

Abstract
The objective of this study was to investigate a possible role of mitochondrial dihydrolipoamide dehydrogenase (DLDH) as a chemical preconditioning target for neuroprotection against ischemic injury. We used 5-methoxyindole-2-carboxylic acid (MICA), a reportedly reversible DLDH inhibitor, as the preconditioning agent and administered MICA to rats mainly via dietary intake. Upon completion of 4 week’s MICA treatment, rats underwent 1 h transient ischemia and 24 h reperfusion followed by tissue collection. Our results show that MICA protected the brain against ischemic stroke injury as the infarction volume of the brain from the MICA-treated group was significantly smaller than that from the control group. Data were then collected without or with stroke surgery following MICA feeding. It was found that in the absence of stroke following MICA feeding, DLDH activity was lower in the MICA treated group than in the control group, and this decreased activity could be partly due to DLDH protein sulfenation. Moreover, DLDH inhibition by MICA was also found to upregulate the expression of NAD(P)H-ubiquinone oxidoreductase 1(NQO1) via the Nrf2 signaling pathway. In the presence of stroke following MICA feeding, decreased DLDH activity and increased Nrf2 signaling were also observed along with increased NQO1 activity, decreased oxidative stress, decreased cell death, and increased mitochondrial ATP output. We also found that MICA had a delayed preconditioning effect four weeks post MICA treatment. Our study indicates that administration of MICA confers chemical preconditioning and neuroprotection against ischemic stroke injury.
Keywords: 5-methoxyindole-2-carboxylic acid (MICA), chemical preconditioning, dihydrolipoamide dehydrogenase, ischemic stroke, neuroprotection
Graphical abstract
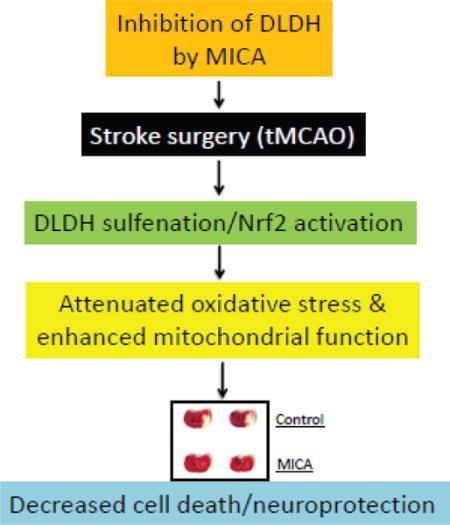
Introduction
Stroke is a leading cause of disability and death in the United States. Fortunately, the brain can be induced to tolerate stroke injury. One promising strategy to achieve this tolerance is called preconditioning [1–3], wherein exposure to non-injurious stimuli affords protection against subsequent injurious ischemic challenges. Preconditioning in the brain can be triggered not only by brief episodes of ischemia-reperfusion [4, 5] but also by administration of certain chemicals or drugs [6–10]. As ethical considerations do not allow for the use of brief repeated occlusion of the cerebral arteries to elicit stroke tolerance in humans, animal models of chemical preconditioning have been actively investigated in hopes to develop a clinically-useful approach to stroke preconditioning in humans. Moreover, chemical preconditioning may also be useful as a prophylactic approach to neuroprotection [11, 12]. The reason for this is that while the onset of stroke is often sudden and unpredictable, individuals with a higher than normal risk of stroke could benefit from prior measures that enhance the brain’s tolerance to potential ischemic injury. Such is the case for patients who are scheduled to undergo cardiovascular procedures during which the brain needs to be prophylactically protected against possible stroke injury.
Mitochondria are a known target for preconditioning against stroke injury [13–15]. When oxygen and nutrients supply to the affected area of tissues come to a halt upon ischemia, mitochondrial ATP production is severely decreased. The decrease in ATP content triggers functional impairment of ATP-dependent calcium channels, leading to overload of cellular and mitochondrial calcium [16, 17] that in turn can trigger glutamate excitotoxicity [18, 19]. On the other hand, as cells have to undergo anaerobic respiration in the absence of oxygen, lactate formed from pyruvate via lactate dehydrogenase accumulates, which results in a decrease in cellular pH and closure of mitochondrial permeability transition pore (MPTP) [20]. Upon reperfusion, a sudden resumption of blood flow can over energize mitochondrial respiration, leading to a spike in mitochondrial generation of reactive oxygen species (ROS) and opening of MPTP [21–24]. This opening can release cytochrome c that then activates cell death pathways and causes tissue infarction [25, 26]. Moreover, ROS production could further accentuate cellular apoptosis as ROS can induce oxidative stress and impair protein functions [27–35]. Therefore, based on the key roles of mitochondria in cell death and ischemic reperfusion injury, numerous mitochondrial proteins have been assessed or suggested as targets for preconditioning against ischemic stroke injury [14, 36].
In this paper, we describe our findings that mitochondrial dihydrolipoamide dehydrogenase (DLDH) could be a target for chemical preconditioning against ischemic stroke injury. DLDH is a family member of flavin-dependent, pyridine dinucleotide oxidoreductases [37]. It is the third component of α-ketoglutarate dehydrogenase complex, pyruvate dehydrogenase complex, and branched chain amino acid dehydrogenase complexes. DLDH is also involved in the glycine cleavage system [38, 39]. Each of these complexes or pathways is fundamental for mitochondrial bioenergetics and cell survival. Yet, mouse with a loss of 50% DLDH protein content is viable and fertile [40], suggesting that DLDH function could be inhibited to an extent to which no harm ensues. Our main findings indicate that dietary inhibition of DLDH for 4 weeks using chow supplemented with 5-methoxyindole-2-carboxylic acid (MICA), a specific inhibitor of DLDH function [41–44], significantly decreased infarct volume after transient middle cerebral artery occlusion (tMCAO), while no detrimental effects on food intake, body weight gain, blood glucose concentrations, and mitochondrial electron transport chain activities were detected in the absence of stroke. The preconditioning mechanism in the absence of stroke appeared to involve decreased DLDH activity and increased NAD(P)H: ubiquinone oxidoreductase-1 (NQO1) expression via activation of the Nrf2 signaling pathway. This mechanism was also found to operate upon stroke after MICA feeding. Our data indicate that decreased oxidative stress and apoptosis and increased mitochondrial ATP output are involved in stroke neuroprotection induced by MICA/DLDH preconditioning.
Materials and methods
Animals
Young male Sprague-Dawley rats (approximately 3 months old) were used in this study. The use of animals was approved by Institutional Care and Use Committee of University of North Texas Health Science Center and the protocol was in accordance with NIH Guidelines for the Care and Use of Laboratory Animals. Rats were randomly grouped for MICA groups and control groups.
Chemicals and reagents
5-methoxyindole-2-carboxylic acid (MICA) was purchased from Fisher Scientific (Hanover Park, IL). Lipoamide, BSA, nitro-blue tetrazolium (NBT) tablets, NADH, NAD+, succinate, ATP, antimycin A, cytochrome c, 2,3,5-triphenyltetrazolium chloride (TTC), and pyruvate were obtained from Sigma (St. Louis, MO). Dihydrolipoamide was synthesized from lipoamide using sodium borohydride as we previously reported [45]. Amino caproic acid was purchased from MP Biochemicals. Acrylamide, bis-acrylamide, Coomassie brilliant blue, Bradford protein assay solution, and streptavidin-HRP were purchased from Bio-Rad. DLDH antibodies and HRP conjugated secondary antibodies were from US Biological (Swampscott, MA) and Invitrogen (San Diego, CA), respectively. Mito-ID extracellular O2 sensor kit for the measurement of mitochondrial respiration was purchased from Enzo Life Sciences (Farmingdale, NY). Immunoblot membranes and ECL immunochemical detection solution were from GE Healthcare (Piscataway, NJ). Protein sulfenic acid probe DCP-Bio1 was purchased from Karafast (Boston, MA). Other antibodies were obtained from Abcam (Cambridge, UK).
Administration of MICA
MICA-containing chow was made by TestDiet (Richmond, IN). For feeding studies, rats were fed with diet supplemented with 0.33% MICA for 4 weeks. Rats were randomly assigned to either control or MICA-treated groups. Control rats were fed a standard rodent diet (Prolab RMH 1800, 5LL2), and the MICA-treated rats consumed a 5LL2 diet containing 0.33% of MICA. All rats had ad libitum access to food and water. Food consumption and body weight were monitored. The average dose of MICA derived from 0.33% diet supplement was equivalent to 200 mg/kg per rat per day, a dosage adopted from a previous study [42]. Dietary feeding of MICA was followed by stroke surgery or decapitation for tissue collection. No death caused by MICA feeding was observed. For MICA injection studies, rats were injected with MICA (200 mg/kg body weight) 24 h before ischemic surgery.
Transitional middle cerebral artery occlusion (tMCAO)
For tMCAO surgery, an intraluminal filament model was adapted as previously described [46, 47]. Rats were anesthetized by 1–3% isoflurane in 30% oxygen using an anesthetic vaporizer and flowmeter. The left MCA was occluded by a 4–0 monofilament suture (coated with silicon to a diameter of 0.30 - 0.33 mm) introduced via internal carotid artery. After a 60 minute occlusion, the suture was withdrawn for reperfusion. Subsequently, rats were maintained in the University of North Texas Health Science Center Vivarium. For sham surgery, anesthesia and surgery were performed as described for tMCAO except that no suture was introduced.
Measurement of infarct size
Brain ischemic injury was assessed by measuring the infarct volume using 2,3,5-triphenyltetrazolium chloride (TTC) staining [46, 47]. Briefly, brain slice was incubated for 30 minutes in a 2% solution of TTC in physiological saline at 37°C, and then fixed in 10% formalin. The stained slice was then digitally scanned and subsequently measured for the ischemic lesion size (AlphaEaseFC) [48]. The percentage of infarction volume over total brain volume was calculated as previously described [49].
Preparation of brain mitochondria
Mitochondria isolation from whole brain was carried out using Percoll gradient centrifugation as previously reported [50] with slight modifications [45, 51]. Brains were removed rapidly and homogenized in 15 ml of ice-cold mitochondrial isolation buffer containing 0.32 M sucrose, 1 mM EDTA and 10 mM Tris-HCl, pH 7.1. The homogenate was centrifuged at 1,330 g for 10 min and the supernatant was saved. The pellet was resuspended in half volume (7.5 ml) of the original isolation buffer and centrifuged again under the same conditions. The two supernatants were combined and centrifuged further at 21,200 g for 10 min. The resulting pellet was resuspended in 12% Percoll solution prepared in mitochondrial isolation buffer followed by centrifugation at 6,900 g for 10 min. The obtained soft pellet was resuspended in 10 ml of the mitochondrial isolation buffer and centrifuged again at 6,900 g for 10 min. All of the mitochondrial pellets obtained after centrifugation were either used immediately or frozen at −80°C until analysis. Protein concentrations were determined by Bradford assay [52].
Measurement of enzyme activities
DLDH dehydrogenase activity was determined using dihydrolipoamide and NAD+ as the substrates as previously described [45, 51]. Quantitation of mitochondrial complexes I, IV and V activities was also conducted as previously described using in-gel based assays [53]. Activities for complexes II and III were determined spectrophotometrically as previously described [54, 55]. NQO1 enzyme activity was assayed using NADH as the electron donor and 2,6-dichloroindophenol (DCPIP) as the electron acceptor as previously described [56]. Briefly, the final volume of reaction was 1 ml, and the mixture contained 50 mM Tris-HCl, pH 7.4, 15 mg/ml BSA, 1.5 mM EDTA, 40 μM DCPIP, and 30 μg brain cytosolic proteins. The reaction was started by addition of 0.2 mM NADH and the decrease in absorbance at 600 nm due to DCPIP reduction by NADH was monitored for 5 min. Dicoumarol (20 μM) was used as an NQO1 inhibitor to test the specificity of the assay [56] and NQO1 activity was deduced by subtracting the residual activity obtained in the presence of dicoumarol.
Polyacrylamide gel electrophoresis and Western blot analysis
Nongradient blue native gel electrophoresis was performed as previously described [45, 57]. For SDS-PAGE and Western blot assays, 10% resolving SDS-PAGE was usually performed. Typically, one of the resulting gels was stained with Coomassie colloid blue [53], and the other gel was subjected to electrophoretic transfer to immunoblot membrane and immunoblotting [58]. Immunochemical signals on the immunoblot membrane were detected with an enhanced chemiluminescence (ECL) kit. All images were documented by an EPSON PERFECTION 1670 scanner with all densitometric quantifications of gel images being analyzed by AlphaEaseFC software.
Other measurements
Nrf2 nuclear translocation was determined by electrophoretic mobility shift assay (EMSA) [59] using a commercially available kit purchased from Signosis Inc (Santa Clara, CA, catalogue number GS-0031). Brain homogenate H2O2 was measured by the Amplex Red method [60] using a kit purchased from Invitrogen (catalog number A22188). Caspase-3 activity (cleaved form), as a cell death parameter [61], was determined by a kit that was purchased from BioAssay (Hayward, CA). Protein carbonyl content was determined by a gel-based Western blot analysis of biotin-hydrazide derivatized proteins [31, 62, 63], whereby Western blot signal intensities were normalized against those of Coomassie blue stained bands. Lipid peroxidation was determined by measuring thiobarbituric acid reactive substances (TBARS) [64] using a kit purchased from BioAssay Systems (Hayward, CA). ATP content was measured spectrophotometrically by an ATP probe kit that was purchased from BioVision (Milpitas, CA, catalog# K354–100). This assay employs phosphorylation of glycerol by ATP to yield ADP, and the byproduct of this reaction eventually oxidizes the probe to form resorufin that can be easily monitored at 570 nm [65, 66]. Mitochondrial function measured as mitochondrial respiration was performed using Mito-ID extracellular oxygen sensor kit as previously described [67].
Data analysis
Statistical data analysis was performed by GraphPad’s 2-tailed unpaired t test (GraphPad, San Diego, CA). P < 0.05 was considered statistically significant.
Results
MICA administration via injection induces ischemic tolerance
We initially tested whether administration of MICA via intraperitoneal injection would increase the brain’s ischemic tolerance or impart preconditioning after tMCAO. In these studies, rats received MICA injection (200 mg/kg, I.P.) [42] once per day for seven days. Following tMCAO performed 24 h after the last MICA injection, brain infarction volume was measured densitometrically after histochemical staining with 2,3,5-triphenyltetrazolium chloride (TTC) [68]. Results in Fig. 1 A and B show that the infarction volume was nearly 60% lower in MICA-treated than in control rats, indicating that MICA had a preconditioning effect.
Fig.1.
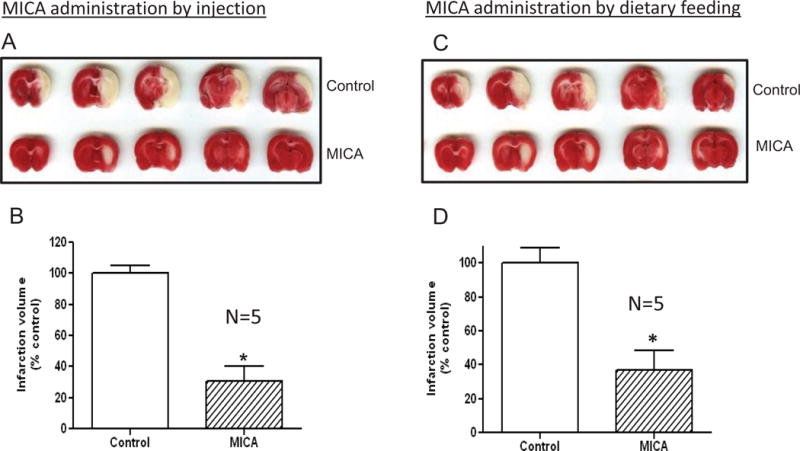
DLDH inhibition by MICA induces preconditioning effects in the brain. (A) Representative TTC stained brain slices from rats at the end of 1 h ischemia and 24 h reperfusion that received daily vehicle or MICA injections for 7 days. (B) Relative infarct volume derived from A. (mean ± SEM, n=5 per group). (C) Representative TTC-stained brain slices from rats fed control or MICA-containing diets for 4 weeks followed by tMCAO. (D) Relative infarct volume derived from C (mean ± SEM, n=5). P< 0.05 indicates statistical significance.
MICA administration via dietary intake also induces ischemic tolerance
Based on the above results of MICA injection studies, we wanted to test whether MICA administered orally and non-invasively could also be used as a preconditioning approach for ischemic tolerance. Results show that there was also a significant decrease in infarction volume in the MICA-treated vs. control rats (Fig. 1 C and D), indicating that 4 weeks’ MICA treatment via dietary administration also induced preconditioning effects in the brain.
MICA diet does not affect body weight, food intake, or blood glucose concentrations in the absence of stroke
To investigate whether MICA dietary administration had any detrimental effects on the animals, we measured body weight gain and food intake as well as blood glucose concentrations before and after the whole feeding procedure. Blood glucose concentrations were measured by a glucose meter (FreeStyle Light, Abbott). As shown in Fig. 2, MICA diet had no effect on body weight gain, food intake, and blood glucose concentrations, indicating that administration of MICA via dietary feeding does not have detectable toxic effects on the animals in the absence of stroke.
Fig. 2.
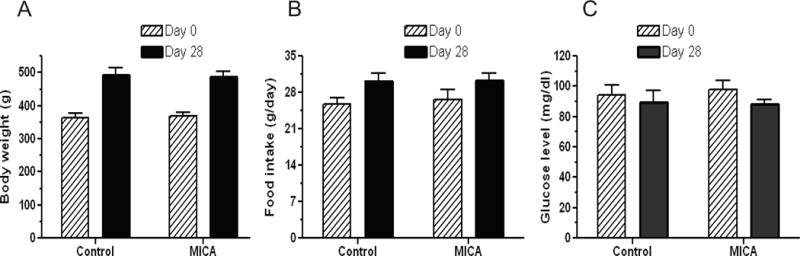
Effects of MICA feeding on body weight increase (A), food intake (B), and blood glucose concentrations (C). Measurements were made on day 0 and day 28 for both control and MICA diets. Values are mean ± SEM, n = 6.
MICA diet does not affect mitochondrial electron transport chain and oxidative phosphorylation in the absence of stroke
To determine whether the 4 weeks’ MICA dietary administration had any negative effects on mitochondrial function, we measured activities of mitochondrial respiratory complexes as previously described [53] and mitochondrial respiration reflected by oxygen consumption. These measurements were conducted on mitochondria isolated from brains without tMCAO. Results indicate that mitochondrial electron transport chain complexes were not affected by MICA diet (Fig. 3 A–D). Additionally, MICA did not affect complex V (F1F0 ATP synthase) activity (Fig. 3E). Moreover, MICA treatment did not impair mitochondrial function as there was no difference between control diet and MICA diet in mitochondrial respiration (Fig. 3F). These results indicate that dietary MICA intake does not impair mitochondrial oxidative phosphorylation in the absence of stroke.
Fig. 3.
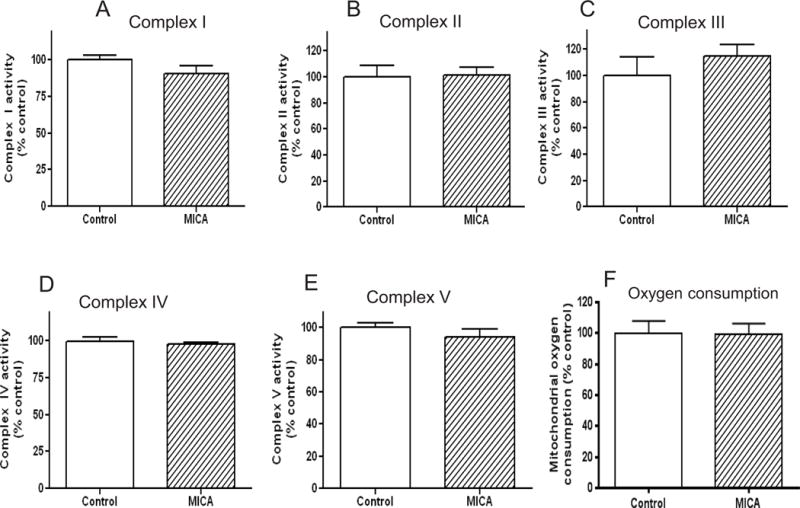
Effects of MICA diet on activities of mitochondrial electron transport chain components (panels A to E for complexes I to V, respectively) and mitochondrial function. Activities were measured as previously described [53, 55]. Panel F shows mitochondrial oxygen consumption between control and MICA diet. For A to E, values are mean ± SEM from 5 rats per group; for F, values are mean ± SEM from 3 rats per group.
Dietary MICA decreases DLDH activity
Based on previous studies that MICA reversibly inhibits DLDH function [41–44], we then investigated whether there was a change in DLDH activity after MICA dietary feeding by measuring DLDH activity using blue native gel analysis. Results in Fig. 4A shows that DLDH activity was lower in the MICA-treated group than in the control group. Interestingly, DLDH protein content did not show detectable changes when measured by anti-DLDH Western blot assay (Fig. 4 B and C), suggesting that the partial loss in DLDH activity in the MICA-treated group was likely due to posttranslational oxidative modification of the enzyme.
Fig. 4. Left panel.
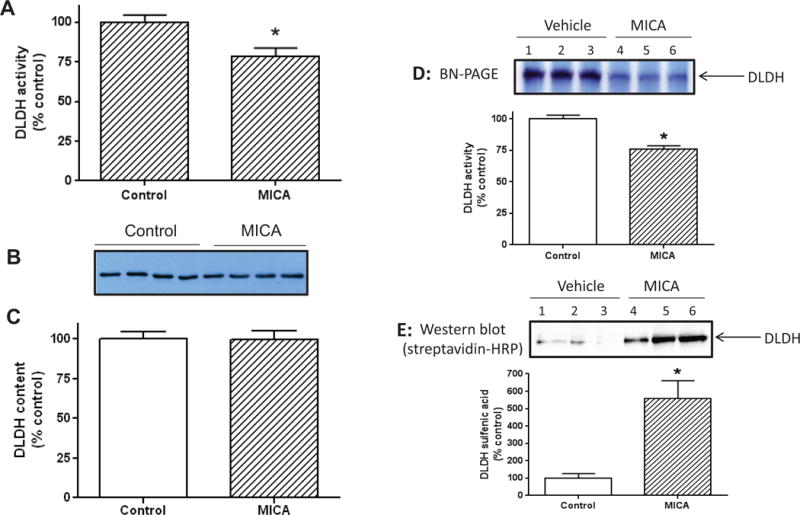
Effects of MICA diet on DLDH activity and protein content. (A): DLDH activity determined spectrophotometrically; (B) DLDH protein content determined by anti-DLDH Western blot; (C) Densitometric quantification of the bands shown in B. (*p<0.05. mean ± SEM, n = 4 for each group). Right panel: Detection of DLDH sulfenation after MICA feeding in the absence of tMCAO. Brain mitochondria were isolated after MICA feeding and were treated by DCP-Bio1, a probe that is specific for sulfenic acids [74]. DLDH was then separated from other mitochondrial proteins by BN-PAGE and visualized by in-gel activity staining as shown in (D), which is a representative gel image. The activity containing band was excised and further subjected to SDS-PAGE followed by Western blot probed with streptavidin-HRP as shown in (E). For both D and E, the densitometric analysis of the corresponding image was presented as a bar graph, respectively. Three rats in each group were analyzed.
Attenuation of DLDH activity is likely partly due to protein sulfenation caused by MICA treatment
As DLDH is known to undergo sulfenation under oxidative stress conditions [69, 70], we investigated whether the loss of DLDH activity observed in Fig. 4A could be also due to protein sulfenation which is a reversible process [71–73]. Mitochondrial proteins were labeled with a specific sulfenation probe DCP-Bio1 [74] followed by BN-PAGE isolation of DLDH and Western blot detection of DLDH sulfenation. Result in Fig. 4E (the upper image and the lower bar graph) shows that DLDH indeed underwent sulfenation after MICA treatment, indicating that the loss of DLDH activity (also confirmed in Fig. 4D, the upper image and the lower bar graph) could be partly caused by sulfenation of its cysteine residues. It should be noted that occurrence of DLDH sulfenation did not incur a big drop in DLDH activity, which indicates that MICA may have other targets in the body that are involved in MICA-induced preconditioning.
Dietary MICA up-regulates NQO1 expression via activation of the Nrf2 signaling pathway
To identify the most critical molecular components of the DLDH/MICA preconditioning pathway, we tested the hypothesis that dietary targeting of DLDH could induce the expression of phase II cytoprotective proteins. Accordingly, we used whole brain homogenate to measure the endogenous antioxidant GSH/GSSG ratio and activities of enzymes including thioredoxin reductase, glutamate-cysteine ligase (GCL), glutathione s-transferase, heme oxygenase-1 (HO-1), and NQO1, all of which can be upregulated by the Nrf2/ARE pathway [75]. It should be pointed out that all these measurements used samples from rats that did not undergo tMCAO. Although there were no differences in GSH/GSSG ratio, thioredoxin reductase activity, GCL activity, HO-1 activity or glutathione s-transferase activity (data not shown); NQO1 activity and protein levels were significantly higher in the MICA-treated rats than in the control rats (Fig. 5 A, B, and C). Thus, NQO1 was specifically upregulated in response to MICA-induced DLDH inhibition. Inasmuch as NQO1 is regulated by the Nrf2 transcriptional signaling pathway, we assessed Nrf2 translocation into the nucleus via electrophoretic mobility shift assay and observed that nuclear Nrf2 content was indeed significantly increased (Fig. 5 D).
Fig. 5.
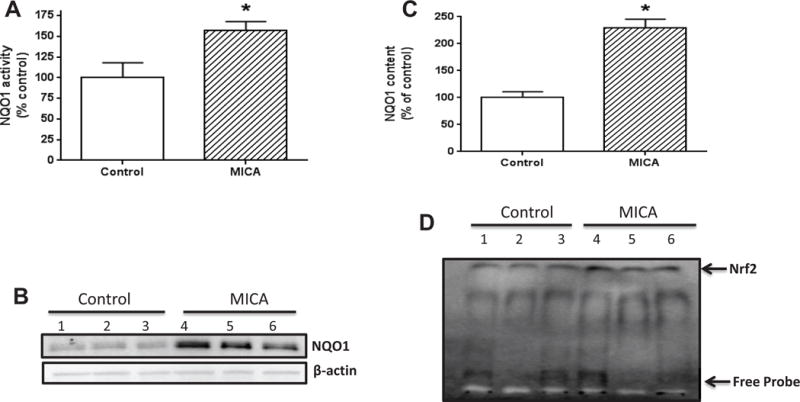
Effects of MICA feeding on NQO1 activity and expression. A to C shows that inhibition of DLDH by MICA upregulates NQO1 expression and function. (A) Higher NQO1 activity in the MICA-treated group than in the control group; (B) Western blot detection of NQO1 protein expression; (C) Densitometric quantitation of NQO1 protein content between control and MICA groups. Data were derived from B. (D) Nuclear Nrf2 content assessed by gel shift assay. For all the experiments in this figure, three animals per group were used (*p<0.05, mean ± SEM).
tMCAO following MICA dietary preconditioning also reveals decreased DLDH activity, increased Nrf2 signaling, increased NQO1 activity, decreased oxidative stress and cell death, and increased mitochondrial ATP output
As most of the above biochemical parameters were measured in the absence of tMCAO following MICA dietary feeding (data in Figs. 2–5), we then evaluated DLDH activity, Nrf2 nuclear translocation, NQO1 activity, oxidative stress, cell death (caspase 3 activity), and mitochondrial function (reflected by ATP content) after tMCAO following MICA feeding. Results in Fig. 6 show that, in the presence of tMCAO following MICA feeding, DLDH activity was lower in the MICA treated group (Fig. 6 A and B, note that the indicated Coomassie blue band was used as loading control; the same for complex I band below); and no complex I activity change was detected (Fig. 6 A and C), nor was complex IV activity (Fig. 6A: lower panel). These results are in agreement with what were observed in the preconditioning phase without tMCAO (Figs. 3 and 4). On the other hand, nuclear content of Nrf2 was increased (Fig. 6D) that was accompanied also with an increase in NQO1 enzymatic activity (Fig. 6F). Moreover, oxidative stress, reflected by cellular hydrogen peroxide content (Fig. 6G) and mitochondrial protein carbonyls (Fig. 6H) and lipid peroxidation measured by TBARS (Fig. 6I), was attenuated by MICA treatment. It should be noted that for the measurement of H2O2 by Amplex red that can be oxidized in H2O2-independent pathways [76], data were presented as fluorescence intensity in the presence of catalase (approximately 10 μM) and the catalase sensitive part of the fluorescence signal was compared (Fig. 6G). Additionally, as we used brain homogenate and Amplex reacts with intermediates generated by reaction of H2O2 with horseradish peroxidase, we think it is residual H2O2 that was measured.
Fig. 6.
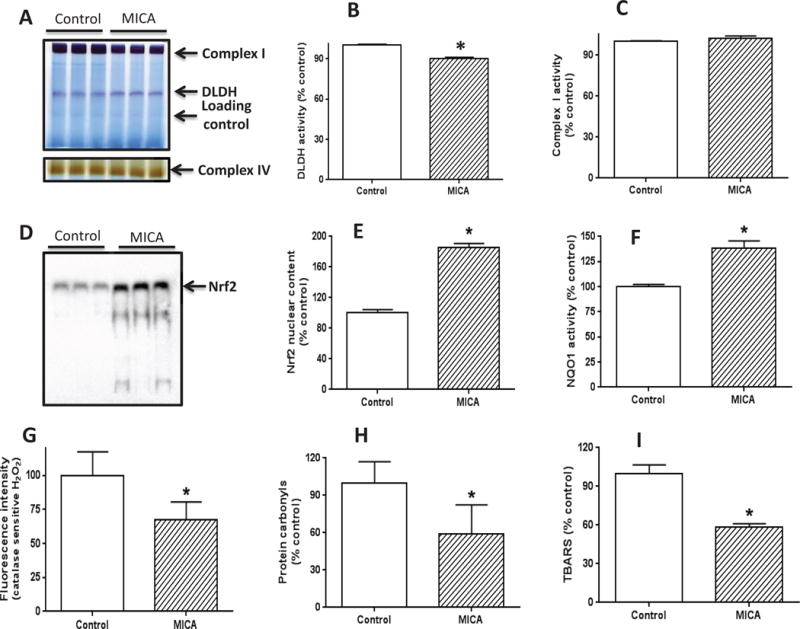
Measurement of DLDH activity, Nrf2 signaling pathways, and oxidative stress after brain ischemia reperfusion following MICA feeding. (A) BN-PAGE analysis of DLDH and complex I activities (upper panel) and complex IV activities (lower panel); (B) Densitometric quantification of DLDH activity derived from (A); (C) Densitometric quantification of complex I activity derived from (A). For both B and C, the densitometric intensity was normalized against the indicated Coomassie blue band as loading control. (D) nuclear content of Nrf2 assessed by gel shift analysis; (E) Densitometric quantification of nuclear Nrf2 content derived from (D); (F) NQO1 activity measured spectrophotometrically; (G) Fluorescence intensity reflecting catalase-sensitive hydrogen peroxide content; (H) Mitochondrial protein carbonyl content determined by biotin-hydrazide labeling followed by Western blot analysis. (I) Lipid peroxidation measured by TBARS; (*p<0.05, mean ± SEM, n=3 for each group).
Furthermore, we also determined cell death by caspase 3 activities and mitochondrial ATP output following MICA dietary administration and tMCAO. Results indicate that caspase 3 activity was significantly lower in the MICA group than in the control group (Fig. 7A), suggesting an attenuated process of cell death by MICA. Conversely, mitochondrial ATP output was greatly increased (Fig. 7B). Taken together, the data collected after stroke surgery further indicate the possibility that increased Nrf2 signaling is involved in MICA preconditioning-induced neuroprotection; and this neuroprotection involves attenuation of oxidative stress, decreased cell death, and increased mitochondrial ATP output. It should be noted that as we did not observe a detrimental effect of MICA on mitochondrial function in the absence of stroke (Fig. 3), the preservation of ATP content observed in Fig. 7B may just reflect improved cell viability and/or decreased ATP consumption.
Fig. 7.
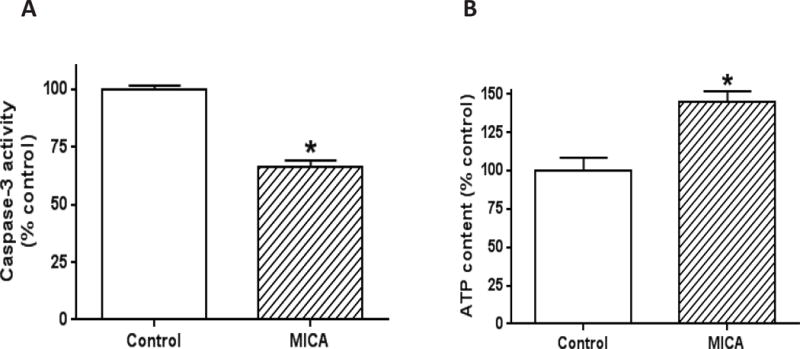
Measurement of cell death and mitochondrial ATP output after brain ischemia reperfusion following MICA feeding. (A) Cell death magnitude reflected by caspase 3 activity; (B) Mitochondrial ATP production. (*p<0.05, mean ± SEM, n=3 for each group).
DLDH inhibition by MICA shows delayed preconditioning effect
We also wanted to test whether MICA administration had any delayed precondition effect. To this end, tMCAO was performed 4 weeks after the end of 7 daily MICA intraperitoneal injections. TTC staining of the infarction volume shown in Fig. 8 shows that there was a neuroprotective effect even 4 weeks post MICA treatments, indicating that there was a prolonged protective effect of possible DLDH inhibition by MICA. Moreover, this delayed neuroprotective effect also involved activation of the Nrf2 signaling pathway (Fig. 8C) and NQO1 upregulation (Fig. 8 D and E).
Fig. 8.
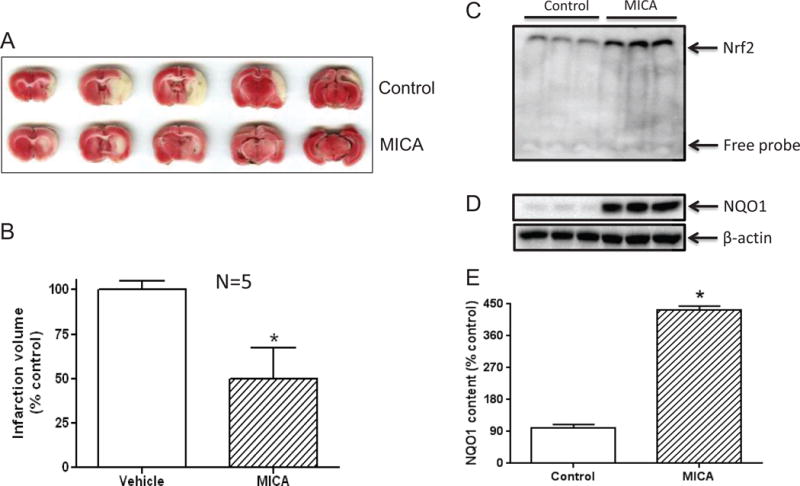
Delayed preconditioning effect of DLDH inhibition by MICA. MICA was administered via intraperitoneal injection for 7 consecutive days (once per day) and tMCAO surgery was performed 4 weeks after the last MICA injection. (A) Representative TTC staining showing the infarction areas of control and MICA; (B) Quantification of infarction volume (*p<0.05, mean ± SEM, n=5 for each group); (C) Nuclear content of Nrf2 assessed by gel shift analysis; (D) Comparison of NQO1 expression between control and MICA groups; (E) Densitometric quantification of NQO1 expression (normalized against the actin bands’ intensities) derived from D.
Discussion
The major findings of the present study are that mitochondrial DLDH, involved in four metabolic pathways, may serve as a preconditioning target for neuroprotection against ischemic stroke injury and that MICA administered via dietary feeding does not exhibit detectable toxicity on the animals. The underlying mechanisms of MICA preconditioning are likely multiple, but decreased DLDH activity and NQO1 upregulation via Nrf2 signaling may contribute to the observed preconditioning effects, which involve attenuated oxidative stress, decreased cell apoptosis, and increased mitochondrial ATP output. Moreover, potential DLDH inhibition by MICA also exhibits delayed preconditioning effects. It should be pointed out that the small decrease in DLDH activity observed after MICA treatment may serve as a trigger for neuroprotection. In other words, DLDH activity decrease may not be sufficient for the observed neuroprotective effects as there might be other signaling pathways contributing to MICA-induced neuroprotection in this model.
How did we become interested in DLDH preconditioning for neuroprotection against ischemic stroke injury? This was really a serendipitous finding. We initially observed that following ischemia reperfusion, there was a loss of DLDH activity when the reperfusion length was 1 hr. Beyond two hours of reperfusion (up to 24 h tested, data not shown), DLDH activity recovered (Fig. 9A). Yet brain damage clearly occurred at the end of 24 h reperfusion as reflected by TTC staining of the infarction volume. This observation suggested that if DLDH activity is prevented from recovering and kept low during the reperfusion process, the infarction volume might be decreased and there might be a neuroprotective effect in stroke. Hence we used MICA to inhibit DLDH activity, taking advantage of the well-established preconditioning concept. Interestingly, when DLDH activity was lost within one hour of reperfusion, DLDH protein content was not altered (Fig. 9B), indicating a reversible post translational modification that had occurred to DLDH upon reperfusion and that the existence of this modification was reperfusion time dependent. The nature of this modification causing rapid loss and recovery of DLDH activity could be partially attributed to protein sulfenation that is known to be a reversible process [77, 78] (Fig. 4E), though the exact site of sulfenation is yet to be determined.
Fig. 9.
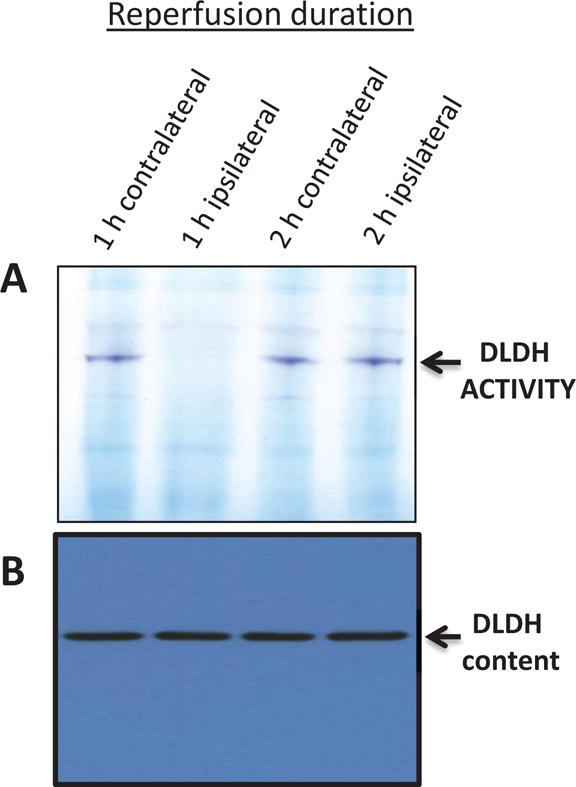
Measurement of DLDH activity (A) and protein content (B) after ischemia reperfusion. Animals were not treated with MICA. One h ischemia was followed by 1 or 2 h reperfusion. This was further followed by brain mitochondria isolation for gel analysis. DLDH activity was measured by in-gel BN-PAGE assay (upper panel) as described in the text, and DLDH protein content was measured by anti-DLDH Western blot assay (lower panel). Shown are contralateral samples and ipsilateral samples after 1 and 2 h reperfusion. As can be seen in the upper panel, DLDH activity was lost after 1 h reperfusion but recovered after 2 h reperfusion, but protein content did not change.
Numerous mitochondrial proteins have been evaluated as preconditioning targets for neuroprotection against stroke injury [36]. For example, two-well studied proteins are succinate dehydrogenase [7] and mitochondrial KATP channels [79]. For succinate dehydrogenase, the effects of 3-nitropropionic acid (3-NPA) have been comprehensively investigated [8]. 3-NPA is an irreversible inhibitor of succinate dehydrogenase and is highly toxic [8]. For mitochondrial KATP channels, diazoxide, as a channel opener, has been well studied [80]. Both 3-NPA and diazoxide target, respectively, one single protein in the mitochondria; and ROS production and membrane depolarization as well as membrane potential alteration have been suggested to be the mechanisms involved in their preconditioning effects [36, 81]. Our model of potential DLDH inhibition by MICA could serve as a unique paradigm of preconditioning as DLDH is involved in four metabolic reactions. Therefore, multiple targets could actually be inhibited by MICA, which also did not show toxicity when chronically administered via dietary intake.
It should be noted that as DLDH is involved in four metabolic pathways or complexes, we assume that all the 4 complexes or pathways can be inhibited to certain degrees by MICA. However, whether or not all the 4 pathways were equally targeted in our experiments remains unknown at this time. It is likely that one complex may be inhibited more by MICA than the others. Regardless, this aspect remains to be investigated.
Also, as DLDH is known to contribute to mitochondrial ROS production under many conditions [70, 82, 83], DLDH sulfenation, by lowering DLDH activity, may also attenuate DLDH contribution to ROS production, thereby leading to decreased ischemia injury after stroke. Indeed, we found that cellular ROS production and oxidative stress reflected by hydrogen peroxide content, protein oxidation and lipid peroxidation were all significantly lower in the MICA-treated group than in the control group (Fig. 6,G–I), demonstrating a decrease in ROS production and oxidative stress in this preconditioning paradigm.
In vitro, when DLDH activity was measured in the presence of 5 mM MICA followed by addition of 25 mM substrate (dihydrolipoamide), there was no inhibitory effect of MICA that could be detected (data not shown), confirming the notion that MICA is a reversible DLDH inhibitor [41–44]. In vivo, when mitochondria were isolated after dietary MICA feeding from the brain, there should be no MICA present as it would not be pelleted with mitochondria after centrifugation due to its small molecular weight and its reversible binding to DLDH. Nonetheless, we have found that DLDH activity was lower in the MICA treated group than in the control group (Fig. 4A) in the preconditioning phase without stroke surgery. This observation, along with the result that DLDH protein content was not altered by MICA feeding, suggests that DLDH undergoes posttranslational modifications. Indeed, sulfenic acid formation (sulfenation) is at least one of the potential mechanisms that decrease DLDH activity (Fig. 4E).
While our study suggests that DLDH sulfenation is likely involved in MICA inhibition of DLDH activity, the mechanism and nature of this modification remains elusive. In mammals, DLDH has 10 cysteine residues [84], and two of which (Cys 45 and Cys 50) are located at the active center and are involved in DLDH catalyzed reactions [51]. Modification of either of the two cysteine residues would inhibit DLDH activity and impair the enzyme function [45, 51]. Unfortunately, we failed to identify which of the two cysteine residues was sulfenated in this study. The reason for this is that each time the protein was analyzed by mass spectrometry, the peptide containing the two cysteine residues failed to be recovered. Therefore, different approaches will need to be taken to investigate the nature of the sulfenation in this preconditioning paradigm. Nonetheless, it is likely that the substrate-receiving cysteine residue (Cys45) underwent modification as previous studies have indicated that this residue is more susceptible to oxidative modifications [85].
MICA can cross the blood brain barrier as it has been reported that this chemical is an extremely poor inhibitor of D-amino acid oxidase in the brain [86]. Also, as literature abounds with studies that MICA is a specific DLDH inhibitor [41–44], we did not verify this inhibitory specificity in vivo in our preconditioning model. We also don’t know whether MICA would also target other proteins in vivo. These are the potential caveats of the present study. Nonetheless, studies in vitro using isolated mitochondria, as described above, clearly show that MICA is a specific and reversible DLDH inhibitor. In addition, there are many questions that remain to be answered. For example, one question arising from the study is that how MICA inhibition of DLDH could enhance DLDH sulfenation. We can only speculate at this point. It is possible that MICA binding of DLDH changes the structure of the protein so that small molecules of reactive oxygen species such as superoxide and nitric oxide could readily get access to the cysteine residues at the active center of the enzyme, resulting in sulfenation. Also we cannot exclude the possibility that other proteins are sulfenated during MICA treatment. Additionally, sulfenation could likely be only one type of modifications that may be involved in DLDH functional change and neuroprotection induced by MICA. Another question is how MICA makes the redox change that leads to DLDH sulfenation. As we have observed that MICA enhanced DLDH sulfenation during the preconditioning phase, MICA would act as an indirect antioxidant as protein sulfenation represents a scavenging process, which can lower the level of ROS. Indeed, it has been reported that sulfenation of a protein target can serve as an electron sink [87, 88]. As mentioned above, DLDH has two cysteine residues at its active center that is known to be very reactive towards hydropoxides. This, together with the fact that sulfenation is a reversible process, makes it possible that DLDH could serve as a scavenger of hydroperoxides as previously reported [89, 90]. Also, how DLDH inhibition leads to Nrf2 activation remains unknown at this time. It is known that the Nrf2/Keap1 pathway is activated by the oxidation and dissociation of Keap1 [75, 91], therefore, it is possible that DLDH sulfenation contributes to Keap1 oxidation and dissociation. It is also possible that redox change caused by DLDH inhibition activates Keap1 dissociation, leading to Nrf2 release and nuclear translocation. Regardless, the detailed mechanisms by which potential DLDH inhibition by MICA activates the Nrf2 signaling pathways remain to be elucidated. Future studies using Nrf2, DLDH, or NQO1 knock out/knock down animal models in conjunction with MICA treatments may provide further insights into the neuroprotective mechanisms of this preconditioning paradigm.
Finally, it should be emphasized that data collected both before and after tMCAO following MICA feeding all indicate the potential involvement of decreased DLDH activity and increased Nrf2 signaling as the underlying mechanisms of MICA preconditioning mechanisms. Our reasoning of doing data collection without stroke surgery after MICA feeding is that the preconditioning stage is the preparation phase during which the brain is reprogrammed and prepared for ischemic stroke challenge. Our data show that there were at least two processes that occurred during the MICA preconditioning stage. One is lowered DLDH activity via protein sulfenation, and the other is upregulation of NOQ1 via the Nrf2 signaling pathway, which is known to be involved in protecting the brain against stroke injury [91–93]. These phenomena continued to exhibit when data were collected and analyzed after stroke surgery. Moreover, our data (Figs. 6 and 7) also indicates that potential mechanisms of MICA-induced neuroprotection against stroke injury involve attenuated oxidative stress, decreased cell death, and increased mitochondrial function as reflected by mitochondrial ATP output. It is conceivable that DLDH functional change, possibly via protein sulfenation, may be one of the initial stressful events in the Nrf2 signaling activation process. It should be pointed out that the involvement of other proteins potentially targeted by MICA in vivo cannot be excluded.
Conclusions
The present study has provided evidence that MICA, potentially targeting mitochondrial DLDH, confers preconditioning effect on neuroprotection against ischemic stroke injury. MICA administration via dietary intake did not show detectable metabolic toxicity on the animals that were studied. The likely mechanisms underlying this neuroprotective effect are decreased DLDH function and augmented Nrf2 signaling process that is well-known for its role in cytoprotection [91, 93–95], together with decreased oxidative stress and cell death as well as increased mitochondrial ATP production. It should be pointed out that we based our study on previous investigations that MICA specifically inhibits DLDH [41–44]. Nonetheless, whether MICA indeed specifically inhibits DLDH in the brain has not been directly demonstrated in the present study. Therefore, our study may only suggest that DLDH could be manipulated for neuroprotective therapies.
Highlights.
Inhibition of dihydrolipoamide dehydrogenase (DLDH) triggers preconditioning effect against stroke injury
DLDH preconditioning involves attenuation of DLDH function, but not DLDH content
DLDH preconditioning involves NQO1 upregulation by the Nrf2 signaling pathway
DLDH preconditioning decreases oxidative stress and cell death and hence confers neuroprotection
Acknowledgments
This work was supported by National Institute of Neurological Disorders and Stroke, the National Institutes of Health (Grant number: R01NS079792).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest
References
- 1.Sharp FR, Ran R, Lu A, Tang Y, Strauss KI, Glass T, Ardizzone T, Bernaudin M. Hypoxic preconditioning protects against ischemic brain injury. NeuroRx. 2004;1(1):26–35. doi: 10.1602/neurorx.1.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7(6):437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 3.Ran R, Xu H, Lu A, Bernaudin M, Sharp FR. Hypoxia preconditioning in the brain. Dev Neurosci. 2005;27(2–4):87–92. doi: 10.1159/000085979. [DOI] [PubMed] [Google Scholar]
- 4.Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, et al. ’Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528(1):21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- 5.Zhan RZ, Fujihara H, Baba H, Yamakura T, Shimoji K. Ischemic preconditioning is capable of inducing mitochondrial tolerance in the rat brain. Anesthesiology. 2002;97(4):896–901. doi: 10.1097/00000542-200210000-00022. [DOI] [PubMed] [Google Scholar]
- 6.Riepe MW, Esclaire F, Kasischke K, Schreiber S, Nakase H, Kempski O, Ludolph AC, Dirnagl U, Hugon J. Increased hypoxic tolerance by chemical inhibition of oxidative phosphorylation: “chemical preconditioning”. J Cereb Blood Flow Metab. 1997;17(3):257–264. doi: 10.1097/00004647-199703000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Riepe MW, Ludolph AC. Chemical preconditioning: a cytoprotective strategy. Mol Cell Biochem. 1997;174(1–2):249–254. [PubMed] [Google Scholar]
- 8.Wiegand F, Liao W, Busch C, Castell S, Knapp F, Lindauer U, Megow D, Meisel A, Redetzky A, Ruscher K, Trendelenburg G, Victorov I, Riepe M, Diener HC, Dirnagl U. Respiratory chain inhibition induces tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19(11):1229–1237. doi: 10.1097/00004647-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29(31):9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakase H, Heimann A, Uranishi R, Riepe MW, Kempski O. Early-onset tolerance in rat global cerebral ischemia induced by a mitochondrial inhibitor. Neurosci Lett. 2000;290(2):105–108. doi: 10.1016/s0304-3940(00)01345-8. [DOI] [PubMed] [Google Scholar]
- 11.Goulton CS, Patten AR, Kerr JR, Kerr DS. Pharmacological Preconditioning with GYKI 52466: A Prophylactic Approach to Neuroprotection. Front Neurosci. 2010;4:1–11. doi: 10.3389/fnins.2010.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bell KFS, Fowler JH, Al-Mubarak B, Horsburgh K, Hardingham GE. Activation of Nrf2-Regulated Glutathione Pathway Genes by Ischemic Preconditioning 10.1155/2011/689524. Oxidative Medicine and Cellular Longevity. 2011;2011 doi: 10.1155/2011/689524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busija DW, Katakam PV. Mitochondrial mechanisms in cerebral vascular control: shared signaling pathways with preconditioning. J Vasc Res. 2014;51(3):175–89. doi: 10.1159/000360765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dirnagl U, Meisel A. Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology. 2008;55(3):334–344. doi: 10.1016/j.neuropharm.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 15.Correia SC, Santos RX, Cardoso SM, Santos MS, Oliveira CR, Moreira PI. Cyanide preconditioning protects brain endothelial and NT2 neuron-like cells against glucotoxicity: role of mitochondrial reactive oxygen species and HIF-1alpha. Neurobiol Dis. 2012;45(1):206–18. doi: 10.1016/j.nbd.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 16.MacDonald JF, Xiong ZG, Jackson MF. Paradox of Ca2+ signaling, cell death and stroke. Trends Neurosci. 2006;29(2):75–81. doi: 10.1016/j.tins.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Culmsee C, Krieglstein J. Ischaemic brain damage after stroke: new insights into efficient therapeutic strategies. International Symposium on Neurodegeneration and Neuroprotection. EMBO Rep. 2007;8(2):129–133. doi: 10.1038/sj.embor.7400892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manzanero S, Santro T, Arumugam TV. Neuronal oxidative stress in acute ischemic stroke: sources and contribution to cell injury. Neurochem Int. 2013;62(5):712–8. doi: 10.1016/j.neuint.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Woodruff TM, Thundyil J, Tang SC, Sobey CG, Taylor SM, Arumugam TV. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener. 2011;6(1):11. doi: 10.1186/1750-1326-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem Int. 2002;40(6):511–526. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 21.Moro MA, Almeida A, Bolanos JP, Lizasoain I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic Biol Med. 2005;39(10):1291–304. doi: 10.1016/j.freeradbiomed.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016;23(2):254–263. doi: 10.1016/j.cmet.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 23.Genova ML, Pich MM, Bernacchia A, Bianchi C, Biondi A, Bovina C, Falasca AI, Formiggini G, Castelli GP, Lenaz G. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann N Y Acad Sci. 2004;1011:86–100. doi: 10.1007/978-3-662-41088-2_10. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307(Pt 1):93–8. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 26.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis, Free. Radic Biol Med. 2010;48(6):749–62. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng H, Wu J, Jin Z, Yan LJ. Protein Modifications as Manifestations of Hyperglycemic Glucotoxicity in Diabetes and Its Complications. Biochem Insights. 2016;9:1–9. doi: 10.4137/BCI.S36141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan LJ, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002;21(19):5164–5172. doi: 10.1093/emboj/cdf528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai Z, Yan LJ. Protein oxidative modifications: Beneficial roles in disease and health. Journal of Biochemical and Pharmacological Research. 2013;1(1):15–26. [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, Luo X, Yan LJ. Two dimensional blue native/SDS-PAGE to identify mitochondrial complex I subunits modified by 4-hydroxynonenal (HNE) Frontiers in Physiology. 2015;6 doi: 10.3389/fphys.2015.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan LJ. Analysis of oxidative modification of proteins. Curr Protoc Protein Sci Chapter. 2009;14 doi: 10.1002/0471140864.ps1404s55. Unit14 4. [DOI] [PubMed] [Google Scholar]
- 32.Anderson EJ, Katunga LA, Willis MS. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin Exp Pharmacol Physiol. 2012;39(2):179–93. doi: 10.1111/j.1440-1681.2011.05641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ames BN, Shigenaga MK. Oxidants are a major contributor to aging. Ann N Y Acad Sci. 1992;663:85–96. doi: 10.1111/j.1749-6632.1992.tb38652.x. [DOI] [PubMed] [Google Scholar]
- 34.Shacter E. Protein oxidative damage. Methods Enzymol. 2000;319:428–436. doi: 10.1016/s0076-6879(00)19040-8. [DOI] [PubMed] [Google Scholar]
- 35.Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, Chan PH. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal. 2011;14(8):1505–17. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin Z, Wu J, Yan LJ. Chemical Conditioning as an Approach to Ischemic Stroke Tolerance: Mitochondria as the Target. Int J Mol Sci. 2016;17(3) doi: 10.3390/ijms17030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams CH., Jr . Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase-a family of flavoenzyme transhydrogenases. In: Muller F, editor. Chemistry and Biochemistry of Flavoenzymes. CRC Press; Boca Raton: 1992. pp. 121–212. [Google Scholar]
- 38.Kikuchi G, Hiraga K. The mitochondrial glycine cleavage system. Unique features of the glycine decarboxylation. Mol Cell Biochem. 1982;45(3):137–149. doi: 10.1007/BF00230082. [DOI] [PubMed] [Google Scholar]
- 39.Neuburger M, Polidori AM, Pietre E, Faure M, Jourdain A, Bourguignon J, Pucci B, Douce R. Interaction between the lipoamide-containing H-protein and the lipoamide dehydrogenase (L-protein) of the glycine decarboxylase multienzyme system. 1. Biochemical studies. Eur J Biochem. 2000;267(10):2882–2889. doi: 10.1046/j.1432-1327.2000.01301.x. [DOI] [PubMed] [Google Scholar]
- 40.Johnson MT, Yang HS, Magnuson T, Patel MS. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci U S A. 1997;94(26):14512–14517. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bauman N, Hill CJ. Inhibition of gluconeogenesis and alpha-keto oxidation by 5-methoxyindole-2-carboxylic acid. Biochemistry. 1968;7(4):1322–1327. doi: 10.1021/bi00844a011. [DOI] [PubMed] [Google Scholar]
- 42.Hanson RL, Ray PD, Walter P, Lardy HA. Mode of action of hypoglycemic agents. I. Inhibition of gluconeogenesis by quinaldic acid and 5-methoxyindole-2-carboxylic acid. J Biol Chem. 1969;244(16):4351–4359. [PubMed] [Google Scholar]
- 43.Haramaki N, Han D, Handelman GJ, Tritschler HJ, Packer L. Cytosolic and mitochondrial systems for NADH- and NADPH-dependent reduction of alpha-lipoic acid, Free. Radic Biol Med. 1997;22(3):535–542. doi: 10.1016/s0891-5849(96)00400-5. [DOI] [PubMed] [Google Scholar]
- 44.Miller JA, Runkle SA, Tjalkens RB, Philbert MA. 1,3-Dinitrobenzene Induced Metabolic Impairment Through Selective Inactivation of the Pyruvate Dehydrogenase Complex. Toxicol Sci. 2011;122(2):502–511. doi: 10.1093/toxsci/kfr102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan LJ, Yang SH, Shu H, Prokai L, Forster MJ. Histochemical staining and quantification of dihydrolipoamide dehydrogenase diaphorase activity using blue native PAGE. Electrophoresis. 2007;28(7):1036–1045. doi: 10.1002/elps.200600574. [DOI] [PubMed] [Google Scholar]
- 46.Yang SH, Liu R, Wen Y, Perez E, Cutright J, Brun-Zinkernagel AM, Singh M, Day AL, Simpkins JW. Neuroendocrine mechanism for tolerance to cerebral ischemia-reperfusion injury in male rats. J Neurobiol. 2005;62(3):341–351. doi: 10.1002/neu.20103. [DOI] [PubMed] [Google Scholar]
- 47.Li R, Luo X, Wu J, Thangthaeng N, Jung ME, Jing S, Li L, Ellis DZ, Liu L, Ding Z, Forster MJ, Yan LJ. Mitochondrial dihydrolipoamide dehydrogenase is upregulated in response to intermittent hypoxic preconditioning. Int J Med Sci. 2015;12(5):432–440. doi: 10.7150/ijms.11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma Y, Lu C, Li C, Li R, Zhang Y, Ma H, Zhang X, Ding Z, Liu L. Overexpression of HSPA12B protects against cerebral ischemia/reperfusion injury via a PI3K/Akt-dependent mechanism. Biochim Biophys Acta. 2013;1832(1):57–66. doi: 10.1016/j.bbadis.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL, Kalbfleisch JH, Browder IW, Li C. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199(1–2):75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sims NR. Methods in Toxicology: Mitochondrial Dysfunction. Academic Press; San Diego: 1993. [Google Scholar]
- 51.Yan LJ, Thangthaeng N, Forster MJ. Changes in dihydrolipoamide dehydrogenase expression and activity during postnatal development and aging in the rat brain. Mech Ageing Dev. 2008;129:282–290. doi: 10.1016/j.mad.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 53.Yan LJ, Forster MJ. Resolving mitochondrial protein complexes using nongradient blue native polyacrylamide gel electrophoresis. Anal Biochem. 2009;389(2):143–149. doi: 10.1016/j.ab.2009.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tripathy MK, Mitra D. Differential modulation of mitochondrial OXPHOS system during HIV-1 induced T-cell apoptosis: up regulation of Complex-IV subunit COX-II and its possible implications. Apoptosis. 2010;15(1):28–40. doi: 10.1007/s10495-009-0408-9. [DOI] [PubMed] [Google Scholar]
- 55.Gusdon AM, Votyakova TV, Reynolds IJ, Mathews CE. Nuclear and mitochondrial interaction involving mt-Nd2 leads to increased mitochondrial reactive oxygen species production. J Biol Chem. 2007;282(8):5171–5179. doi: 10.1074/jbc.M609367200. [DOI] [PubMed] [Google Scholar]
- 56.Lind C, Cadenas E, Hochstein P, Ernster L. DT-diaphorase: purification, properties, and function. Methods Enzymol. 1990;186:287–301. doi: 10.1016/0076-6879(90)86122-c. [DOI] [PubMed] [Google Scholar]
- 57.Luo X, Wu J, Jin Z, Yan LJ. Non-Gradient Blue Native Polyacrylamide Gel Electrophoresis. Curr Protoc Protein Sci. 2017;87:19 29 1–19 29 12. doi: 10.1002/cpps.21. [DOI] [PubMed] [Google Scholar]
- 58.Yan LJ, Orr WC, Sohal RS. Identification of oxidized proteins based on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, immunochemical detection, isoelectric focusing, and microsequencing. Anal Biochem. 1998;263(1):67–71. doi: 10.1006/abio.1998.2799. [DOI] [PubMed] [Google Scholar]
- 59.Favreau LV, Pickett CB. Transcriptional regulation of the rat NAD(P)H:quinone reductase gene. Identification of regulatory elements controlling basal level expression and inducible expression by planar aromatic compounds and phenolic antioxidants. J Biol Chem. 1991;266(7):4556–4561. [PubMed] [Google Scholar]
- 60.Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem. 1997;253(2):162–8. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]
- 61.Codaccioni JL, Velly LJ, Moubarik C, Bruder NJ, Pisano PS, Guillet BA. Sevoflurane preconditioning against focal cerebral ischemia: inhibition of apoptosis in the face of transient improvement of neurological outcome. Anesthesiology. 2009;110(6):1271–8. doi: 10.1097/ALN.0b013e3181a1fe68. [DOI] [PubMed] [Google Scholar]
- 62.Yan LJ, Forster MJ. Chemical probes for analysis of carbonylated proteins: A review. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879(17–18):1308–1315. doi: 10.1016/j.jchromb.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu J, Luo X, Jing S, Yan LJ. Two-dimensional gel electrophoretic detection of protein carbonyls derivatized with biotin-hydrazide. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1019:128–31. doi: 10.1016/j.jchromb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yan LJ, Lodge JK, Traber MG, Packer L. Apolipoprotein B carbonyl formation is enhanced by lipid peroxidation during copper-mediated oxidation of human low-density lipoproteins. Arch Biochem Biophys. 1997;339(1):165–71. doi: 10.1006/abbi.1996.9867. [DOI] [PubMed] [Google Scholar]
- 65.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–8. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 66.Wu J, Jin Z, Yan LJ. Redox imbalance and mitochondrial abnormalities in the diabetic lung. Redox Biol. 2017;11:51–59. doi: 10.1016/j.redox.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chu DS, Bocek MJ, Shi J, Ta A, Ngambenjawong C, Rostomily RC, Pun SH. Multivalent display of pendant pro-apoptotic peptides increases cytotoxic activity. J Control Release. 2015;205:155–61. doi: 10.1016/j.jconrel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang SH, Shi J, Day AL, Simpkins JW. Estradiol exerts neuroprotective effects when administered after ischemic insult. Stroke. 2000;31(3):745–749. doi: 10.1161/01.str.31.3.745. discussion 749–50. [DOI] [PubMed] [Google Scholar]
- 69.Yan LJ, Liu L, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by Angeli’s salt. Acta Biophysica Sinica (Sheng Wu Wu Li Hsueh Bao) 2012;28(4):341–350. [PMC free article] [PubMed] [Google Scholar]
- 70.Yan LJ, Sumien N, Thangthaeng N, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic Res. 2013;47(2):123–133. doi: 10.3109/10715762.2012.752078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chauvin JR, Pratt DA. On the Reactions of Thiols, Sulfenic Acids, and Sulfinic Acids with Hydrogen Peroxide. Angew Chem Int Ed Engl. 2017;56(22):6255–6259. doi: 10.1002/anie.201610402. [DOI] [PubMed] [Google Scholar]
- 72.Hutson SM, Poole LB, Coles S, Conway ME. Redox regulation and trapping sulfenic Acid in the peroxide-sensitive human mitochondrial branched chain aminotransferase. Methods Mol Biol. 2009;476:135–148. doi: 10.1007/978-1-59745-129-1_10. [DOI] [PubMed] [Google Scholar]
- 73.Gupta V, Paritala H, Carroll KS. Reactivity, Selectivity, and Stability in Sulfenic Acid Detection: A Comparative Study of Nucleophilic and Electrophilic Probes. Bioconjug Chem. 2016;27(5):1411–8. doi: 10.1021/acs.bioconjchem.6b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, Daniel LW, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85(4):241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 76.Debski D, Smulik R, Zielonka J, Michalowski B, Jakubowska M, Debowska K, Adamus J, Marcinek A, Kalyanaraman B, Sikora A. Mechanism of oxidative conversion of Amplex(R) Red to resorufin: Pulse radiolysis and enzymatic studies. Free Radic Biol Med. 2016;95:323–32. doi: 10.1016/j.freeradbiomed.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 78.Roos G, Messens J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med. 2011;51(2):314–326. doi: 10.1016/j.freeradbiomed.2011.04.031. [DOI] [PubMed] [Google Scholar]
- 79.Busija DW, Lacza Z, Rajapakse N, Shimizu K, Kis B, Bari F, Domoki F, Horiguchi T. Targeting mitochondrial ATP-sensitive potassium channels–a novel approach to neuroprotection. Brain Res Brain Res Rev. 2004;46(3):282–94. doi: 10.1016/j.brainresrev.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 80.Blanco M, Lizasoain I, Sobrino T, Vivancos J, Castillo J. Ischemic preconditioning: A novel target for neuroprotective therapy. Cerebrovasc Dis. 2006;21(Suppl 2):38–47. doi: 10.1159/000091702. [DOI] [PubMed] [Google Scholar]
- 81.Horiguchi T, Kis B, Rajapakse N, Shimizu K, Busija DW. Opening of mitochondrial ATP-sensitive potassium channels is a trigger of 3-nitropropionic acid-induced tolerance to transient focal cerebral ischemia in rats. Stroke. 2003;34(4):1015–20. doi: 10.1161/01.STR.0000063404.27912.5B. [DOI] [PubMed] [Google Scholar]
- 82.Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-Oxoacid Dehydrogenase Complexes in Mitochondria Can Produce Superoxide/Hydrogen Peroxide at Much Higher Rates than Complex I. J Biol Chem. 2014 doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ambrus A, Tretter L, Adam-Vizi V. Inhibition of the alpha-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. J Neurochem. 2009;109(Suppl 1):222–229. doi: 10.1111/j.1471-4159.2009.05942.x. [DOI] [PubMed] [Google Scholar]
- 84.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, Zeeberg B, Buetow KH, Schaefer CF, Bhat NK, Hopkins RF, Jordan H, Moore T, Max SI, Wang J, Hsieh F, Diatchenko L, Marusina K, Farmer AA, Rubin GM, Hong L, Stapleton M, Soares MB, Bonaldo MF, Casavant TL, Scheetz TE, Brownstein MJ, Usdin TB, Toshiyuki S, Carninci P, Prange C, Raha SS, Loquellano NA, Peters GJ, Abramson RD, Mullahy SJ, Bosak SA, McEwan PJ, McKernan KJ, Malek JA, Gunaratne PH, Richards S, Worley KC, Hale S, Garcia AM, Gay LJ, Hulyk SW, Villalon DK, Muzny DM, Sodergren EJ, Lu X, Gibbs RA, Fahey J, Helton E, Ketteman M, Madan A, Rodrigues S, Sanchez A, Whiting M, Young AC, Shevchenko Y, Bouffard GG, Blakesley RW, Touchman JW, Green ED, Dickson MC, Rodriguez AC, Grimwood J, Schmutz J, Myers RM, Butterfield YS, Krzywinski MI, Skalska U, Smailus DE, Schnerch A, Schein JE, Jones SJ, Marra MA. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A. 2002;99(26):16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thorpe C, Williams CH., Jr Differential reactivity of the two active site cysteine residues generated on reduction of pig heart lipoamide dehydrogenase. J Biol Chem. 1976;251(12):3553–3557. [PubMed] [Google Scholar]
- 86.Heefner DL, Currie MG, Rossi RF, Zepp CM. D-amino acid oxidase inhibitors for leaning and memory. United States Patent Application Publication, Sepracor Inc; USA: 2003. pp. 1–17. [Google Scholar]
- 87.Kaiserova K, Srivastava S, Hoetker JD, Awe SO, Tang XL, Cai J, Bhatnagar A. Redox activation of aldose reductase in the ischemic heart. J Biol Chem. 2006;281(22):15110–15120. doi: 10.1074/jbc.M600837200. [DOI] [PubMed] [Google Scholar]
- 88.Kaiserova K, Tang XL, Srivastava S, Bhatnagar A. Role of nitric oxide in regulating aldose reductase activation in the ischemic heart. J Biol Chem. 2008;283(14):9101–9112. doi: 10.1074/jbc.M709671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Igamberdiev AU, Bykova NV, Ens W, Hill RD. Dihydrolipoamide dehydrogenase from porcine heart catalyzes NADH-dependent scavenging of nitric oxide. FEBS Lett. 2004;568(1–3):146–150. doi: 10.1016/j.febslet.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 90.Korotchkina LG, Yang H, Tirosh O, Packer L, Patel MS. Protection by thiols of the mitochondrial complexes from 4-hydroxy-2-nonenal. Free Radic Biol Med. 2001;30(9):992–999. doi: 10.1016/s0891-5849(01)00491-9. [DOI] [PubMed] [Google Scholar]
- 91.Alfieri A, Srivastava S, Siow RC, Modo M, Fraser PA, Mann GE. Targeting the Nrf2-Keap1 antioxidant defence pathway for neurovascular protection in stroke. J Physiol. 2011;589(Pt 17):4125–36. doi: 10.1113/jphysiol.2011.210294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alfieri A, Srivastava S, Siow RC, Cash D, Modo M, Duchen MR, Fraser PA, Williams SC, Mann GE. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med. 2013;65:1012–22. doi: 10.1016/j.freeradbiomed.2013.08.190. [DOI] [PubMed] [Google Scholar]
- 93.Bell KF, Al-Mubarak B, Fowler JH, Baxter PS, Gupta K, Tsujita T, Chowdhry S, Patani R, Chandran S, Horsburgh K, Hayes JD, Hardingham GE. Mild oxidative stress activates Nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proc Natl Acad Sci U S A. 2011;108(1):E1–2. doi: 10.1073/pnas.1015229108. author reply E3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang XS, Chen HP, Yu HH, Yan YF, Liao ZP, Huang QR. Nrf2-dependent upregulation of antioxidative enzymes: a novel pathway for hypoxic preconditioning-mediated delayed cardioprotection. Mol Cell Biochem. 2014;385:33–41. doi: 10.1007/s11010-013-1812-6. [DOI] [PubMed] [Google Scholar]
- 95.Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 2010;501(1):116–123. doi: 10.1016/j.abb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]