

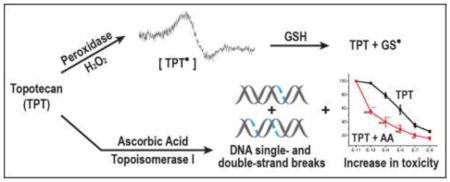
Abstract
Topotecan, a derivative of camptothecin, is an important anticancer drug for the treatment of various human cancers in the clinic. While the principal mechanism of tumor cell killing by topotecan is due to its interactions with topoisomerase I, other mechanisms, e.g., oxidative stress induced by reactive free radicals, have also been proposed. However, very little is known about how topotecan induces free radical-dependent oxidative stress in tumor cells. In this report we describe the formation of a topotecan radical, catalyzed by a peroxidase-hydrogen peroxide system. While this topotecan radical did not undergo oxidation-reduction with molecular O2, it rapidly reacted with reduced glutathione and cysteine, regenerating topotecan and forming the corresponding glutathiyl and cysteiniyl radicals. Ascorbic acid, which produces hydrogen peroxide in tumor cells, significantly increased topotecan cytotoxicity in MCF-7 tumor cells. The presence of ascorbic acid also increased both topoisomerase I-dependent topotecan-induced DNA cleavage complex formation and topotecan-induced DNA double-strand breaks, suggesting that ascorbic acid participated in enhancing DNA damage induced by topotecan and that the enhanced DNA damage is responsible for the synergistic interactions of topotecan and ascorbic acid. Cell death by topotecan and the combination of topotecan and ascorbic acid was predominantly due to necrosis of MCF-7 breast tumor cells.
Keywords: Topotecan; Ascorbic Acid; Topoisomerase; Electron Spin Resonance; Reduced Glutathione; Oxidized Glutathione; 5,5-Dimethyl Pyrroline-1-N-oxide; Reactive Oxygen Species
Graphical Abstract
INTRODUCTION
Topoisomerase I (topo I) is a nuclear enzyme responsible for maintaining DNA topology and functions [1–5]. Camptothecin and its analog topotecan (TPT) are important anticancer agents for the treatment of various human malignancies in the clinic [6–8]. The major mechanism of action of camptothecin and TPT (Figure 1) is due to the stabilization of transient complexes formed between topo I and DNA (cleavable complexes), resulting in the formation of highly toxic double-strand breaks in tumor cells, causing cell death [2, 3, 5]. While other mechanisms of actions of TPT that are independent of topo I have also been suggested, including induction of oxidative stress [9, 10] and inhibition of hypoxia-inducible factor [11, 12], there is very little known about how TPT induces oxidative stress in tumor cells. Akbas et al. [9] have shown that treatment of MCF-7 tumor cells with TPT leads to increased formation of reactive oxygen species (ROS) and nitrite, indicating increased oxidative stress. In addition, Timur et al. [13] have shown that there is a significant decrease in glutathione levels following TPT treatment of MCF-7 breast cancer cells with concomitant increases in lipid peroxidation and levels of antioxidant enzymes, superoxide dismutase, glutathione peroxidase and catalase, suggesting TPT-induced oxidative stress in MCF-7 tumor cells. It is interesting to note that Sordet et al. [14, 15] have shown that ROS generated by certain compounds, e.g., arsenic trioxide, induce formation of DNA-topo I complexes, which may play a role in the mechanism of drug cytotoxicity. Moreover, Daroui et al. [16] have shown that H2O2 cytotoxicity is partly mediated by topo I. Thus, it is possible that ROS formed from TPT may also contribute to topo I-induced DNA damage and cytotoxicity.
Figure 1.

Structure of Topotecan and the proposed Topotecan Radical
It has been shown that ascorbic acid (AA) induces tumor cell death by generating extracellular hydrogen peroxide (H2O2) which is taken up by tumor cells and leads to the formation of reactive hydroxyl radical (•OH) in the presence of metal ions [17] [18, 19]. The role of ascorbic acid (commonly known as Vitamin C) in cancer is extremely controversial [20–23]. Many investigators have found that ascorbic acid is highly toxic to tumor cells and combinations of ascorbic acid with several anticancer drugs are more than additive in tumor cell killing [19, 24–26], while other investigators have found no effect in cell culture and animal models [27, 28].
Because of the lack of understanding of how TPT produces reactive free radical species in tumor cells that can significantly affect topo I-induced DNA cleavage formation and modulate the toxicity of topo I drugs, we have investigated the mechanisms of free radical formation from TPT and examined the effects of ascorbic acid on TPT cytotoxicity in MCF-7 breast cancer cells. We have found that in the presence of H2O2 and peroxidases, TPT generates a TPT radical (TPT•), which is very stable and does not react with O2 to redox cycle and generate superoxide/H2O2. However, TPT• is extremely reactive with glutathione and cysteine, forming the corresponding thiyl (GS•and Cys•) radicals and regenerating TPT. Moreover, we found that ascorbic acid (a cellular H2O2 generator) modulates the cytotoxicity of TPT, and the combinations are highly synergistic in MCF-7 breast cancer cells.
Materials and Methods
Camptothecin was a gift of the Drug Synthesis and Chemistry Branch, Developmental Therapeutic Program of NCI, NIH. Topotecan hydrochloride was obtained from Cayman Chemicals (Ann Arbor, MI). A stock solution of camptothecin was prepared in DMSO, TPT was dissolved in doubly distilled water, and solutions were stored at −80°C. Ascorbic acid and horseradish peroxidase (HRP) were obtained from Sigma-Aldrich Chemical Company (St. Louis, MO). Ascorbic acid (100 mM) was dissolved in chelex-treated phosphate buffer and the pH of the solution was adjusted to 7.2 and stored at −80°C. Human myeloperoxidase was purchased from Lee Biosolutions (St. Louis, MO). Human topo I, antibody to human topo I, and SDS/KCl precipitation assay kits were obtained from Topogen (Buena Vista, CO). Primary antibody for the analysis of p53 was obtained from Santa Cruz Biochemicals (Dallas, TX). Primary antibody anti-gamma H2AX, for the detection of DNA double-strand breaks, anti-p21, and anti-β-actin were obtained from Abcam (Cambridge, MA). DMPO was purchased from Dojindo (Rockville, MD) and used without any further purification.
Electron Spin Resonance Studies
The ESR studies for the detection of TPT radicals were carried out as described previously [29, 30]. Briefly, reaction mixtures were in 1 ml chelex-treated phosphate buffer containing 25 μM DETPAC (pH 7.4), TPT (250–500 μM), HRP or human myeloperoxidase (250 μg/ml), and H2O2 (250–500 μM). For the detection of glutathione and cysteine radicals, the TPT radical was generated from TPT and HRP/H2O2 as above and the mixture treated for 5 min with excess catalase (2500 U/incubation) to remove unreacted H2O2. DMPO (100 mM) and GSH (1 mM) or cysteine (1 mM) were added, the mixture was transferred into an ESR flat cell, and the spectra recorded with an ELEXSYS E 500 ESR spectrometer (Brucker Biospin, Billerica, MA, USA) equipped with an ER4122SHQ cavity operating at 9.76 GHz at room temperature. The ESR settings were as follows: scan range 100 G; modulation frequency 100 KHz; modulation amplitude 1.0 G; microwave power 20 mW; receiver gain 2 × 104; time constant 250 ms and conversion time 250 ms.
Cell Culture
Human breast MCF-7 cancer cells (ATCC, Rockville, MD) were grown in Phenol Red-free RPMI 1640 media supplemented with 10% fetal bovine serum and antibiotics. MCF-7 cells were routinely used for 20–25 passages, after which the cells were discarded and a new cell culture was started from the frozen stock.
Cytotoxicity Studies
The cytotoxicity studies were carried out by using a cell growth inhibition assay. Briefly, 150,000–200,000 MCF-7 cells/well were plated in 2 ml of complete medium onto a 6-well plate (in triplicate) and allowed to attach for 18 h. The medium was removed and fresh warm medium (2 ml) was added. Cells were treated with various concentrations of TPT, and incubated for 48 h in the complete medium. Effects of ascorbic acid on TPT cytotoxicity were evaluated by adding ascorbic acid (1 mM) to MCF-7 breast cancer cells in complete medium (2 ml) under various conditions, adding TPT to the cells, and incubating for 48 h. Cells were trypsinized and the numbers of surviving cells were determined by counting the cells in a cell counter (Beckman, Brea, CA).
SDS-KCl Precipitation Assay
The formation of covalent topo I-DNA complexes with TPT and with and without ascorbic acid in MCF-7 breast cancer cells was quantitated by the SDS-KCl precipitation assay as described previously [31]. Briefly, the DNA of cells growing in the logarithmic phase (1 × 105 cells/ml), seeded into six-well plates, was labeled with [methyl-3H]-thymidine (1.0 μci, 20Ci/mmol; Perkin-Elmer, Waltham, MA) for 18–24 h, and washed twice with the medium, then TPT or ascorbic acid (1 mM) or combinations of TPT and ascorbic acid were added in 2 ml of complete medium and incubated for 1 h. Cells were washed with PBS (twice), and lysed with 1 ml of prewarmed lysis solution (Topogen). After lysis and shearing of DNA, DNA-TPT-topo I-complexes were precipitated with KCl. The precipitate was collected by centrifugation, and washed extensively (four times) with the washing solution (Topogen) per the manufacturer’s instructions. The radioactivity was counted in a scintillation counter after adding 5 ml of scintillation fluid.
Western Blot Assay
MCF-7 cancer cells were treated with TPT or ascorbic acid (1 mM) or a combination of TPT and ascorbic acid for various times. Cells were washed with ice-cold PBS and collected by centrifugation. Samples (10–40 μg of total protein) were electrophoresed under reducing conditions through 4–12% Bis-Tris NuPage acrylamide gels (Invitrogen, Carlsbad, CA). After electrophoresis, proteins were transferred onto a nitrocellulose membrane and probed with anti-p53, -p21, -H2AX and -beta actin antibodies. An Odssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE) was used to acquire images.
Analysis of Plasma Membrane Integrity and Cell Size by Flow Cytometry
Plasma membrane integrity was measured by adding propidium iodide (PI, Invitrogen, Waltham, MA) to cells at a final concentration of 10 μg/ml immediately prior to flow cytometric analysis. Ten thousand single cells were analyzed using a BD LSRII flow cytometer (San Jose, CA) equipped with FACSDiVa software. Cells were excited with a 561 nm laser and PI fluorescence was detected at 585 nm. A gate was drawn on a PI histogram for the control sample to determine the percent of viable and dead experimental cells. Changes in cell size were examined using forward-scatter (FSC) light. An increase in FSC indicates an increase in cell size.
RESULTS
ESR STUDIES
Incubation of TPT with rat microsomes in the presence or absence of NADPH gave no ESR detectable radical. However, when TPT was incubated with HRP in the presence of H2O2, a strong ESR signal was detected (Figure-2 Panel A). This single line spectrum had a g value of 2.006. It was found to be stable under aerobic conditions, demonstrating that the radical did not react with molecular O2. The same radical was also detected when human myeloperoxidase was used in the presence of H2O2. While the exact identity of this TPT• is not known currently, we assign it as a melanin-like polymer radical resulting from a phenoxyl radical of TPT (Figure 2a, Panel A). Such radicals have been previously observed from the phenoxyl radical of acetaminophen [32]. Formation of this radical is due to the one-electron oxidation of the 10-OH group of the quinoline ring of TPT (Figure-1). This is further confirmed by the failure of camptothecin, the parent drug which lacks the 10-OH group, to form any ESR-detectable radical (Panel A, b) under the conditions that resulted in the formation of TPT•, indicating that the 10-OH group was responsible for the TPT• formation.
Figure 2.

Panel A: The ESR spectra (a) of the TPT radical obtained following incubation of TPT (250 μM) with HRP (0.25 μg/ml) and H2O2 (500 μM) and (b) from camptothecin under similar conditions. Panel B: Spin trapping of Glutathione (a) and Cysteine (c) radicals by ESR in the presence of DMPO (100 mM). The TPT radical obtained as described above was treated with catalase to remove any H2O2 and treated with glutathione (1 mM) or cysteine (1 mM) in the presence of DMPO and the resulting spectra were recorded. The GS-DMPO radical (a) had the following hyperfine coupling constants aN = 15.1 G and aH = 16.1 G; (b) Simulation of GS-DMPO. The Cys-DMPO (c) had the following coupling constants: aN = 15.1 G; and aH = 17.4 G (d) Simulation of Cys-DMPO radical. The ESR studies were carried out as described in the methods section.
While this TPT• was stable under aerobic conditions, it reacted rapidly with GSH, and in the presence of the spin trap DMPO, a DMPO adduct was formed which was detected by ESR. The characteristic ESR spectrum of this adduct (shown in Figure-2, a, Panel B) consists of four lines with hyperfine coupling constants of aN = 15.1 G, and aH = 16.1 G. These splittings are similar to those of a known GSH adduct of DMPO (DMPO-SG) [33–35]. Similarly, TPT•, also reacted with cysteine to form a DMPO adduct (Figure-2 Panel- B, c; six-line spectrum) with coupling constants of aN = 15.1 G, aH = 17.4 G. These coupling constants are similar to those assigned to a DMPO-cysteine adduct [33, 35, 36]
CYTOTOXICITY STUDIES
Because TPT formed a free radical species in the presence of H2O2 that reacted with glutathione, we evaluated the cytotoxicity of TPT in MCF-7 breast cancer cells in the presence of ascorbic acid. Ascorbic acid is known to generate H2O2 in cells [17, 18]. As shown in Figure-3, TPT induced significant cell death in MCF-7 cells (IC50 = 5.0 ±1.0 × 10−7 M) following 48h of treatment. The presence of ascorbic acid significantly enhanced the cell death induced by TPT and lowered IC50 values to 6.0 ±1.0 × 10−9 M, about a 75-fold enhancement of TPT cytotoxicity. We next examined the schedule dependency of ascorbic acid with TPT and found that the significant enhancement of cell death by ascorbic acid was similar whether ascorbic acid was added first and TPT added 1h later (Figure-3C), TPT added for 1 h, before ascorbic acid was added (Figure-3C) or both were added simultaneously (Figure-3A). We chose 1 mM ascorbic acid because it induced 15–20% cell killing under our experimental conditions; the IC50 value was 2.5 ± 0.25 mM.
Figure 3.

Cytotoxicity (A) of TPT alone (■-■) and in the presence of ascorbic acid (●-●,1 mM). Mixtures were allowed to incubate for 48 h and the surviving cells were counted as described in the methods section. (B) Synergy plot at lower concentration of TPT. The synergy plot was calculated as described by Jonsson et al. [37]. The green line (▲-▲) in Figure 3A represents calculated values for additive interactions between TPT and ascorbic acid. TPT alone (■-■, black line) and TPT + AA (●-●, red line, added simultaneously) are actual values obtained experimentally. (C) Cytotoxicity of TPT under various conditions in the presence of ascorbic acid (1 mM). Ascorbic acid added (▲-▲) first; TPT added (●-●) first. Values represent four separate experiment carried out in triplicates. ***, ** and * p values ≤ 0.001, 0.005, and ≤ 0.05 compared with concentration-matched samples. ###, ##, and # values ≤ 0.001, 0.005 and ≤ 0.05 from concentration-matched predicated additive values.
Analysis for additive or synergistic interactions was carried out as described by Jonsson et al. [37]. In this model of analysis, if drug A has survival indices of 20% and drug B has survival indices of 40%, the combination of A and B is expected to produce survival indices of 8% (0.2 × 0.4 = .08 or 8%); any value lower than that predicted by this additive model suggests synergy. Under our experimental conditions, and using this method of analysis, the combination of ascorbic acid and TPT was found to be synergistic at all concentrations of TPT as shown in Figure-3A and the highest synergy was observed at the lower concentrations (e.g., 10−10 M) of TPT as shown in Figure-3B.
DNA DAMAGE STUDIES
As TPT is considered a topo I poison and the main mechanism of tumor cell death induced by TPT results from its interactions with topo I protein, leading to protein-associated DNA damage (single- and double-strand breaks), we examined the effects of ascorbic acid on both cleavable complex formation and induction of double-strand breaks in the presence of TPT. Data presented in Figure-4 (panel A) show that TPT alone was extremely active in inducing cleavable complex formation as detected by the SDS-KCl precipitation assay. While ascorbic acid alone was not effective in inducing topo I-dependent cleavage complex formation, the presence of ascorbic acid significantly increased TPT-induced SDS-KCl precipitate formation over a range of TPT concentrations (Figure-4). This would suggest that ascorbic acid increases TPT-dependent topo I-induced DNA damage in MCF-7 breast cancer cells.
Figure 4.

Panel A Formation of cleavage complexes in MCF-7 breast cancer cells by TPT alone (□) and TPT in the presence of ascorbic acid (■). Cells were treated with TPT (0.25 and 1.0 μM) for 1 h in complete medium (2 ml) in the presence or in the absence of ascorbic acid (1 mM). The SDS-KCl precipitation assay was carried out as described in the methods section. Data represent three independent experiments carried out in duplicates. ** and, * p values ≤ 0.005 and ≤ 0.05 compared with concentration-matched samples. Panel B: Western blots analysis for γ-H2AX, p53 and p21 following treatment with TPT (1 μM) alone and in the presence of ascorbic acid (1 mM) for 2 h. Panel C: Quantification of H2AX proteins. *, p value ≤ 0.05 compared to TPT alone.
We next examined the effects of ascorbic acid on TPT-induced double-strand break formation using the γ-H2AX assay, as it has been shown to be a marker for DNA double-strand breaks [38]. Treatment with TPT significantly increased γ-H2AX in MCF-7 breast cancer cells over the control (no treatment) at 2 h (Figure-4B). While ascorbic acid alone had no effect on double-strand breaks, it significantly enhanced TPT-induced γ-H2AX (Figure-4B), suggesting that ascorbic acid increases DNA double-strand break formation by TPT.
Increases in DNA damage caused by TPT and the combinations of TPT and ascorbic acid also induced both wild-type p53 and p21 proteins in MCF-7 cancer cells as noted before with topo I-active drugs [39, 40] (Figure-4B). It is interesting to note that significantly (2–3-fold) more p53 was induced with TPT in the presence of ascorbic acid than with TPT alone, most likely from the increased DNA damage observed with TPT in the presence of ascorbic acid.
APOTOSIS/NECROSIS STUDIES
Analyses of cell death caused by TPT, and the combination of TPT and ascorbic acid, clearly show that these drugs induced significant necrosis of MCF-7 tumor cells at 48 h (Figure 5). At 48 h both swelling and significant cell death was observed with TPT alone and the combinations of TPT and AA (Figure-5). Furthermore, a small increase in TPT-induced cell size was also observed in the presence of AA. These observations indicate that in MCF-7 breast cancer cells, cell death by TPT and the combination of TPT and ascorbic acid results mostly from necrosis.
Figure 5.

Necrosis induced by TPT and TPT + ascorbic acid in MCF-7 breast cancer cells at 48 h. MCF-7 cells were treated with TPT (0.5 μM) in the presence or absence of ascorbic acid (1 mM). Cells were collected and washed twice with ice-cold PBS and analyzed. Panel 1, control, no treatment; panel 2, ascorbic acid; panel 3, TPT; and panel 4, TPT + ascorbic acid. A representative scan is shown here.
DISCUSSION
Studies described in this report show that one-electron oxidation of TPT, catalyzed by a peroxidase-hydrogen peroxide system, readily forms a TPT radical. Because camptothecin, the parent drug of TPT, lacks a 10-OH group in the quinolone ring, and did not form any radical under the same experimental conditions that generated TPT•, we believe that the 10-OH of the TPT is required for this free radical formation, and the radical detected, therefore, is a phenoxyl radical-derived polymer radical [32] of TPT. We found that TPT• was stable under aerobic conditions, indicating that TPT• did not react with molecular O2 to generate oxygen-based reactive species, e.g., O2•−, H2O2 and •OH. However, TPT• reacted with GSH and cysteine, forming the corresponding thiyl radicals and regenerating the parent drug. While the main mechanism of tumor cell killing of TPT is believed to result from its interactions with topo I and inhibition of DNA functions [1–5], TPT also induces oxidative stress in tumor cells by depleting reduced GSH and it has been suggested that this oxidative stress may induce tumor cell death [13]. However, little has been known about how TPT generates oxygen free radicals and induces GSH depletion in tumor cells. Our present study now clearly provides a mechanism for the depletion of GSH in tumor cells induced by TPT radical that may lead to an imbalance of the redox state in tumor cells (i.e., oxidative stress).
We measured both glutathione depletion and oxidized glutathione formation following TPT and TPT + AA treatment in MCF-7 cells by using Elman’s reagent as described by Rahman et al. [41] and a Promega Glo (luminescent assay) assay kit. We did not to find significant changes in total glutathione in MCF-7 breast cells following TPT and TPT and AA combinations for either 1 h or 6 h incubations. It should be noted that Kagan et al. [42, 43] have shown that VP-16 causes more topo II-dependent DNA damage following depletion of cellular glutathione, resulting in increased VP-16 cytotoxicity.
High-dose ascorbic acid has been reported to cause tumor cell death and interact synergistically with various chemotherapy drugs in tumor cells and in vivo [26, 44–47]. Furthermore, certain tumor cells have been found to be susceptible to H2O2 toxicity [48]. Our present study clearly shows that ascorbic acid significantly increased (>75-fold) TPT cytotoxicity in MCF-7 breast cancer cells, and it was synergistic at all concentrations of TPT, especially at lower and biologically more relevant concentrations of TPT. It is interesting to note that this synergy was observed under various conditions of treatment, and there was no dependence on the TPT or ascorbic acid treatment schedule in MCF-7 breast cancer cells. We also found that both TPT and combinations of ascorbic acid and TPT increased cell size, a hallmark of necrosis, in MCF-7 breast cancer cells. MCF-7 cancer cells are known to be resistant to apoptosis to topo I drugs [49].
Mechanisms of this synergistic interaction between ascorbic acid and TPT in MCF-7 breast tumor cells were also investigated. TPT induces topo I-dependent cleavable complex formation in tumor cells that are converted to both single- and double-strand breaks, leading to tumor cell death. The presence of ascorbic acid significantly enhanced both cleavable complex formation and double-strand break formation by TPT in MCF-7 breast cancer cells. Since ascorbic acid produces H2O2 intracellularly, the possibility exists that peroxidase activity converted TPT into its free radical form (TPT•) which then participated in increased DNA damage, resulting in synergistic tumor cell death from combinations of TPT and ascorbic acid. Alternatively, Sordet et al. [14] have shown that oxygen free radicals generated from arsenic trioxide or hydrogen peroxide induce a topo I-mediated increase in cleavable complex formation; thus, the increased DNA damage observed with TPT in the presence of ascorbic acid, topo I may have participated in enhanced H2O2-dependent cytotoxicity. Pourquier et al. [50] have reported that oxidized DNA bases, especially after 8-oxo-guanine modification, result in a significant trapping of topo I-DNA complexes when present close to topo I cleavage sites (+1 or +2) in DNA. 8-Oxo-guanine formation in DNA is believed to be a hallmark of oxidative stress, resulting from the oxidation of DNA by H2O2-derived hydroxyl radicals [51, 52]. Thus, it is possible that H2O2 formed from ascorbic acid generates reactive •OH radicals in the presence of trace metal ions (Fe/Cu), resulting in the formation of 8-oxo-guanine near the topo I cleavage site, causing enhanced formation of both cleavage complexes and DNA double-strand-breaks in the presence of TPT, and resulting in increased cell death. Bruzzese et al. [53] have also reported a synergistic anticancer effect of TPT and vorinostate in small cell lung cancer cells which is mediated by generation of ROS, resulting in an increase in DNA-topo I covalent complexes and DNA double-strand breaks. Their observations are similar to the results reported in this study.
Sane et al. [54] have shown that in Jukart cells, high dose ascorbate antagonizes camptothecin cytotoxicity, while results presented in this study show that high dose ascorbate increases cytotoxicity from topotecan synergistically. It is interesting to note that although we were unable to detect free radical species from camptothecin during peroxidase-H2O2 catalysis, topotecan readily generated semiquinone polymer radicals. Taken together, this would suggest that free radical species formed intracellularly from ascorbate-generated H2O2 play an important role in topotecan-induced DNA damage and cell death.
Finally, it is interesting to note that the chemistry of TPT closely resembles that of another topo-poison, VP-16. VP-16 is a topo II-poison and contains a phenolic OH group that is easily oxidized to a phenoxyl radical by an HRP (or myeloperoxidase)- system [29, 30, 43]. Like TPT•, the VP-16• is stable at physiological pH; however, H2O2 it is still extremely reactive with glutathione, depleting glutathione in cells and in vivo [35, 55]. VP-16•, however, also undergoes significant metabolism forming various reactive products that bind to DNA and proteins [30, 56], including topo II, inhibiting its function [57]. At present, we do not know whether TPT• also undergoes metabolism to generate other species that contribute to TPT cytotoxicity. However, TPT is known to form some (less than 5%) N-demethylated products in human patients [58], and it has been suggested that the metabolism of TPT does not contribute significantly to the antitumor activities of TPT as the response rate was similar in patients with and without compromised liver functions [6].
In conclusion, we have shown that TPT undergoes one-electron oxidation to form a TPT-derived polymer radical that reacts with glutathione, resulting in the formation of glutathiyl radical. We found that inclusion of ascorbic acid, which produces H2O2 in tumor cells, significantly increases TPT cytotoxicity in MCF-7 breast cancer cells. Furthermore, ascorbic acid significantly enhanced the DNA damage induced by TPT, indicating that the synergistic cytotoxicity of the combination observed in this study is related to this increased DNA damage.
Highlights.
One-electron oxidation of topotecan generates topotecan phenoxy radical
Topotecan radical rapidly reacts with glutathione to generate glutathiyl radical
Ascorbic acid (AA) significantly enhances topotecan cytotoxicity in MCF-7 tumor cells
Ascorbic acid significantly increased topoisomerase I-dependent DNA damage
Synergistic cytotoxicity of topotecan and AA may result from increased DNA damage
Acknowledgments
We thank Ms. Mary Mason for her invaluable help in editing the manuscript. We also thank Drs. Maria Kadiiska and Douglas Ganini for their critical evaluation of the manuscript. We also thank Brett Wagner for his helpful discussions.
Funding: This research was supported by the intramural research program of the National Institute of Environmental Health Sciences, NIH. Statements contained herein do not necessarily represent the statements, opinions, or conclusions of NIEHS, NIH, or the US Government.
Abbreviations
- TPT
Topotecan
- AA
Ascorbic Acid
- Topo
Topoisomerase
- ESR
Electron Spin Resonance
- HRP
Horseradish Peroxidase
- GSH
Reduced Glutathione
- GSSG
Oxidized Glutathione
- DMPO
5,5-Dimethyl Pyrroline-1-N-oxide
- ROS
Reactive Oxygen Species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pommier Y, Leteurtre F, Fesen MR, Fujimori A, Bertrand R, Solary E, Kohlhagen G, Kohn KW. Cellular determinants of sensitivity and resistance to DNA topoisomerase inhibitors. Cancer investigation. 1994;12(5):530–42. doi: 10.3109/07357909409021413. [DOI] [PubMed] [Google Scholar]
- 2.Pommier Y, Tanizawa A, Kohn KW. Mechanisms of topoisomerase I inhibition by anticancer drugs. Advances in pharmacology. 1994;29B:73–92. doi: 10.1016/s1054-3589(08)61132-1. [DOI] [PubMed] [Google Scholar]
- 3.Froelich-Ammon SJ, Osheroff N. Topoisomerase poisons: harnessing the dark side of enzyme mechanism. The Journal of biological chemistry. 1995;270(37):21429–32. doi: 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- 4.Sinha BK. Topoisomerase inhibitors. A review of their therapeutic potential in cancer. Drugs. 1995;49(1):11–9. doi: 10.2165/00003495-199549010-00002. [DOI] [PubMed] [Google Scholar]
- 5.Stewart L, Redinbo MR, Qiu X, Hol WG, Champoux JJ. A model for the mechanism of human topoisomerase I. Science. 1998;279(5356):1534–41. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 6.Rowinsky EK, Verweij J. Review of phase I clinical studies with topotecan. Seminars in oncology. 1997;24(6 Suppl 20):S20-3–S20-10. [PubMed] [Google Scholar]
- 7.Muderspach LI, Blessing JA, Levenback C, Moore JL., Jr A Phase II study of topotecan in patients with squamous cell carcinoma of the cervix: a gynecologic oncology group study. Gynecologic oncology. 2001;81(2):213–5. doi: 10.1006/gyno.2000.6024. [DOI] [PubMed] [Google Scholar]
- 8.Robati M, Holtz D, Dunton CJ. A review of topotecan in combination chemotherapy for advanced cervical cancer. Therapeutics and clinical risk management. 2008;4(1):213–8. doi: 10.2147/tcrm.s1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akbas SH, Timur M, Ozben T. The effect of quercetin on topotecan cytotoxicity in MCF-7 and MDA-MB 231 human breast cancer cells. The Journal of surgical research. 2005;125(1):49–55. doi: 10.1016/j.jss.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 10.Kisa U, Caglayan O, Kacmaz M. The effects of topotecan on lipid peroxidation and antioxidant enzyme levels in rabbit liver tissue. Redox report: communications in free radical research. 2005;10(2):79–82. doi: 10.1179/135100005X21705. [DOI] [PubMed] [Google Scholar]
- 11.Rapisarda A, Uranchimeg B, Sordet O, Pommier Y, Shoemaker RH, Melillo G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: mechanism and therapeutic implications. Cancer research. 2004;64(4):1475–82. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- 12.Puppo M, Battaglia F, Ottaviano C, Delfino S, Ribatti D, Varesio L, Bosco MC. Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1alpha and -2alpha. Molecular cancer therapeutics. 2008;7(7):1974–84. doi: 10.1158/1535-7163.MCT-07-2059. [DOI] [PubMed] [Google Scholar]
- 13.Timur M, Akbas SH, Ozben T. The effect of Topotecan on oxidative stress in MCF-7 human breast cancer cell line. Acta biochimica Polonica. 2005;52(4):897–902. [PubMed] [Google Scholar]
- 14.Sordet O, Khan QA, Plo I, Pourquier P, Urasaki Y, Yoshida A, Antony S, Kohlhagen G, Solary E, Saparbaev M, Laval J, Pommier Y. Apoptotic topoisomerase I-DNA complexes induced by staurosporine-mediated oxygen radicals. The Journal of biological chemistry. 2004;279(48):50499–504. doi: 10.1074/jbc.M410277200. [DOI] [PubMed] [Google Scholar]
- 15.Sordet O, Liao Z, Liu H, Antony S, Stevens EV, Kohlhagen G, Fu H, Pommier Y. Topoisomerase I-DNA complexes contribute to arsenic trioxide-induced apoptosis. The Journal of biological chemistry. 2004;279(32):33968–75. doi: 10.1074/jbc.M404620200. [DOI] [PubMed] [Google Scholar]
- 16.Daroui P, Desai SD, Li TK, Liu AA, Liu LF. Hydrogen peroxide induces topoisomerase I-mediated DNA damage and cell death. The Journal of biological chemistry. 2004;279(15):14587–94. doi: 10.1074/jbc.M311370200. [DOI] [PubMed] [Google Scholar]
- 17.Clement MV, Ramalingam J, Long LH, Halliwell B. The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxidants & redox signaling. 2001;3(1):157–63. doi: 10.1089/152308601750100687. [DOI] [PubMed] [Google Scholar]
- 18.Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, Khosh DB, Drisko J, Levine M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(32):11105–9. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, Taghiyev AF, Du C, Knudson CM, Cullen JJ. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(2):509–20. doi: 10.1158/1078-0432.CCR-09-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conklin KA. Cancer chemotherapy and antioxidants. The Journal of nutrition. 2004;134(11):3201S–3204S. doi: 10.1093/jn/134.11.3201S. [DOI] [PubMed] [Google Scholar]
- 21.Park S. The effects of high concentrations of vitamin C on cancer cells. Nutrients. 2013;5(9):3496–505. doi: 10.3390/nu5093496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs C, Hutton B, Ng T, Shorr R, Clemons M. Is there a role for oral or intravenous ascorbate (vitamin C) in treating patients with cancer? A systematic review. The oncologist. 2015;20(2):210–23. doi: 10.1634/theoncologist.2014-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffer LJ, Robitaille L, Zakarian R, Melnychuk D, Kavan P, Agulnik J, Cohen V, Small D, Miller WH., Jr High-dose intravenous vitamin C combined with cytotoxic chemotherapy in patients with advanced cancer: a phase I–II clinical trial. PloS one. 2015;10(4):e0120228. doi: 10.1371/journal.pone.0120228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du J, Cieslak JA, 3rd, Welsh JL, Sibenaller ZA, Allen BG, Wagner BA, Kalen AL, Doskey CM, Strother RK, Button AM, Mott SL, Smith B, Tsai S, Mezhir J, Goswami PC, Spitz DR, Buettner GR, Cullen JJ. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer research. 2015;75(16):3314–26. doi: 10.1158/0008-5472.CAN-14-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cieslak JA, Cullen JJ. Treatment of Pancreatic Cancer with Pharmacological Ascorbate. Current pharmaceutical biotechnology. 2015;16(9):759–70. doi: 10.2174/138920101609150715135921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fromberg A, Gutsch D, Schulze D, Vollbracht C, Weiss G, Czubayko F, Aigner A. Ascorbate exerts anti-proliferative effects through cell cycle inhibition and sensitizes tumor cells towards cytostatic drugs. Cancer chemotherapy and pharmacology. 2011;67(5):1157–66. doi: 10.1007/s00280-010-1418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Creagan ET, Moertel CG, O’Fallon JR, Schutt AJ, O’Connell MJ, Rubin J, Frytak S. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. The New England journal of medicine. 1979;301(13):687–90. doi: 10.1056/NEJM197909273011303. [DOI] [PubMed] [Google Scholar]
- 28.Heaney ML, Gardner JR, Karasavvas N, Golde DW, Scheinberg DA, Smith EA, O’Connor OA. Vitamin C antagonizes the cytotoxic effects of antineoplastic drugs. Cancer research. 2008;68(19):8031–8. doi: 10.1158/0008-5472.CAN-08-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haim N, Roman J, Nemec J, Sinha BK. Peroxidative free radical formation and O-demethylation of etoposide(VP-16) and teniposide(VM-26) Biochemical and biophysical research communications. 1986;135(1):215–20. doi: 10.1016/0006-291x(86)90965-4. [DOI] [PubMed] [Google Scholar]
- 30.Haim N, Nemec J, Roman J, Sinha BK. Peroxidase-catalyzed metabolism of etoposide (VP-16-213) and covalent binding of reactive intermediates to cellular macromolecules. Cancer research. 1987;47(22):5835–40. [PubMed] [Google Scholar]
- 31.Liu LF, Rowe TC, Yang L, Tewey KM, Chen GL. Cleavage of DNA by mammalian DNA topoisomerase II. The Journal of biological chemistry. 1983;258(24):15365–70. [PubMed] [Google Scholar]
- 32.West PR, Harman LS, Josephy PD, Mason RP. Acetaminophen: enzymatic formation of a transient phenoxyl free radical. Biochemical pharmacology. 1984;33(18):2933–6. doi: 10.1016/0006-2952(84)90222-3. [DOI] [PubMed] [Google Scholar]
- 33.Harman LS, Carver DK, Schreiber J, Mason RP. One- and two-electron oxidation of reduced glutathione by peroxidases. The Journal of biological chemistry. 1986;261(4):1642–8. [PubMed] [Google Scholar]
- 34.Saez G, Thornalley PJ, Hill HA, Hems R, Bannister JV. The production of free radicals during the autoxidation of cysteine and their effect on isolated rat hepatocytes. Biochimica et biophysica acta. 1982;719(1):24–31. doi: 10.1016/0304-4165(82)90302-6. [DOI] [PubMed] [Google Scholar]
- 35.Katki AG, Kalyanaraman B, Sinha BK. Interactions of the antitumor drug, etoposide, with reduced thiols in vitro and in vivo. Chemico-biological interactions. 1987;62(3):237–47. doi: 10.1016/0009-2797(87)90025-1. [DOI] [PubMed] [Google Scholar]
- 36.Harman LS, Mottley C, Mason RP. Free radical metabolites of L-cysteine oxidation. The Journal of biological chemistry. 1984;259(9):5606–11. [PubMed] [Google Scholar]
- 37.Jonsson E, Fridborg H, Nygren P, Larsson R. Synergistic interactions of combinations of topotecan with standard drugs in primary cultures of human tumor cells from patients. European journal of clinical pharmacology. 1998;54(7):509–14. doi: 10.1007/s002280050505. [DOI] [PubMed] [Google Scholar]
- 38.Nagelkerke A, Span PN. Staining Against Phospho-H2AX (gamma-H2AX) as a Marker for DNA Damage and Genomic Instability in Cancer Tissues and Cells. Advances in experimental medicine and biology. 2016;899:1–10. doi: 10.1007/978-3-319-26666-4_1. [DOI] [PubMed] [Google Scholar]
- 39.Liu W, Zhang R. Upregulation of p21WAF1/CIP1 in human breast cancer cell lines MCF-7 and MDA-MB-468 undergoing apoptosis induced by natural product anticancer drugs 10-hydroxycamptothecin and camptothecin through p53-dependent and independent pathways. International journal of oncology. 1998;12(4):793–804. [PubMed] [Google Scholar]
- 40.Deptala A, Li X, Bedner E, Cheng W, Traganos F, Darzynkiewicz Z. Differences in induction of p53, p21WAF1 and apoptosis in relation to cell cycle phase of MCF-7 cells treated with camptothecin. International journal of oncology. 1999;15(5):861–71. doi: 10.3892/ijo.15.5.861. [DOI] [PubMed] [Google Scholar]
- 41.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nature protocols. 2006;1(6):3159–65. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 42.Yalowich JC, Tyurina YY, Tyurin VA, Allan WP, Kagan VE. Reduction of phenoxyl radicals of the antitumour agent etoposide (VP-16) by glutathione and protein sulfhydryls in human leukaemia cells: Implications for cytotoxicity. Toxicology in vitro: an international journal published in association with BIBRA. 1996;10(1):59–68. doi: 10.1016/0887-2333(95)00106-9. [DOI] [PubMed] [Google Scholar]
- 43.Kagan VE, Kuzmenko AI, Tyurina YY, Shvedova AA, Matsura T, Yalowich JC. Pro-oxidant and antioxidant mechanisms of etoposide in HL-60 cells: role of myeloperoxidase. Cancer research. 2001;61(21):7777–84. [PubMed] [Google Scholar]
- 44.Sarna S, Bhola RK. Chemo-immunotherapeutical studies on Dalton’s lymphoma in mice using cisplatin and ascorbic acid: synergistic antitumor effect in vivo and in vitro. Archivum immunologiae et therapiae experimentalis. 1993;41(5–6):327–33. [PubMed] [Google Scholar]
- 45.Verrax J, Calderon PB. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free radical biology & medicine. 2009;47(1):32–40. doi: 10.1016/j.freeradbiomed.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 46.Yeom CH, Lee G, Park JH, Yu J, Park S, Yi SY, Lee HR, Hong YS, Yang J, Lee S. High dose concentration administration of ascorbic acid inhibits tumor growth in BALB/C mice implanted with sarcoma 180 cancer cells via the restriction of angiogenesis. Journal of translational medicine. 2009;7:70. doi: 10.1186/1479-5876-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohno S, Ohno Y, Suzuki N, Soma G, Inoue M. High-dose vitamin C (ascorbic acid) therapy in the treatment of patients with advanced cancer. Anticancer research. 2009;29(3):809–15. [PubMed] [Google Scholar]
- 48.Doskey CM, Buranasudja V, Wagner BA, Wilkes JG, Du J, Cullen JJ, Buettner GR. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox biology. 2016;10:274–284. doi: 10.1016/j.redox.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nieves-Neira W, Pommier Y. Apoptotic response to camptothecin and 7-hydroxystaurosporine (UCN-01) in the 8 human breast cancer cell lines of the NCI Anticancer Drug Screen: multifactorial relationships with topoisomerase I, protein kinase C, Bcl-2, p53, MDM-2 and caspase pathways. International journal of cancer. 1999;82(3):396–404. doi: 10.1002/(sici)1097-0215(19990730)82:3<396::aid-ijc13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 50.Pourquier P, Ueng LM, Fertala J, Wang D, Park HJ, Essigmann JM, Bjornsti MA, Pommier Y. Induction of reversible complexes between eukaryotic DNA topoisomerase I and DNA-containing oxidative base damages. 7, 8-dihydro-8-oxoguanine and 5-hydroxycytosine. The Journal of biological chemistry. 1999;274(13):8516–23. doi: 10.1074/jbc.274.13.8516. [DOI] [PubMed] [Google Scholar]
- 51.Grollman AP, Moriya M. Mutagenesis by 8-oxoguanine: an enemy within. Trends in genetics: TIG. 1993;9(7):246–9. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 52.Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2′-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis, Journal of environmental science and health. Part C. Environmental carcinogenesis & ecotoxicology reviews. 2009;27(2):120–39. doi: 10.1080/10590500902885684. [DOI] [PubMed] [Google Scholar]
- 53.Bruzzese F, Rocco M, Castelli S, Di Gennaro E, Desideri A, Budillon A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Molecular cancer therapeutics. 2009;8(11):3075–87. doi: 10.1158/1535-7163.MCT-09-0254. [DOI] [PubMed] [Google Scholar]
- 54.Sane AT, Cantin AM, Paquette B, Wagner JR. Ascorbate modulation of H(2)O(2) and camptothecin-induced cell death in Jurkat cells. Cancer chemotherapy and pharmacology. 2004;54(4):315–21. doi: 10.1007/s00280-004-0828-8. [DOI] [PubMed] [Google Scholar]
- 55.Fan Y, Schreiber EM, Giorgianni A, Yalowich JC, Day BW. Myeloperoxidase-catalyzed metabolism of etoposide to its quinone and glutathione adduct forms in HL60 cells. Chemical research in toxicology. 2006;19(7):937–43. doi: 10.1021/tx0600595. [DOI] [PubMed] [Google Scholar]
- 56.Haim N, Nemec J, Roman J, Sinha BK. In vitro metabolism of etoposide (VP-16-213) by liver microsomes and irreversible binding of reactive intermediates to microsomal proteins. Biochemical pharmacology. 1987;36(4):527–36. doi: 10.1016/0006-2952(87)90362-5. [DOI] [PubMed] [Google Scholar]
- 57.Gantchev TG, Hunting DJ. The ortho-quinone metabolite of the anticancer drug etoposide (VP-16) is a potent inhibitor of the topoisomerase II/DNA cleavable complex. Molecular pharmacology. 1998;53(3):422–8. doi: 10.1124/mol.53.3.422. [DOI] [PubMed] [Google Scholar]
- 58.Rosing H, Herben VM, van Gortel-van Zomeren DM, Hop E, Kettenes-van den Bosch JJ, ten Bokkel Huinink WW, Beijnen JH. Isolation and structural confirmation of N-desmethyl topotecan, a metabolite of topotecan. Cancer chemotherapy and pharmacology. 1997;39(6):498–504. doi: 10.1007/s002800050605. [DOI] [PubMed] [Google Scholar]