

Abstract
The human RAD51 recombinase possesses DNA pairing and strand exchange activities that are essential for the error-free, homology-directed repair of DNA double-strand breaks. The recombination activities of RAD51 are activated upon its assembly into presynaptic filaments on single-stranded DNA at resected DSB ends. Defects in filament assembly caused by mutations in RAD51 or its regulators such as BRCA2 are associated with human cancer. Here we describe two novel RAD51 missense variants located in the multimerization/BRCA2 binding region of RAD51. F86L is a breast tumor-derived somatic variant that affects the interface between adjacent RAD51 protomers in the presynaptic filament. E258A is a germline variant that maps to the interface region between the N-terminal and RecA homology domains of RAD51. Both variants exhibit abnormal biochemistry including altered DNA strand exchange activity. Both variants inhibit the DNA strand exchange activity of wild-type RAD51, suggesting a mechanism for negative dominance. The inhibitory effect of F86L on wild-type RAD51 is surprising since F86L alone exhibits robust DNA strand exchange activity. Our findings indicate that even DNA strand exchange-proficient variants can have negative functional interactions with wild-type RAD51. Thus heterozygous F86L or E258 mutations in RAD51 could promote genomic instability, and thereby contribute to tumor progression.
Keywords: RAD51, recombination, homology-directed repair, cancer, variant, presynaptic filament
1. Introduction
Human RAD51 protein is a member of the highly conserved RecA/Rad51 family of recombinase enzymes. RAD51 catalyzes DNA pairing and strand exchange reactions that are central to the processes of homologous recombination and homology-directed repair of DNA double-strand breaks (1). Defects in RAD51-dependent HR/HDR pathways lead to genomic instability in the affected cells and are directly associated with human cancer (2,3).
RAD51 biochemical properties include sequence-nonspecific binding to ssDNA and dsDNA, ssDNA-stimulated ATP hydrolysis, and ATP-dependent pairing and exchange of homologous ssDNA/dsDNA segments (4). The catalytic activities of RAD51 are activated upon its assembly into presynaptic filaments. These nucleoprotein filaments form on the ssDNA that is generated at the resected ends of DNA double-strand breaks. Presynaptic filaments undergo dynamic changes in association, dissociation, and conformation in response to ATP binding and hydrolysis (4–6). The active, ATP-bound form of the filament has a high helical pitch of 90–130Å and contains ssDNA that is significantly stretched (5,6). The ADP-bound form has a helical pitch of 65–85Å.
Structurally, RAD51 is organized into two domains (Fig. 1A): Major determinants of RAD51 catalytic activity reside in the C-terminal RecA homology domain (RHD), also referred to as the catalytic core domain, which contains the Walker A and B motifs as well as DNA binding loops L1 and L2. The α-helical N-terminal domain (NTD) is connected to the RHD by a linker peptide. Both NTD and linker are important for protomer-protomer interactions within the RAD51 filament (7,8) and this region is also the site of interactions with the BRC repeat motifs of tumor suppressor protein BRCA2 (9) (Figs. 2–3). Interactions between protomers in the filament, and between RHD and NTD domains, are dynamic and are regulated by the cycles of DNA binding and ATP hydrolysis as well as by exogenous factors including BRCA2 (4–9).
Figure 1. Locations of F86L and E258A mutations in the primary structure of human RAD51 protein.

(A) Positions of mutations are denoted by red asterisks. Walker A and B motifs and DNA binding loops L1 and L2 are shown in yellow and black, respectively. F86L occurs in the linker between the N-terminal (NTD, cyan) and RecA homology domains (RHD, gray), while E258A occurs in the RHD upstream of L2. (B) Protein sequence alignment in the vicinity of Phe-86 (red) in the linker region between NTD and RHD. (C) Alignment in the vicinity of Glu-258 (red) in the L2 region of RAD51.
Figure 2. Sites of the F86L and E258A mutations in the RAD51 protomer.
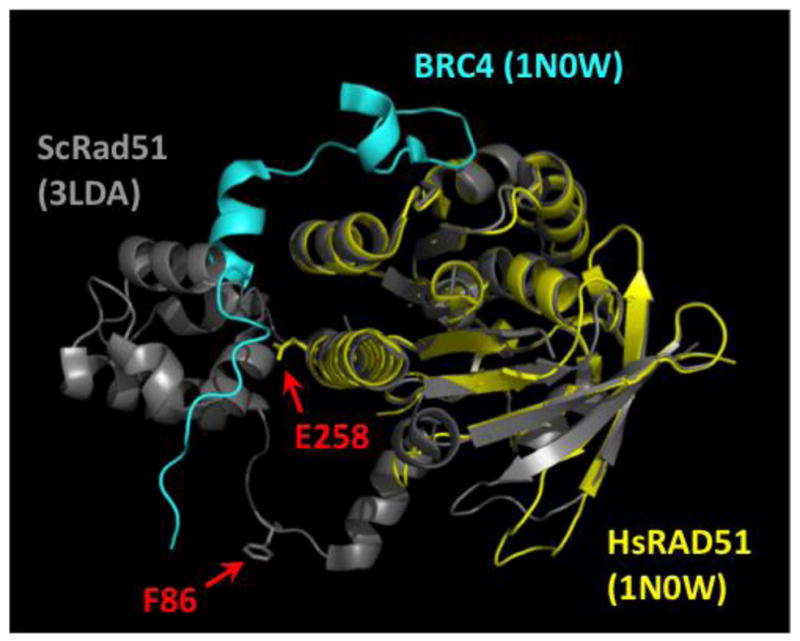
The structure of the human RAD51 RecA homology domain (yellow) in complex with the BRC4 peptide from BRCA2 (cyan) (PDB ID no. 1N0W) is shown overlaid onto the structure of one protomer from the S. cerevisiae Rad51 filament (PDB ID no. 3LDA), with the positions of human RAD51 residues Phe-86 and Glu-258 indicated by red arrows. The position of Phe-86 is inferred from the alignment with ScRad51, where the conserved residue is Phe-144 (Fig. 1A–B).
Figure 3. Structural contexts of the F86L and E258A mutations in the RAD51 filament.

(A) Structural context of human RAD51 Phe-86 residue inferred by alignment to a model of the protomer-protomer interface region in the yeast Rad51 filament (PDB ID no. 3LDA). Human RAD51 residue numbering is indicated. Phe-86 from one protomer (cartoon, cyan) binds to a hydrophobic pocket on the adjacent protomer (surface) comprised of residues Met-158’, Ile-160’, Leu-203’, and Met-210’. All of these residues are conserved between human and yeast, except for Met-158’ which is replaced by Leu-216’ in yeast Rad51. (B) Structural context of human RAD51 Glu-258 residue is informed by the structure of the NTD/RHD interface within one protomer of the yeast Rad51 filament (PDB ID no. 3LDA). Yeast Rad51 residue numbering is indicated. Red spheres represent ordered water molecules. A water-mediated hydrogen bond network connects side chains of Gln-316 (equivalent of Glu-258 in human RAD51) and Arg-312 from the RHD to main chain CO and NH functionalities in the NTD.
Changes in RAD51 recombination activity are a common feature of many human cancers. Many tumors exhibit changes in RAD51 expression levels (10–19). Others contain deleterious mutations in RAD51 itself (20–23), or in any of several tumor suppressor proteins (BRCA2, PALB2, p53 and others) that interact with RAD51 and regulate its activity (2–4,24–26). Changes in RAD51 expression may correlate with the progress of cancer and/or with the development of tumor resistance or sensitivity to chemotherapeutic drugs (10–12). RAD51 overexpression occurs in malignant prostate cancer and small cell lung carcinoma. In invasive ductal breast cancer RAD51 expression correlates directly with the histological grading of the tumor (11). On the other hand ~30% of sporadic breast tumors and cell lines exhibit down-regulation of RAD51 expression (14–17). RAD51 down-regulation increases the radiosensitivity of prostate cancer and malignant glioma cells (18,19).
Recent studies have identified several RAD51 missense variants that are associated with cancer. Three breast cancer-associated variants—germline R150Q (20–22) and somatic D149N and G151D (21), map to a Schellman loop or “paperclip” motif in RAD51 that interacts with a reported p53 binding site (24–26). Two somatic variants, Q268P and Q272L, from lung and kidney tumors respectively, map to the DNA binding loop 2 (L2) region of RAD51 (23). All five of these variants exhibit abnormal DNA binding properties, and several have altered catalytic properties including decreased ATPase activity in R150Q and G151D and decreased DNA strand exchange activity in Q268P and Q272L (21,23). Likewise all of these variants appear to form mixed presynaptic filaments with wild-type RAD51, which have intermediate or variant-dominant biochemical properties. Intriguingly, expression of the G151D variant in human breast epithelial cell line MCF10A induces hyper-recombination plus doxorubicin- and IR-resistance (27). Two dominant-negative RAD51 variants, T131P and A293T, are associated with a novel FA-R subtype of Fanconi Anemia (28,29). Both T131P and A293T are genetically dominant, and cells expressing either variant are deficient in DNA interstrand crosslink repair. Together, the data indicate that disease-associated RAD51 missense variants have functional phenotypes, and that heterozygous expression of RAD51 variants can affect DNA recombination/repair functions through co-integration of the variant and wild-type proteins into hybrid presynaptic filaments (21,23,27–29).
In the current study we describe the biochemical properties of two new sequence variants of RAD51 protein—a somatic F86L variant from a breast carcinoma, and a germline E258A variant. Both mutations affect the multimerization/BRCA2 binding region of RAD51. Both variants exhibit altered thermal stability, reduced DNA binding affinity, and reduced ATPase activity compared to wild-type. F86L efficiently catalyzes DNA strand exchange reactions in vitro, whereas the strand exchange activity of E258A is severely reduced compared to wild-type. However, mixtures of either variant with wild-type RAD51 exhibit strong defects in DNA strand exchange activity compared to wild-type protein alone. The data support the hypothesis that incompatibilities between variant and wild-type RAD51 proteins may lead to “recombinationopathies” through the formation of hybrid presynaptic filaments with altered biochemical properties. Our findings extend our knowledge of the range of RAD51 dysfunctions that may occur in cancer and normal cells, and which could potentially serve as drivers of genomic instability and tumor progression.
2. Materials and Methods
2.1. Reagents
Chemicals, biochemicals, and enzymes were purchased from Sigma-Aldrich unless specifically stated. Restriction enzymes and T4 polynucleotide kinase were purchased from New England Biolabs. All reagents were analytical grade and solutions were made with Barnstead NANO-pure water. TE buffer contained 10 mM Tris-HCl, pH 8.0, and 1 mM EDTA.
2.2. Nucleic acids
Oligonucleotides were purchased from Operon. Circular M13mp18 ssDNA (7.3 kb) was prepared as described (30). Circular φX174 ssDNA (5.4 kb) was purchased from New England Biolabs. Supercoiled φX174 dsDNA (5.4 kbp) was purchased from Promega and was linearized by digestion with PstI restriction endonuclease. Linearized dsDNA molecules were purified using a DNA Clean-up Kit from Thermo Scientific. The concentrations of ssDNA and dsDNA stock solutions were determined by UV absorbance in a Nanodrop Spectrophotometer (Thermo Scientific), according to manufacturer’s instructions, and are expressed in units of micromoles of nucleotide residues per liter. All DNA molecules were stored at −20°C in TE buffer.
2.3. Site-directed mutagenesis of human RAD51 protein
Plasmid pET-15b expressing a His6-tagged version of the human RAD51 protein was a generous gift from Dr. Hitoshi Kurumizaka at Waseda University, Japan. The F86L and E258A mutations were introduced separately using the Q5 (New England Biolabs) site-directed mutagenesis protocol. The primers used are shown in Table 1 (oligos 1 and 2 for E258A; oligos 3 and 4 for F86L). PCR reactions were carried out according to the Q5 protocol and the resulting plasmids were sequenced at the University of Vermont Cancer Center DNA Analysis Facility to verify successful mutagenesis.
Table 1.
Lengths and sequences of oligonucleotide primers and substrates.
| Oligo No. | Length (b) | Sequence (5’ → 3’) | Use |
|---|---|---|---|
| 1 | 23 | CTCGCTGATGCGTTTGGTGTAGC | E258A forward primer |
| 2 | 18 | TCGCAGAAGCATCCGCAG | E258A reverse primer |
| 3 | 21 | TCCAATGGGTCTCACCACTGC | F86L forward primer |
| 4 | 20 | ACTAATTTAGCTGCCTCAGC | F86L reverse primer |
| 5 | 63 | ACAGCACCAGATTCAGCAATTAAGCTCTAAGCC ATCCGCAAAAATGACCTCAAAACAAAAGGA |
Strand exchange partial duplex |
| 6 | 32 | TCCTTTTGTTTTGAGGTCATTTTTGCGGATGG | Strand exchange partial duplex |
| 7 | 31 | ACAGCACCAGATTCAGCAATTAAGCTCTAAG | Strand exchange homol. duplex |
| 8 | 31 | CTTAGAGCTTAATTGCTGAATCTGGTGCTGT | Strand exchange homol. duplex |
| 9 | 25 | AAAAAAAAAAAAAAAAAAAAAAA*A (*denotes AF546 label position) |
DNA binding w/AF546 label |
| 10 | 25 | TTTTTTTTTTTTTTTTTTTTTTTTT | Compl. of #9 for dsDNA binding |
2.4. Purification of human RPA and RAD51 Proteins
Human RPA protein expression plasmid p11d-tRPA was a kind gift from Dr. Marc S. Wold at the University of Iowa. RPA was expressed in E. coli strain BL21 (DE3) and the protein was purified as described (31,32). Human RAD51 wild-type, F86L, and E258A proteins were expressed and purified from plasmid pET-15b in Rosetta2 (DE3) E. coli cells (Novagen, Inc., Madison, WI) as described (21). Purified RAD51 proteins were dialyzed into storage buffer consisting of 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.1 mM EDTA, 2 mM 2-mercaptoethanol, and 10% glycerol, frozen in liquid nitrogen and stored at −80°C. Protein concentrations were determined by UV absorbance using a Nanodrop Spectrophotometer (Thermo Scientific) and an extinction coefficient of 0.34 A280 = 1 mg/mL. All protein stock solutions were >95% pure as determined by SDS-PAGE, nucleic acid-free as determined by A280/A260 ratio, and nuclease-free as determined by agarose gel electrophoresis of treated DNA samples.
2.5. ATPase assays
Steady-state rates of ATP hydrolysis were measured using thin-layer chromatography (TLC) assays. Reactions at 37°C contained 30 mM MES (pH 6.2), 6 mM MgCl2, 30 mM KCl, 1 mM DTT, and 0.1 mg/mL BSA in a final volume of 50 μL. In addition the reaction mixtures contained 2.0 μM wild-type or variant RAD51, 12 μM M13mp18 ssDNA, and 1 mM ATP (containing 10 μCi/mL of α-[32P]-ATP). Reactions were initiated by the addition of ATP + α-[32P]-ATP. Aliquots (8 μL) were removed at various time points and quenched with 8 mM EDTA and 1% SDS (final concentrations). Quenched samples were spotted out at 1 μL volume onto PEI-cellulose TLC plates (20 X 20 cm) at 1 cm intervals. After all the samples were spotted and dried the TLC plates were developed with 0.75 M KH2PO4 and allowed to air dry. The TLC plates were exposed for 1.5 hours to a K-screen (Kodak) and scanned by a Bio-Rad Personal Molecular Imager-FX (University of Vermont Cancer Center DNA Analysis Facility). Quantification of the phosphorimage was performed by Quantity One v4.5.1 (Bio-Rad) software and subsequently fit using Graphpad Prism v5.0 (Graphpad Software Inc).
2.6. DNA strand exchange assays with φX174 substrates
DNA strand exchange reactions using φX174 ssDNA and dsDNA substrates were carried out as described (23). All reaction steps were carried out at 37ºC. The final volume of each reaction was 50 μL. All concentrations given are final concentrations. RAD51 wild-type, F86L, or E258A protein (7.5 μM) was preincubated with 30 μM φX174 ssDNA for 5 min in buffer containing 40 mM Tris-HCl (pH 7.8), 1 mM MgCl2, 100 mM KCl, 1 mM DTT, 2 mM ATP, 8 mM creatine phosphate, and 28 μg/mL creatine phosphokinase. Human RPA protein (2 μM) was added followed by a 5 min incubation, then 100 mM ammonium sulfate was added followed by a 1 min incubation. Reactions were initiated by the simultaneous addition of 30 μM (15 μM basepairs) linear φX174 dsDNA and 4 mM spermidine. At the indicated times, 6.5 μL aliquots were removed and brought to a final concentration of 0.8% SDS and 0.8 mg/mL Proteinase K, then incubated at 37ºC for 15 min. Following addition of 10% glycerol and a trace of bromophenol blue, the samples were loaded onto a 0.8% agarose gel in TAE buffer, and electrophoresed for 16 hrs at 25 V. Gels were stained with 1 μg/mL Sybr Gold and digitally photographed to record a negative image. Results were quantified by measuring the intensity of the joint molecule and nicked circle product bands relative to total DNA by using a Bio-Rad Personal Molecular Imager-FX with Quantity One (Bio-Rad) software (University of Vermont Cancer Center DNA Analysis Facility). Some reactions were modified to contain mixtures of wild-type and variant RAD51 proteins at ratios and total concentrations given in the text.
2.7. DNA strand exchange assays with oligonucleotide substrates
Oligo 8 was 5’-[32P] labeled using T4 polynucleotide kinase (NEB) according to manufacturer’s instructions, and excess label was removed using the Probe Quant G-50 microcolumn kit (GE Healthcare). The partial duplex DNA substrate was constructed by annealing oligo 5 to oligo 6 (11.7 μM molecules, each), and the homologous duplex DNA substrate was constructed by annealing [32P]-oligo 8 to oligo 7 (11.7 μM molecules, each), both in 30 mM HEPES, pH 7.0. In a PCR thermal cycler, annealing mixtures were heated at 95ºC for 5 min, followed by 60 min at 50ºC, and then cooled to 20ºC in 5ºC increments over 30 min. Strand exchange reactions were carried out at 30ºC under the following conditions (stated concentrations represent final concentrations in a final reaction volume of 50 μL): Preincubation mixture contained 30 mM HEPES, pH 7.0, 20 mM KCl, 0.35 μM oligo 5/6 partial duplex DNA, 10.5 μM RAD51 (WT, F86L, or E258A), 6.2 mM ATP, and 2 mM MgCl2. Control reactions contained an equivalent volume of RAD51 storage buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.1 mM EDTA, 2 mM 2-mercapotethanol, 10% glycerol) in place of protein. After a 5 min preincubation, the concentration of MgCl2 was increased to 18 mM, and the reaction was allowed to preincubate for another 15 min. The reaction was started by adding 0.35 μM [32P]-oligo 8/7 homologous duplex DNA. Aliquots of 10 μL each were removed at 0, 30, 60, and 90 min and quenched by adding to 10 μL of stopping solution to yield final concentrations of 60 mM Tris-HCl, pH 7.5, 2% SDS, 50 mM EDTA, 2.5 mg/mL proteinase K, and 1x Promega Blue/Orange Loading Dye, then kept at room temperature prior to electrophoresis. Samples were run on a 4–20% TBE PAGEr Gold Precast Polyacrylamide gel (Lonza) and exposed to a K-screen for visualization. Images were taken using Bio-Rad Personal Molecular Imager-FX (Vermont Cancer Center DNA Analysis Facility), and data was quantified using Quantity One v4.5.1 (Bio-Rad).
2.8. Electrophoretic mobility shift assays
The properties of RAD51 and variant complexes with circular φX174 ssDNA and linear φX174 dsDNA were examined by electrophoretic mobility shift. Protein-dsDNA complexes were assembled in buffer containing 24 mM HEPES (pH 7.5), 1 mM MgCl2, 30 mM NaCl, 1 mM dithiothreitol, 0.4 mM 2-mercaptoethanol, 0.02 mM EDTA, 0.1 mg/mL BSA, 2% glycerol, and 1 mM ATP. For protein-ssDNA complex assembly, the NaCl concentration was increased to 45 mM. RAD51 wild-type, F86L, or E258A proteins (0–8 μM depending on experiment) were incubated with 20 μM φX174 ssDNA or 20 μM (10 μM basepairs) φX174 dsDNA for 20 min at 37ºC. Samples were loaded onto a 0.8% agarose gel in Tris-acetate-EDTA buffer, then electrophoresed for 16 hrs at 25 V. The gel was stained with 1 μg/mL Sybr Gold, trans-illuminated with UV light, and digitally photographed.
2.9. AlexaFluor 546 DNA binding assay
RAD51-DNA interactions were measured using DNA substrates labeled with the fluorophore AlexaFluor 546 where a fluorescence quenching effect occurs upon recombinase binding (21,33). Titration experiments were conducted by adding increasing amounts of RAD51 WT, F86L, or E258A protein to either 2 μM (nucleotides) of AlexaFlour 546-labeled ssDNA (oligo 9) or 2 μM basepairs of AlexaFluor 546-labeled dsDNA (oligo 9/10). Titrations were carried out at room temperature (21ºC) and in the presence/absence of 2 mM ATP in binding buffer containing 30 mM Tris, pH 7.5, 10 mM MgCl2, and 0.1 mM DTT. After each addition of protein, the mixture was allowed to incubate for 30 seconds before taking the fluorescence reading. Amplitudes of fluorescence quenching were quantified by determining the fraction of the fluorescence signal that was quenched by the addition of RAD51 at each concentration (dilution factors were taken into account). Amplitude values were plotted against concentration of RAD51. The binding curves were fitted to the Hill equation using Graphpad. For all binding experiments, the fluorophore was excited at 554 nm and fluorescence was monitored between 560–600 nm. All slit widths were set at 5 nm.
2.10. Thermofluor assay for thermal stability of proteins
The thermofluor stability assay was conducted as previously described with minor changes (34,35). Samples were assembled directly into a 96-well plate (USA Scientific) where each well contained assay buffer (30 mM Tris, pH 7.5; 10 mM magnesium chloride; 0.1 mM dithiothreitol), 5 μM RAD51, 25x Sypro Orange (Invitrogen), and either 0 or 2 mM ATP. Once assembled, the plate was sealed with an optical adhesive film (USA Scientific). Temperature was controlled using an AB 7500 Fast Sequence Detection System (UVM Advanced Genome Technologies Core) where the temperature ranged from 25°C to 95°C with a 1°C increment and a 1 min dwell time per cycle. Normalized fluorescence intensities were plotted versus temperature and melting temperatures (Tm) were calculated using the build-in Boltzmann equation in GraphPad (35). The data presented here are representative of three independently conducted trials with similar observations.
2.11. Bioinformatic and structural analyses
Human RAD51 variants were catalogued and annotated by the University of Vermont Molecular Bioinformatics Shared Resource. Variants were identified using Ensembl Variation, which pulls data from 36 data sources, such as dbSNP, COSMIC, and ESP. Variants are predicted to be of low, moderate, or high impact using SNPEff (36). As missense variants, F86L and E258A were predicted to be of moderate impact. Zygosity and validation status were taken from the COSMIC database. Sequences of Homo sapiens RAD51 (HsRAD51; CAG38796.1), Saccharomyces cervisiae Rad51 (ScRad51; CAA45563.1), Drosophila melanogaster Rad51 (DmRad51; BAA04580.1), Ustilago maydis Rad51 (UmRad51; AAC61878.1), Methanococcus voltae RadA (MvRadA; AAC23499.1), and Sulfolobus solfataricus RadA (SsRadA; AAC44123.1) proteins in Figure 1 were aligned using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). All structural figures of human RAD51 and yeast Rad51 proteins were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.). Structures of monomeric human RAD51 core domain and core domain-BRC4 complex were derived from PDB ID no. 1N0W (9). Structures of filamentous S. cerevisiae Rad51 protein were derived from PDB ID no. 3LDA (8).
3. Results
3.1. Descriptions of RAD51 somatic variant F86L and germline variant E258A
A survey of 999 breast invasive carcinoma biopsies identified one with a c.258C>A substitution mutation in the coding sequence of the RAD51 gene, resulting in an F86L missense mutation in the RAD51 protein (sample ID TCGA-BH-A1FN-01; COSMIC ID COSM1478082) (37,38). The mutation was confirmed somatic but its zygosity was unknown. The source was a tumor biopsy sample taken from a 34 year-old white (not Hispanic or Latino) female who was diagnosed with stage II-A breast cancer, and who later died of metastatic disease at age 40. The tumor was positive for both estrogen and progesterone receptors (ER+ PR+) but its HER2 status was unknown.
The germline RAD51 missense variant E258A (dbSNP ID rs191297852) was detected in 5/66,740 genomes in the European (non-Finnish) population, but was not found in other populations (39). The allele frequency in the European (non-Finnish) population is therefore 7.492e-5 (or approximately 1/13,000), while in the general population the allele frequency is ≤ 4.118e-5 (~1/24,000). All positive individuals are presumed to be heterozygous for the mutant allele. To date, there are no reports of the E258A variant from tumor tissues (38).
3.2. Sequence and structural contexts of F86L and E258A mutations
The F86L and E258A mutations both affect a region of RAD51 protein that is important for the formation and regulation of presynaptic filaments. Specifically, both mutations occur within the area of connection between the RecA homology domain (RHD; catalytic core) and the N-terminal domain (NTD) of RAD51. Phe-86 is located in the physical linker between the NTD and RHD, and it is highly conserved in eukaryotes and archaea (Fig. 1A–B; Fig. 2). Phe-86 also forms part of the interface between RAD51 protomers in the filament, by interacting with a hydrophobic pocket on the opposite protomer, as shown by alignments to the yeast Rad51 filament structure (Fig. 3A) (8). Therefore it is reasonable to hypothesize that missense mutations of Phe-86 could affect the stability or other properties of RAD51 presynaptic filaments.
Glu-258 is located in the conserved RecA homology domain of RAD51 protein, upstream of DNA binding loop L2, in an α-helix that makes up part of the repeating α/β motif of the RecA fold (Fig. 1A,C; Fig. 2). Glu-258 is highly conserved among vertebrates, invertebrates, and some fungi, but it is substituted by glutamine (Gln-316) in S. cerevisiae Rad51 protein (Fig. 1C). Comparison to the yeast Rad51 filament structure (Fig. 3B) suggests that Glu-258 forms part of a water-mediated hydrogen bond network at the interface between the RHD and NTD of the RAD51 protomer. The NTD is known to move relative to the RHD in response to DNA and/or nucleotide binding (5,40). Comparison of the human RAD51 RHD/BRC4 crystal structure to that of the yeast Rad51 filament indicates that the BRC4 motif from BRCA2 clashes sterically with the predicted position of the NTD (Fig. 2), indicating that the NTD must move relative to the RHD in order to accommodate BRCA2 binding. This is consistent with cryo-EM data showing that binding of the BRCA2 BRC3 and BRC4 repeats bind to different surfaces of the NTD and RHD, respectively (41) Therefore it is reasonable to hypothsize that missense mutations of Glu-258 could affect RAD51 filament dynamics or interactions with the BRCA2 tumor suppressor protein.
3.3. Thermal stability defects of RAD51 F86L and E258A variant proteins
The thermofluor assay (34,35) was used to compare the thermal stabilities of human RAD51 F86L and E258A with that of WT (Fig. 4, Table 2). The fluorescence intensity of Sypro Orange dye is related to the hydrophobicity of its environment. Temperature-induced unfolding of RAD51 allows the dye to interact with the interior hydrophobic regions of the protein, yielding a melting curve from the increased fluorescence. As reported previously (23), RAD51 WT exists in two stable and distinguishable conformational states depending on the presence of bound ATP. The thermal stability of the ATP-bound form is increased by 15°C relative to the apo enzyme (Fig. 4, Table 2). Both transitions are monophasic, suggesting that unfolding occurs cooperatively throughout the molecule both in the absence and presence of ATP.
Figure 4. Thermal stabilities of RAD51 wild-type (WT), F86L and E258A variant proteins in the presence and absence of ATP.

Thermofluor assays to determine melting curves and Tm values (± standard deviation) were as described in Materials and Methods. Relative fluorescence is normalized to the highest signal in each curve. Solid lines represent melting curves in the absence of ATP for RAD51 WT (blue), E258A (red), and F86L (green), respectively. Dotted lines represent melting curves in the presence of 2 mM ATP for RAD51 WT (blue), E258A (red), and F86L (green), respectively.
Table 2.
Melting temperatures (Tm) in the presence and absence of ATP.a
| Variant | ATP | Tm1b (°C) | Tm2b (°C) |
|---|---|---|---|
| WT | 0 | 44.4 ± 0.2 | None |
| F86L | 0 | 30.9 ± 0.9 | 55.1 ± 0.9 |
| E258A | 0 | (47.8 ± 1.8)c | (N.A.)c |
| WT | 2 mM | N.A. | 59.4 ± 0.9 |
| F86L | 2 mM | N.A. | 54.7 ± 0.1 |
| E258A | 2 mM | N.A. | 56.5 ± 0.7 |
Tm values were estimated from curve fits of thermofluor data in Figure 4, as described in Materials and Methods. N.A. – not applicable.
The low temperature transition observed with WT RAD51 in the absence of ATP is referred to as Tm1, and the high-temperature transition observed in the presence of ATP is referred to as Tm2. The low and high temperature transitions observed with RAD51 variants under various conditions are categorized as Tm1 or Tm2 based on an arbitrary cutoff of 50°C.
The melting curve for E258A in the absence of ATP likely includes two transitions, but these were poorly resolved (Fig. 4). The reported value of Tm1 represents the overall midpoint of the melting curve, which may be considered a composite or average of the different transitions.
In the presence of ATP the variants E258A and F86L also exhibit monophasic melting, however their Tm values reflect thermal destabilization of −2.9°C and −4.7°C, respectively compared to WT (Fig. 4, Table 2). In the absence of ATP the thermal profiles of the variants are quite different from that of WT; both appear to contain two melting transitions instead of one. In F86L the two transitions are clearly resolved (Fig. 4, Table 2): The first occurs at 30.9°C, which is destabilized by −13.5°C compared to the single transition of WT in the absence of ATP; the second occurs at 55.1°C, which is statistically indistinguishable from the Tm of ATP-bound F86L. In E258A there is a broad transition with an apparent Tm of 47.8°C in the absence of ATP (Fig. 4, Table 2). However this could be interpreted as consisting of two or more poorly resolved transitions with different Tm values, one similar to the less stable apo form of WT, and one similar to the more stable ATP-bound form. The presence of two transitions in a melting curve is indicative of a high degree of flexibility between two conformational states (35). Therefore the protein dynamics of F86L and E258A are different from those of WT. Unlike WT, in which the transition between the two conformations is ATP-dependent, F86L and E258A appear to sample both conformational states in the absence of ATP. The addition of ATP captures the variants in the more stable conformation.
3.4. DNA binding defects of F86L and E258A variant proteins
The DNA binding properties of F86L and E258A were investigated and compared to WT using two complementary assays: A qualitative electrophoretic mobility shift assay was used to characterize the interactions of RAD51 proteins with long, mixed-sequence ϕX174 ssDNA and dsDNA molecules (Fig. 5). Also, a quantitative flourescence quenching assay was used to determine the affinities of RAD51 proteins for homopolymeric ssDNA and dsDNA olignucleotides that are labeled with AlexaFluor 546 (23,33). The amplitude of fluorescence change is a measure of the fractional saturation of DNA with protein, which allows calculation of apparent Kd values from titration data (Fig. 6; Table 3).
Figure 5. Electrophoretic mobility shift assays for the binding of RAD51 variant and wild-type proteins to ϕX174 ssDNA and dsDNA in the presence of 1 mM ATP.

(A–B) Concentrations of RAD51 WT or E258A as indicated were incubated with 20 μM ϕX174 circular ssDNA (CSS) or linear dsDNA (LDS) then electrophoresed as described in Materials and Methods. (C–D) Binding reactions identical to Panels A–B except that RAD51 F86L replaced E258A.
Figure 6. Quantitative ssDNA and dsDNA binding data for RAD51 variants and wild-type in the presence and absence of ATP.

Binding of RAD51 protein to AlexaFluor 546-labeled ssDNA and dsDNA homopolymers was monitored by fluorescence quenching as described in Materials and Methods. Blue circles denote wild-type RAD51, red triangles denote E258A, and green squares denote F86L. (A) Protein binding to ssDNA in the presence of 1 mM ATP. (B) Protein binding to ssDNA in the absence of ATP. (C) Protein binding to dsDNA in the presence of 1 mM ATP. (D) Protein binding to dsDNA in the absence of ATP. Details of AF546-labeled ssDNA and dsDNA lattices plus other reaction conditions are described in Materials and Methods.
Table 3.
Apparent dissociation constants for RAD51-DNA interactionsa.
| Kd (μM) | WT | F86L | E258A |
|---|---|---|---|
| ssDNA | 1.9 ± 0.2 | N.D.b | N.D.b |
| ssDNA + ATP | 0.6 ± 0.1 | 4.9 ± 0.4 | 3.1 ± 0.3 |
| dsDNA | 0.8 ± 0.1 | 1.7 ± 0.1 | 2.1 ± 0.1 |
| dsDNA + ATP | 0.7 ± 0.1 | 1.8 ± 0.2 | 1.3 ± 0.1 |
Apparent Kd values were estimated from curve fits of data in Figure 6, as described in Materials and Methods.
Not Determined-- Measuring Kd required unattainably high protein concentrations.
As previously reported (21,23), RAD51 WT interacts with ϕX174 ssDNA in the presence of ATP to form complexes with both high and low electrophoretic mobility, resulting in a bimodal smear pattern on non-denaturing electrophoretic gels as the ssDNA is saturated with protein (Fig. 5A,C). E258A exhibits a similar pattern, and resembles WT in its ability to form high mobility complexes on ϕX174 ssDNA in the presence of ATP (Fig. 5A). The formation of low mobility complexes by E258A appears to require higher protein concentrations compared to WT, however. This result suggests either that E258A either has a reduced ability to saturate ssDNA, or that it has a reduced ability to form higher order structures on ssDNA, or both. For F86L, results indicate a more severe ssDNA binding defect (Fig. 5C). Over the same concentration range as WT, F86L barely shifts the electrophoretic mobility of ϕX174 ssDNA in the presence of ATP. Possible explanations for this result include that F86L has a low affinity for ssDNA, that F86L-ssDNA filaments are unstable during electrophoresis, that F86L-ssDNA filaments have morphological defects, or some combination of these factors. E258A and F86L also exhibit large differences from WT in their interactions with ϕX174 dsDNA in the presence of ATP (Fig. 5B,D). Compared to WT, higher concentrations of E258A are required to shift the electrophoretic mobility of dsDNA (Fig. 5B). E258A-dsDNA complexes run as diffuse smears on gels, unlike the relatively well-defined bands formed by the WT-dsDNA complexes (Fig. 5B). These observations suggest that E258A has lower affinity for dsDNA than WT. F86L has even lower affinity for ϕX174 dsDNA in the presence of ATP, since little if any mobility shift is observed at the protein concentrations used in the EMSA assay (Fig. 5D).
The EMSA method used here involved pre-incubation of complexes at 37ºC prior to gel loading. This temperature causes significant unfolding of the nucleotide-free form of F86L (Fig. 4, Table 2), which could lead to destabilization of filaments during some stages of the ATPase cycle. Therefore we repeated the F86L EMSA experiments while omitting the 37ºC pre-incubation step, replacing it with a 21ºC pre-incubation step prior to gel loading. This resulted in no improvement in F86L DNA-binding activity under the conditions of the EMSA assay. We conclude that F86L complexes on ϕX174 ssDNA and dsDNA molecules are inherently unstable during the multi-hour agarose gel electrophoresis runs that are required for the EMSA experiments.
Fluorescence quenching assays for oligonucleotide binding are performed at 21ºC, a temperature at which both variants should be folded (Fig. 4, Table 2). Indeed, both variants exhibit significant binding to ssDNA and dsDNA oligonucleotides under these conditions, however both have dramatically lower affinity for ssDNA and moderately lower affinity for dsDNA than WT (Fig. 6, Table 3). The affinities of WT, F86L, and E258A for ssDNA are strongly ATP-dependent (Fig. 6A,B; Table 3). In the presence of ATP, the apparent dissociation constant for of F86L-ssDNA is increased by approximately 8-fold compared to WT-ssDNA, while that of E258A-ssDNA is increased by approximately 5-fold compared to WT-ssDNA (Fig. 6A, Table 3). The ssDNA binding defects of both variants are exacerbated in the absence of ATP, where the apparent dissociation constants could not be calculated due to the very low saturation of ssDNA by F86L or E258A (Fig. 6B, Table 3). Duplex oligonucleotide binding data demonstrate that both E258A and F86L retain significant dsDNA binding activity under the lower temperature conditions of the fluorescence quenching assay, however both variants exhibit weaker affinity for dsDNA than WT (Fig. 6C,D; Table 3). The affinities of WT, F86L, and E258A for dsDNA are not greatly affected by ATP. In the presence of ATP the apparent dissociation constants of F86L-dsDNA and E258A-dsDNA are 2.6- and 1.9-fold higher than WT-dsDNA, respectively. In the absence of ATP, the apparent Kd values of F86L-dsDNA and E258A-dsDNA are 2.1- and 2.5-fold higher than WT-dsDNA, respectively. Thus even under the permissive conditions of lower temperature and simple DNA ligands, F86L and E258A exhibit global defects in DNA binding. Protein-ssDNA interactions are affected most severely in terms of affinity loss, but the ATP-dependence of ssDNA binding remains in both variants.
3.5. ATPase defect of F86L and E258A variant proteins
In addition to altered DNA binding properties, both F86L and E258A variants exhibit enzymatic deficiencies compared to WT. Wild-type RAD51 protein has an intrinsic ATPase activity; at saturating ATP concentration, the addition of a 2-fold excess of ssDNA (assuming a binding site size of 3 nucleotide residues per protein monomer) stimulates this activity by approximately 5-fold (Fig. 7). The intrinsic ATPase activity of F86L is barely detectable above background—a drastic reduction compared to WT (Fig. 7). However the addition of ssDNA activates ATP hydrolysis by F86L, to approximately 45% of the activity seen with WT under identical conditions. Therefore F86L exhibits an almost complete dependence on ssDNA for ATPase activity, compared to the 5-fold stimulation of WT ATPase activity by ssDNA. In contrast, E258A shows a reduced response to ssDNA in terms of its ATPase activity. The intrinsic ATPase activity of E258A is statistically slightly higher than that of WT (Fig. 7). The addition of ssDNA stimulates the ATPase activity of E258A by approximately 2-fold (Fig. 7), a substantially weaker response than WT and nothing like the ssDNA-dependence of ATP hydrolysis by F86L. With ssDNA, the ATPase activity of E258A rises to approximately 60% that of WT under identical conditions. Thus F86L has little intrinsic ATPase activity but a strong response to ssDNA, while E258A has robust intrinsic ATPase activity but a weak ssDNA response.
Figure 7. Intrinsic and ssDNA-stimulated ATPase activities of RAD51 wild-type, E258A, and F86L proteins.

ATPase assays were carried out as described in Materials and Methods. Dark gray blocks represent intrinsic ATPase activities in the absence of ssDNA. Light gray blocks represent activities observed upon addition of a 2-fold excess of ϕX174 ssDNA (assuming 3 nucleotide residues per monomer).
3.6. DNA strand exchange properties of F86L and E258A variant proteins
The intrinsic recombination activities of the variants were measured under optimized conditions (30ºC, absence of RPA protein and mediator proteins, oligonucleotide substrates lacking stable ssDNA secondary structure) as shown schematically in Figure 8A. Results demonstrate that F86L and E258A both retain intrinsic recombination activity (Fig. 8B). E258A exhibits a partial deficiency, as it catalyzes this reaction at a 2-fold lower rate than WT (Fig. 8B). Amazingly, F86L is hyperactive under these conditions, since it catalyzes DNA strand exchange at a rate that is at least 3-fold faster than WT (Fig. 8B).
Figure 8. Intrinsic DNA strand exchange activities of RAD51 variants at 30ºC and 37°C.

Oligonucleotide substrates have been optimized for sequence and length so that reactions do not require RPA protein. (A) In the presence of RAD51 and ATP, radiolabeled homologous duplex and tailed duplex substrates are converted into heteroduplex and ssDNA products. Tailed duplex (63/32-mer) was constructed from oligos 5/6, and homologous duplex (31/31-mer) was constructed from oligos 7/8 (Table 1). (B) Results of reactions performed at 30ºC. (C) Results of reactions performed at 37ºC. Error bars in panels (B) and (C) represent results from 3 identical experiments. The signal from a background control reaction containing no protein was subtracted from all data.
Dramatic differences are observed when the same reactions are performed at the slightly elevated temperature of 37ºC (Fig. 8C). For both WT and E258A, the initial rates increase at 37ºC compared to 30ºC. The activity of E258A at 37ºC is still much weaker than that of WT however. The hyperactivity of F86L observed at 30ºC disappears at 37ºC, where the activity of this variant is approximately equal to that of WT (Fig. 8C). Thus the hyperactivity of F86L’s intrinsic DNA strand exchange activity appears to be very heat-labile. We note that all reactions at 37ºC are attenuated at longer reactions times (Fig. 8C): For both WT and F86L, reactions arrest by the 60 minute time point, before they reach 50% completion. The amount of strand exchange product actually decreases from the 60’ to the 90’ time point, which may be due to a faster rate of spontaneous thermal denaturation of the heteroduplex product at the elevated temperature. The rate of the reaction catalyzed by E258A also appears to decrease after the 60’ time point (Fig. 8C). Thus all three RAD51 proteins experience time-dependent loss of intrinsic recombination activity at 37ºC, with F86L the most labile of the three.
DNA strand exchange assays with full-length (5.4 kb/kbp) ϕX174 DNA substrates provide a more stringent test of RAD51 recombination activity. A schematic of this assay system is shown in Figure 9A. In addition to RAD51, these reactions require human RPA protein, which must be added to reaction mixtures after pre-incubation of RAD51 with ssDNA and ATP at 37ºC, and prior to the addition of homologous dsDNA to start the reaction. Figure 9B (left) shows the reaction with 7.50 μM RAD51 WT which generates joint molecule (JM) products that increase steadily over time. The equivalent reaction with 7.50 μM E258A generates no JM products over the same time period (Fig. 9B, center). Therefore E258A protein lacks DNA strand exchange activity under stringent conditions of DNA length and RPA presence.
Figure 9. DNA strand exchange activity of RAD51 E258A versus WT with homologous ϕX174 DNA substrates and RPA protein.

(A) Reaction schematic. See Materials and Methods for details. (B) Timecourses of reactions containing a total of 7.50 μM RAD51 protein (WT and/or E258A) at the indicated concentrations. ssDNA (SS), dsDNA (LDS), nicked (NC), and supercoiled (SC) ϕX174 DNA markers (lanes 1–3, 22–24) and joint molecule (JM) products are indicated next to the gel. Left (lanes 4–9)-- Reaction with WT only; Center (lanes 10–15)-- Reaction with E258A only; Right (lanes 16–21)-- Reaction with 1:1 mixture of WT and E258A. All other experimental conditions were as described in Materials and Methods. (C) Quantitative analysis of joint molecule formation based on image scans were performed as described in Materials and Methods. Data are normalized relative to the total density of the Joint Molecules products formed by wild-type RAD51 at the 40 min time point. Results in panels (B) – (C) are typical of those obtained in three separate experiments performed under identical conditions.
Under the same stringent conditions, the F86L variant does promote a robust DNA strand exchange reaction when it is present at 7.50 μM concentration (Fig. 10, center). The rate of joint molecule production is only slightly reduced compared to the rate seen with 7.50 μM RAD51 WT (Fig. 10, left). Therefore the F86L variant alone retains a large fraction of wild-type DNA strand exchange activity under stringent conditions of DNA length and RPA presence.
Figure 10. DNA strand exchange activity of RAD51 F86L versus WT with homologous ϕX174 DNA substrates and RPA protein.

(A) Reactions were carried out as described in Materials and Methods and as shown schematically in Figure 9A. Timecourses of reactions containing a total of 7.50 μM RAD51 protein (WT and/or F86L) at the indicated concentrations. The positions of SS, LDS, NC, and SC (supercoiled ϕX174 DNA) markers (lanes 1–3, 22–24) are indicated next to the gel, along with the position of JM products. Left (lanes 4–9)-- Reaction with WT only; Center (lanes 10–15)-- Reaction with F86L only; Right (lanes 16–21)-- Reaction with 1:1 mixture of WT and F86L. (B) Quantitative analysis of joint molecule formation based on image scans were performed as described in Materials and Methods. Data are normalized relative to the total density of the Joint Molecules products formed by wild-type RAD51 at the 40 min time point. Results in panels (A) – (B) are typical of those obtained in three separate experiments performed under identical conditions. (C) Electrophoretic mobility shift assays of mixed RAD51 WT/F86L versus WT complexes formed with circular ϕX174 ssDNA (lanes 1–10) and linear ϕX174 dsDNA (lanes 11–20). Experimental conditions were identical to those in Fig. 5C–D except that in lanes 6–10 and in lanes 16–20 the DNA molecules were titrated with a 1:1 mixture of RAD51 WT and F86L proteins.
3.7. Defective DNA strand exchange in reactions with mixtures of wild-type and variant RAD51 proteins
As shown in Figures 9B (right) and 9C, a DNA strand exchange reaction containing equimolar E258A and WT (3.75 μM each, 7.50 μM total enzyme concentration) proceded at a detectable but greatly reduced rate compared to the reactions with 7.50 μM WT alone (Fig. 9B, left; Fig. 9C). 7.50 μM RAD51 is sufficient to saturate all of the input ssDNA in the reaction, while 3.75 μM RAD51 is sub-saturating given a binding site size of 3 nucleotided per monomer. E258A alone is inactive in this assay (Fig. 9B, center; Fig. 9C), so the data in lanes 16–21 demonstrate that E58A recombination activity cannot be rescued by incorporation into mixed filaments of variant and WT protein on ssDNA. In fact the activity observed in the mixed E258A/WT reaction is much less than 50% of the WT activity (Fig. 9B–C), suggesting that the presence of E258A in a mixed complex exerts a poisoning or dominant negative effect on the reaction.
As shown in Figures 10A (right) and 10B, a DNA strand exchange reaction containing equimolar F86L and WT (3.75 μM each, 7.50 μM total enzyme concentration) also proceeded at a detectable but greatly reduced rate compared to the reaction with 7.50 μM WT alone (Fig. 10A, left; Fig. 10B). The recombination defect of the F86L/WT mixture is even more severe than that of the E258A/WT mixture shown in Figure 9. This is a surprising result since F86L alone exhibits strong partial DNA strand exchange activity under these reaction conditions (Fig. 10A, center; Fig. 10B). Apparently the activities of both WT and F86L are poisoned in the mixture. To further investigate this mutual poisoning effect, we repeated the electrophoretic mobility shift assays in Figure 5C–D, but this time we used a 1:1 mixture of F86L and WT to titrate the ϕX174 ssDNA and dsDNA molecules rather than F86L alone (Fig. 10C). Results were compared to titrations with WT alone on the same gel. We find that the extent of electrophoretic mobility shift of both ssDNA and dsDNA molecules is severely reduced when the DNA is titrated with a 1:1 mixture of F86L/WT compared to a titration with WT RAD51 alone. (Fig. 10C). These results indicate that the presence of the F86L variant interferes with the formation of stable RAD51 filaments on long mixed-sequence ssDNA and dsDNA molecules, a finding that could partially explain the mutual poisoning of WT and F86L DNA strand exchange activities. The data demonstrate that even a partially active variant of RAD51 can have negative functional interactions with the wild-type enzyme. Possible models for the behavior of both F86L and E258A variants are discussed below.
4. Discussion
Two missense variants of human RAD51 protein, a somatic F86L variant from breast carcinoma and a germline E258A variant, both affect the filament interface/BRCA2 binding region of RAD51 (Figs. 1–3). Both variants exhibit strongly aberrant biochemical properties compared to the wild-type enzyme, including defects in thermal stability, DNA binding activity, ATPase activity, and DNA strand exchange activities. Mixtures of either variant with wild-type also exhibit greatly reduced DNA strand exchange activity compared to wild-type alone, suggesting that heterozygous expression of either variant could lead to recombination defects in cells.
As previously reported (23), wild-type RAD51 undergoes an all-or-none allosteric transition from a thermolabile apo conformation to a thermostable ATP-bound conformation. ATP binding also induces relatively thermostable conformations of F86L and E258A. However both variants are less stable than WT in the presence of ATP (Fig. 4, Table 2). Furthermore both variants exhibit biphasic thermal melting curves in the absence of ATP, suggesting two or more conformational states in equilibrium. Therefore both variants appear to have lost the tight linkage between ATP binding status and conformation that is a normal hallmark of wild-type RAD51. In this property F86L and E258A resemble the previously characterized RAD51 somatic variants Q268P and Q272L, which constitutively sample both apo-like and ATP-bound-like conformations in the absence of added nucleotide (23). In F86L the labile phase has Tm = 30.9°C (Fig. 4, Table 2), which would lead to thermal instability at in vivo temperature and under temperature conditions that are frequently encountered in enzymatic assays.
F86L and E258A exhibit lower affinities for both ssDNA and dsDNA compared to wild-type RAD51. Their DNA binding defects are evident both in EMSA assays for filament formation on long, mixed-sequence ϕX174 DNA molecules (Fig. 5) and in fluorescence assays for binding to short homopolymeric oligonucleotides (Fig. 6, Table 3). In the EMSA assay RAD51 is preincubated with DNA in the presence of ATP at 37°C, then complexes are separated by agarose gel electrophoresis. Thus the degree of electrophoretic mobility shift depends on RAD51-DNA binding affinity as well as on filament stability and morphology. The fluorescence assay, performed at 21°C, follows titrations of DNA with protein in real time without preincubation. This assay allows direct calculations of the apparent Kd values for RAD51-DNA interactions at a permissive temperature, and it is less sensitive to issues of filament stability and morphology. The combined results indicate that in the presence of ATP, E258A forms stable nucleoprotein filaments on ssDNA and dsDNA, however it exhibits reduced affinity for both ssDNA and dsDNA compared to WT (Figs. 5–6, Table 3). These findings suggest that lower intrinsic affinity, not thermal instability, is primarily responsible for the DNA binding defect of E258A. F86L also exhibits reduced DNA binding affinity compared to WT in fluorescence assays (Fig. 6, Table 3), however F86L-DNA filaments appear to be unstable under the EMSA assay conditions, since very little electrophoretic mobility shift of either ssDNA or dsDNA is observed (Fig. 5). The same is true whether complexes are pre-incubated at 37°C or 21°C (data not shown). These findings suggest that F86L-DNA filaments are intrinsically less stable than WT RAD51 filaments. Both lower intrinsic affinity and thermal instability are important components of the DNA binding defect of F86L.
Interestingly both E258A and F86L exhibit severe defects in ssDNA binding in the absence of ATP, but recover significant ssDNA binding activity in the presence of ATP, albeit with 5- to 8-fold reduced affinity compared to WT (Fig. 6, Table 3). Thus both variants are more dependent on ATP for ssDNA binding than is wild-type RAD51. The ATP-dependent binding of F86L and E258A to ssDNA is sufficient to activate their enzymatic activities, since both variants catalyze ssDNA-dependent ATP hydrolysis and intrinsic DNA strand exchange reactions (Figs. 7–8). The rates of ssDNA-stimulated ATP hydrolysis catalyzed by RAD51 WT, E258A, and F86L correlate well with their relative affinities for ssDNA (Fig. 7, Table 3). Similar to WT, E258A also has an intrinsic ATPase activity that does not depend on ssDNA (Fig. 7). The relatively weak stimulation of E258A’s ATPase activity by ssDNA probably reflects its lower affinity for ssDNA compared to WT. F86L completely lacks intrinsic ATPase activity, but recovers ~45% of wild-type ATPase activity in the presence of ssDNA (Fig. 7). Thus F86L and E258A are partially deficient in ATPase activity but for opposite reasons: F86L has little intrinsic ATPase activity but a strong response to ssDNA; E258A has robust intrinsic ATPase activity but a weak ssDNA response. The absence of intrinsic ATPase activity in F86L may be caused by thermal instability in its apo form as discussed above. Therefore the formation of an ATPase-competent F86L-ssDNA complex likely involves synergistic stabilizing effects of bound ATP and ssDNA ligands on the enzyme structure.
The E258A variant is severely defective in DNA strand exchange activity. E258A retains intrinsic DNA strand exchange activity under highly optimized conditions, (absence of RPA protein, oligonucleotide ssDNA and dsDNA substrates designed to minimize ssDNA secondary structure), but its activity is greatly reduced compared to WT (Fig. 8). E258A is completely deficient in DNA strand exchange under stringent conditions (ϕX174 DNA substrates, presence of RPA protein) (Fig. 9). Furthermore a 1:1 mixture of E258A and WT promotes the stringent DNA strand exchange reaction at a rate that is greatly reduced compared to WT alone at the same total enzyme concentration. The rate of joint molecule product formation in the mixed reaction is approximately 30% of the rate in the WT-only reaction (Fig. 9C). Thus biochemically E258A acts as a classic recombination hypomorph, which may exert dominant negative effects on RAD51 WT through the formation of partially active, hybrid presynaptic filaments (23). As a relatively rare germline variant (38,39), E258A would be expected to be heterozygous in individual carriers. Our biochemical data suggest that these individuals might have reduced homology-directed repair activity similar to carriers of germline mutations in BRCA2 or BRCA1.
By comparison to the yeast Rad51 filament structure, Glu-258 in human RAD51 is located at the interface between the RecA homology and N-terminal domains (Fig. 2, Fig. 3B). The all-or-none conformational changes in Rad51 enzymes that occur in response to ligand binding involve movements of the NTD in relation to the RHD (5,40). The effects of the E258A mutation on RAD51 properties make sense when we consider that the loss of electrostatic and hydrogen bonding capacity at this locus disrupts a water-mediated hydrogen bond network that connects the NTD to the RHD (Fig. 3B). In the absence of Glu-258, these domains may have fewer conformational constraints, which could lead to a less efficient allosteric transition from the inactive ADP-bound or apo forms of RAD51 filaments to their active ATP-bound form. This is one possible mechanism to explain the reduced DNA-binding affinity, ATPase activity, and DNA strand exchange activity of E258A compared to wild-type. The apparent dominant effect of E258A in mixed WT/E258A filaments could also be explained by a defect in the allosteric transition of E258A subunits, which might interfere with allosteric signaling by nearby wild-type subunits. Additionally, overlaying structures of the yeast Rad51 filament and human RAD51-BRCA2 complex (Fig. 2) reveals that to accommodate BRC peptide binding to the RHD of RAD51, the NTD would have to shift position relative to the RHD. Since the E258A mutation likely changes the dynamics of the NTD-RHD interactions within each protomer of the filament, regulation of RAD51 activity by BRCA2 could be altered as well.
F86L would normally be considered a conservative mutation based on the similar hydrophobicity indices of Phe (+2.8) and Leu (+3.8) on the Kyte and Doolittle scale (42). However the biochemical recombination activities of this variant diverge unexpectedly and dramatically from wild-type RAD51. F86L exhibits hyperactivity in intrinsic DNA strand exchange assays at 30°C (Fig. 8). We postulate that the hyperactivity of F86L is caused by rapid release of the heteroduplex product, allowing faster turnover of the oligonucleotide substrates. Further work will be necessary to test this hypothesis. In stringent DNA strand exchange reactions containing ϕX174 DNA substrates and RPA, F86L exhibits activity that is only slightly lower than wild-type (Fig. 10). However we were amazed to observe that mixing F86L with WT in these reactions poisons the activities of both—DNA strand exchange activity of the 1:1 mixture is only 15% – 20% of that observed for WT alone or for F86L alone (Fig. 10B). This finding demonstrates that even conservative substitutions in critical residues of the RAD51 filament interface region may create incompatibilities within presynaptic filaments that compromise recombination. A possible mechanism for the mutual-poisoning phenomenon is described below and in Figure 11:
Figure 11. Possible mechanism for poisoning of DNA strand exchange in RAD51 F86L/wild-type mixtures.

Inactive forms of RAD51 WT and F86L are represented by yellow circles and pink ovals, respectively. Active forms of RAD51 WT and F86L are represented by yellow parallelograms and pink polygons, respectively. RPA heterotrimers are shown in cyan. During presynaptic filament assembly on ssDNA, the activation of RAD51’s DNA strand exchange activity requires a minimal filament length or cluster size. This is assumed to be true for both WT and F86L versions of RAD51. (A) When the enzyme concentration is sub-saturating with respect to binding sites on ssDNA, clusters of RAD51 (WT or F86L) are interspersed with clusters of RPA protein. Only a fraction of the RAD51 filaments achieve sufficient length to support the allosteric transition to the enzyme’s active form. DNA strand exchange activity is low. (B) When the enzyme concentration is saturating with respect to binding sites on ssDNA, both WT and F86L enzymes are capable of forming long filaments in the active conformation. DNA strand exchange activity is high. (C) The data indicate that in mixtures, F86L and WT inhibit each other’s DNA strand exchange activities. Since the F86L mutation alters the filament interface, we propose that when F86L and WT subunits occupy adjacent sites on ssDNA, they form hybrid interfaces that do not transmit allosteric signals correctly. Therefore neither WT nor F86L forms clusters that are of sufficient size to undergo activation. As a result, DNA strand exchange activity is extremely low in the mixed reaction, whereas either form alone is active. See text for further discussion.
As shown in Figure 3A, Phe-86 is essential for normal hydrophobic interactions across the interface between two RAD51 subunits within a presynaptic filament. Thus the incorporation of F86L into mixed filaments with WT would create three classes of interfaces—WT/WT, F86L/WT, and F86L/F86L, which might have different stabilities. Our data demonstrate that filaments containing only F86L/F86L interfaces are competent for strand exchange, as are filaments containing only WT/WT interfaces (Fig. 10). However we propose that hybrid F86L/WT interfaces found in mixed filaments disrupt the normal intrafilament allosteric signaling that is needed for strand exchange between long homologous ssDNA and dsDNA substrates. The schematic in Figure 11 shows the essential feature of this model: That recombination between long DNA homologs requires relatively long uninterrupted tracts of either WT or F86L subunits to maintain structural/functional integrity within presynaptic filaments. If F86L and WT at equal concentrations incorporate into filaments randomly, and if F86L/WT interfaces do not transmit allosteric signals correctly, then the occurrence of tracts of either form that are of sufficient length to promote strand exchange would be greatly reduced. Thus two otherwise active forms of the RAD51 enzyme could poison each other through their catalytic incompatibility and competition for binding sites on DNA within the filament.
Data from EMSA experiments with ϕX174 ssDNA and dsDNA molecules appear to support our model for the DNA strand exchange defects of F86L. The EMSA data show that F86L-DNA filaments are unstable compared to WT RAD51-DNA filaments (Fig. 5C–D). Moreover, in a 1:1 mixture of F86L and WT, RAD51-DNA filaments are greatly destabilized (Fig. 10C). This supports the notion that DNA strand exchange fails in WT/F86L mixtures due to the inability of either protein to form stable critical clusters on DNA substrates (Fig. 11). This in turn begs the question: How can F86L form unstable filaments on DNA and yet perform robust DNA strand exchange reactions in the absence of WT RAD51? In the oligonucleotide-based strand exchange assays (Fig. 8), substrates and reaction conditions are optimized for short DNA sequences free of secondary structure, low temperature, and absence of RPA protein. Thus filament stability on substrates may be less important in these assays than filament turnover from products. The relatively low affinity of F86L for DNA may allow for rapid turnover of the postsynaptic filament, allowing the variant to participate in more rounds of strand exchange than we would normally see with WT. This principle could also facilitate the activity of F86L in ϕX174-based strand exchange assays (Fig. 10A–B). However the length and sequence complexity of the DNA substrates necessitates the presence of RPA protein to remove secondary structure from ssDNA. Recalling that the ϕX174 assays are carefully assembled in the order RAD51+ATP → ssDNA → RPA → dsDNA, it seems evident that F86L responds to RPA similarly to WT RAD51, resulting in more complete saturation of ssDNA by enzyme. Nevertheless RPA does not overcome the incompatibility between F86L and WT in mixed reactions.
Breast carcinoma-derived F86L and germline E258A join a growing list of biochemically characterized RAD51 missense variants with demonstrated or suspected links to human cancer and genetic disease. Others include D149N, R150Q, and G151D (breast cancer), Q268P (lung cancer), Q272L (kidney cancer), T131P and A293T (Fanconi Anemia) (21,23,28,29). RAD51 variants in cancer and FA-R map to three distinct functional regions of the enzyme: A Schellman loop or “paperclip” motif located on the outer surface of RAD51 filaments (D149N, R150Q, G151D); DNA binding and ATP hydrolytic sites within the filament (Q268P, Q272L, T131P, A293T); and filament or domain interface surfaces (F86L, E258A – this study). All of these variants exhibit some spectrum of physical and functional defects in vitro, and many of them interfere with the biochemical functions of wild-type RAD51, suggesting molecular mechanisms for genetic dominance. For example the presence of G151D alters DNA binding and strand exchange properties of wild-type RAD51 (21), and expression of G151D in a wild-type RAD51 background in MCF10A cells induces hyper-recombination, drug and radiation resistance (27). Tumor variants F86L, Q268P, and Q272L, germline variant E258A, and Fanconi Anemia variants T131P and A293T all interfere with the DNA homologous pairing and strand exchange activities of wild-type RAD51 in vitro (23,28,29; this study). In vivo, T131P and A293T display genetically dominant defects in DNA interstrand crosslink (ICL) repair (28,29). Thus there is a strong tendency for disease-associated variants of RAD51 to exhibit biochemical and/or genetic dominance. This follows from the essential role of the RAD51-DNA filament in DNA repair activities and in the maintenance of genomic stability via biological functions including homology-directed repair and ICL repair. The RAD51 variants characterized to date all retain the ability to form mixed filaments on DNA with wild-type RAD51. Within these mixed filaments, variants poison wild-type activity by destabilizing the filaments, altering allosteric transitions, or altering interactions with substrates or protein partners. The principle of filament poisoning leading to defects in DNA homologous recombination/repair functions was first established with mutants of the E. coli RecA recombinase (43,44); this principle must now be recognized as central to the pathologies of RAD51 variants in human disease.
Highlights.
Human RAD51 somatic F86L and germline E258A variants are characterized.
Both variants map to the filament interface/BRCA2 binding region of RAD51.
F86L and E258A have abnormal stability, DNA binding, and catalytic properties.
E258A inhibits the DNA strand exchange activity of wild-type RAD51.
F86L and wild-type RAD51 exhibit mutual poisoning of DNA strand exchange.
Acknowledgments
This work was supported by the National Institutes of Health [grant no. P01CA098993], and by an Internal Grant Program award from the Larner College of Medicine at the University of Vermont. The sponsors had no involvement in study design, the collection, analysis and interpretation of data, in the writing of the report, or in the decision to submit the article for publication. The authors thank Dr. Stephen Everse for assistance in the design of structural biology figures.
Abbreviations
- HR
homologous recombination
- HDR
homology-directed repair
- DSB
DNA double-strand break
- dsDNA
double-stranded DNA
- ssDNA
single-stranded DNA
- WT
wild-type
- NTD
N-terminal domain
- RHD
RecA homology domain
Footnotes
Conflicts of interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morrical S. DNA pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harbor Perspect Biol. 2015;7:a016444. doi: 10.1101/cshperspect.a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 3.Richardson C. RAD51, genomic stability, and tumorigenesis. Cancer Lett. 2005;218:127–139. doi: 10.1016/j.canlet.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Liu J, Ehmsen KT, Heyer WD, Morrical SW. Presynaptic filament dynamics in homologous recombination and DNA repair. Crit Rev Biochem Mol Biol. 2011;46:240–270. doi: 10.3109/10409238.2011.576007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu X, Jacobs SA, West SC, Ogawa T, Egelman EH. Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc Natl Acad Sci USA. 2001;98:8419–8424. doi: 10.1073/pnas.111005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.VanLoock MS, Yu X, Yang S, Lai AL, Low C, Campbell MJ, Egelman EH. ATP-mediated conformational changes in the RecA filament. Structure. 2003;11:187–196. doi: 10.1016/s0969-2126(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 7.Conway AB, Lynch TW, Zhang GS, Fortin CW, Fung LS, Symington PA, Rice PA. Crystal structure of a Rad51 filament. Nat Struct Mol Biol. 2004;11:791–796. doi: 10.1038/nsmb795. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Villanueva N, Rould MA, Morrical SW. Insights into the mechanism of Rad51 recombinase from the structure and properties of a filament interface mutant. Nucleic Acids Res. 2010;38:4889–4906. doi: 10.1093/nar/gkq209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- 10.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002;62:219–225. [PubMed] [Google Scholar]
- 11.Maacke H, Opitz S, Jost K, Hamdorf W, Henning W, Krüger S, Feller AC, Lopens A, Diedrich K, Schwinger E, Stürzbecher HW. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int J Cancer. 2000;88:907–913. doi: 10.1002/1097-0215(20001215)88:6<907::aid-ijc11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 12.Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair (Amst) 2002;7:686–693. doi: 10.1016/j.dnarep.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maacke H, Hundertmark C, Miska S, Voss M, Kalthoff H, Stürzbecher HW. Autoantibodies in sera of pancreatic cancer patients identify recombination factor Rad51 as a tumour-associated antigen. J Cancer Res Clin Oncol. 2002;128:219–222. doi: 10.1007/s00432-001-0321-2. [DOI] [PubMed] [Google Scholar]
- 14.Wilson CA, Ramos L, Villaseñor MR, Anders KH, Press MF, Clarke K, Karlan B, Chen JJ, Scully R, Livingston D, et al. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat Genet. 1999;21:236–240. doi: 10.1038/6029. [DOI] [PubMed] [Google Scholar]
- 15.Yoshikawa K, Honda K, Inamoto T, Shinohara H, Yamauchi A, Suga K, Okuyama T, Shimada T, Kodama H, Noguchi S, et al. Reduction of BRCA1 protein expression in Japanese sporadic breast carcinomas and its frequent loss in BRCA1-associated cases. Clin Cancer Res. 1999;5:1249–1261. [PubMed] [Google Scholar]
- 16.Yoshikawa K, Ogawa T, Baer R, Hemmi H, Honda K, Yamauchi A, Inamoto T, Ko K, Yazumi S, Motoda H, et al. Abnormal expression of BRCA1 and BRCA1-interactive DNA-repair proteins in breast carcinomas. Int J Cancer. 2000;88:28–36. [PubMed] [Google Scholar]
- 17.Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat Genet. 1995;9:444–450. doi: 10.1038/ng0495-444. [DOI] [PubMed] [Google Scholar]
- 18.Collis SJ, Tighe A, Scott SD, Roberts SA, Hendry JH, Margison GP. Ribozyme minigene-mediated RAD51 down-regulation increases radiosensitivity of human prostate cancer cells. Nucleic Acids Res. 2001;29:1534–1538. doi: 10.1093/nar/29.7.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohmori T, Yang JL, Price JO, Arteaga CL. Blockade of tumor cell transforming growth factor-betas enhances cell cycle progression and sensitizes human breast carcinoma cells to cytotoxic chemotherapy. Exp Cell Res. 1998;245:350–359. doi: 10.1006/excr.1998.4261. [DOI] [PubMed] [Google Scholar]
- 20.Kato M, Yano K, Matsuo F, Saito H, Katagiri T, Kurumizaka H, Yoshimoto M, Kasumi F, Akiyama F, Sakamoto G, et al. Identification of Rad51 alteration in patients with bilateral breast cancer. J Hum Genet. 2000;45:133–137. doi: 10.1007/s100380050199. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Morrical MD, Donigan KA, Weidhaas JB, Sweasy JB, Averill AM, Tomczak JA, Morrical SW. Tumor-associated mutations in a conserved structural motif alter physical and biochemical properties of human RAD51 recombinase. Nucleic Acids Res. 2015;43:1098–1111. doi: 10.1093/nar/gku1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishida T, Takizawa Y, Sakane I, Kurumizaka H. Altered DNA binding by the human Rad51-R150Q mutant found in breast cancer patients. Biol Pharm Bull. 2007;30:1374–1378. doi: 10.1248/bpb.30.1374. [DOI] [PubMed] [Google Scholar]
- 23.Silva MC, Morrical MD, Bryan KE, Averill AM, Dragon J, Bond JP, Morrical SW. RAD51 variant proteins from human lung and kidney tumors exhibit DNA strand exchange defects. DNA Repair (Amst) 2016;42:44–55. doi: 10.1016/j.dnarep.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedler A, Veprintsev DB, Rutherford T, von Glos KI, Fersht AR. Binding of Rad51 and other peptide sequences to a promiscuous, highly electrostatic binding site in p53. J Biol Chem. 2005;280:8051–8059. doi: 10.1074/jbc.M411176200. [DOI] [PubMed] [Google Scholar]
- 25.Yoon D, Wang Y, Stapleford K, Wiesmüller L, Chen J. P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J Mol Biol. 2004;336:639–654. doi: 10.1016/j.jmb.2003.12.050. [DOI] [PubMed] [Google Scholar]
- 26.Bucchop S, Gibson MK, Wang XW, Wagner P, Sturzbecher HW, Harris CC. Interaction of p53 with the human Rad51 protein. Nucleic Acids Res. 1997;25:3868–3874. doi: 10.1093/nar/25.19.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marsden CG, Jensen RB, Zagelbaum J, Rothenberg E, Morrical SW, Wallace SS, Sweasy JB. The tumor-associated variant RAD51 G151D induces a hyper-recombination phenotype. PLoS Genet. 2016;12:e1006208. doi: 10.1371/journal.pgen.1006208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, Abhyankar A, Sougnez C, Gabriel SB, Auerbach AD, Kowalczykowski SC, Smogorzewska A. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol Cell. 2015;59:478–490. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ameziane N, May P, Haitjema A, van de Vrugt HJ, van Rossum-Fikkert SE, Ristic D, Williams GJ, Balk J, Rockx D, Li H, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun. 2015;6:8829. doi: 10.1038/ncomms9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritsch EF, Miniatis T. Molecular cloning: A Laboratory Manual. Cold spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 31.Binz SK, Dickson AM, Haring SJ, Wold MS. Functional assays for replication protein A (RPA) Methods Enzymol. 2006;409:11–38. doi: 10.1016/S0076-6879(05)09002-6. [DOI] [PubMed] [Google Scholar]
- 32.Henricksen LA, Umbricht CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem. 1994;269:11121–11132. Erratum in: J. Biol. Chem.269: 16519. [PubMed] [Google Scholar]
- 33.Maher RL, Morrical SW. Coordinated binding of single-stranded and double-stranded DNA by UvsX recombinase. PLoS One. 2013;8:e66654. doi: 10.1371/journal.pone.0066654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 2006;357:289–298. doi: 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 35.Fang J, Rand KD, Silva MC, Wales TE, Engen JR, Beuning PJ. Conformational dynamics of the Escherichia coli DNA polymerase manager proteins UmuD and UmuD'. J Mol Biol. 2010;398:40–53. doi: 10.1016/j.jmb.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:11. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.1000 Genomes Project Consortium. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galkin VE, Wu Y, Zhang XP, Qian X, He Y, Yu X, Heyer WD, Luo Y, Egelman EH. The Rad51/RadA N-terminal domain activates nucleoprotein filament ATPase activity. Structure. 2006;14:983–992. doi: 10.1016/j.str.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Galkin VE, Esashi F, Yu X, Yang S, West SC, Egelman EH. BRCA2 BRC motifs bind RAD51-DNA filaments. Proc Natl Acad Sci U S A. 2005;102:8537–8542. doi: 10.1073/pnas.0407266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 43.Kowalczykowski SC, Krupp RA. Biochemical events essential to the recombination activity of Escherichia coli RecA protein. II. Co-dominant effects of RecA142 protein on wild-type RecA protein function. J Mol Biol. 1989;207:735–747. doi: 10.1016/0022-2836(89)90240-4. [DOI] [PubMed] [Google Scholar]
- 44.Lauder SD, Kowalczykowski SC. Negative co-dominant inhibition of recA protein function. Biochemical properties of the recA1, recA13 and recA56 proteins and the effect of recA56 protein on the activities of the wild-type recA protein function in vitro. J Mol Biol. 1993;234:72–86. doi: 10.1006/jmbi.1993.1564. [DOI] [PubMed] [Google Scholar]