted in NVT ensemble. The protein and lipid molecules were restrained using harmonic approximation with gradually decreasing force constants (kcal/mol*Å2): 10, 5, 5, 5, 2.5, and 1.5. Starting from 150 K, each heating step was followed by a step of unrestrained relaxation MD simulation of 250 ps in NPT ensemble.
MD equilibration
After the system was heated to 310 K, allowing the density of the system to equilibrate, we ran 10 repeated MD restarts each of 500 ps with a time step of 2 fs (SHAKE constraint) under Langevin thermostat and NPT ensemble with pressure of 1 atm. No positional restraints were applied. Random seeds by pseudorandom number generator were used to restart the simulations in repeated segments.
Production MD stage
In the previous equilibration stage, the temperature of 310 K and stable density were reached. In this last stage, we ran production dynamics [Supporting Information Fig. S1(e)] with Langevin thermostat and NPT ensemble under pressure of 1 atm. The total run time of 450 ns consisted of nine 50 ns steps, each of 2 fs (SHAKE constraint). Every MD step was restarted from the previous one applying random seeds generator.
Supporting information
Supporting Information
References
- 1. Lee JR, Urban S, Garvey CF, Freeman M (2001) Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell 107:161–171. [DOI] [PubMed] [Google Scholar]
- 2. Urban S, Lee JR, Freeman M (2001) Drosophila rhomboid‐1 defines a family of putative intramembrane serine proteases. Cell 107:173–182. [DOI] [PubMed] [Google Scholar]
- 3. Kinch LN, Grishin NV (2013) Bioinformatics perspective on rhomboid intramembrane protease evolution and function. Biochim Biophys Acta 1828:2937–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lemberg MK (2013) Sampling the membrane: function of rhomboid‐family proteins. Trends Cell Biol 23:210–217. [DOI] [PubMed] [Google Scholar]
- 5. Freeman M (2014) The rhomboid‐like superfamily: molecular mechanisms and biological roles. Annu Rev Cell Dev Biol 30:235–254. [DOI] [PubMed] [Google Scholar]
- 6. Strisovsky K (2016) Why cells need intramembrane proteases ‐ a mechanistic perspective. febs J 283:1837–1845. [DOI] [PubMed] [Google Scholar]
- 7. Strisovsky K (2016) Rhomboid protease inhibitors: emerging tools and future therapeutics. Sem Cell Dev Biol (2016) 60:52–62. [DOI] [PubMed] [Google Scholar]
- 8. Wang Y, Zhang Y, Ha Y (2006) Crystal structure of a rhomboid family intramembrane protease. Nature 444:179–180. [DOI] [PubMed] [Google Scholar]
- 9. Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Urban S, Shi Y (2006) Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol 13:1084–1091. [DOI] [PubMed] [Google Scholar]
- 10. Vinothkumar KR, Strisovsky K, Andreeva A, Christova Y, Verhelst S, Freeman M (2010) The structural basis for catalysis and substrate specificity of a rhomboid protease. EMBO J 29:3797–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zoll S, Stanchev S, Began J, Skerle J, Lepšík M, Peclinovská L, Majer P, Strisovsky K (2014) Substrate binding and specificity of rhomboid intramembrane protease revealed by substrate‐peptide complex structures. EMBO J 33:2408–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dickey SW, Baker RP, Cho S, Urban S (2013) Proteolysis inside the membrane is a rate‐governed reaction not driven by substrate affinity. Cell 155:1270–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Uritsky N, Shokhen M, Albeck A (2016) Stepwise versus concerted mechanisms in general‐base catalysis by serine proteases. Angew Chem Int Ed 55:1680–1684. [DOI] [PubMed] [Google Scholar]
- 14. Cho S, Dickey SW, Urban S (2016) Crystal structures and inhibition kinetics reveal a two‐stage catalytic mechanism with drug design implications for rhomboid proteolysis. Mol Cell 61:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stein RL, Strimpler AM (1987) Slow‐binding inhibition of chymotrypsin and cathepsin G by the peptide aldehyde chymostatin. Biochemistry 26:2611–2615. [DOI] [PubMed] [Google Scholar]
- 16. Hedstrom L (2002) Serine protease mechanism and specificity. Chem Rev 102:4501–4524. [DOI] [PubMed] [Google Scholar]
- 17. Strisovsky K, Sharpe HJ, Freeman M (2009) Sequence‐specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell 36:1048–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moin SM, Urban S (2012) Membrane immersion allows rhomboid proteases to achieve specificity by reading transmembrane segment dynamics. eLife 1:e00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Urban S (2010) Taking the plunge: integrating structural, enzymatic and computational insights into a unified model for membrane‐immersed rhomboid proteolysis. Biochem J 425:501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brooks CL, Lemieux MJ (2013) Untangling structure‐function relationships in the rhomboid family of intramembrane proteases. Biochim Biophys Acta 1828:2862–2872. [DOI] [PubMed] [Google Scholar]
- 21. Tichá A, Stanchev S, Škerle J, Began J, Ingr M, Švehlová K, Polovinkin L, Růžička M, Bednárová L, Hadravová R, Poláchová E, Rampírová P, Březinová J, Kašička V, Majer P, Strisovsky K (2017) Sensitive versatile fluorogenic transmembrane peptide substrates for rhomboid intramembrane proteases. J Biol Chem 292:2703–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.YASARA is a molecular‐graphics, ‐modeling and ‐simulation program. http://www.yasara.org/
- 23.The I‐TASSER suite: protein structure and function prediction by homology modeling. http://zhanglab.ccmb.med.umich.edu/I-TASSER/ [DOI] [PMC free article] [PubMed]
- 24. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I‐TASSER suite: protein structure and function prediction. Nature Methods 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang J, Zhang Y (2015) I‐TASSER server: new development for protein structure and function predictions. Nuc Acids Res 43:W174–W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.AMBER14 Software Suite. http://ambermd.org
- 27. Case DA, Betz RM, Botello‐Smith W, Cerutti DS, Cheatham III TE, Darden TA, Duke RE, Giese TJ, Gohlke HA, Goetz W, Homeyer N, Izadi S Janowski P Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen HT, Nguyen I, Omelyan A, Onufriev DR, Roe A, Roitberg C, Sagui CL, Simmerling H, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, York DM, Kollman PA (2014) AMBER 2014. San Francisco: University of California. [Google Scholar]
- 28. Rawlings ND, Waller M, Barrett AJ, Bateman A (2014) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 42:D503–D509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MEROPS the peptidase database. https://merops.sanger.ac.uk/index.shtml
- 30. Schilling O, Overall CM (2008) Proteome‐derived, database‐searchable peptide libraries for identifying protease cleavage sites. Nature Biotech 26:685–694. [DOI] [PubMed] [Google Scholar]
- 31. Mobley DL, Dill KA (2009) Binding of small‐molecule ligands to proteins: “What you see” is not always “what you get”. Structure 17:489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xue Y, Ha Y (2013) Large lateral movement of transmembrane helix S5 is not required for substrate access to the active site of rhomboid intramembrane protease. J Biol Chem 288:16645–16654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shokhen M, Hirsch M, Khazanov N, Ozeri R, Perlman N, Traube T, Vijayakumar S, Albeck A (2014) From catalytic mechanism to rational design of reversible covalent inhibitors of serine and cysteine hydrolases. Isr J Chem 54:1137–1151. [Google Scholar]
- 34.CHARMM‐GUI a web‐based graphical user interface to generate various molecular simulation systems. http://www.charmm-gui.org/
- 35. Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks IIICL, MacKerellJr AD, Klauda JB, Im W (2016) CHARMM‐GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J Chem Theory Comput 12:405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Epand RF, Savage PB, Epand RM (2007) Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (ceragenins). Biochim Biophys Acta 1768:2500–2509. [DOI] [PubMed] [Google Scholar]
- 37. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, KE, Hauser KE, Simmerling K (2015) FF14SB: Improving the accuracy of protein side chain and backbone parameters from FF99SB. J Chem Theory Comput 11:3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dickson C, Madej B, Skjevik A, Betz R, Teigen K, Gould I, Walker RC (2014) Lipid14: the amber lipid force field. Chem Theory Compu 10:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information