

Abstract
Rationale
Hydrogen peroxide (H2O2) is a stable reactive oxygen species (ROS) that has long been implicated in insulin signal transduction in adipocytes. However, H2O2’s role in mediating insulin’s effects on the heart are unknown.
Objective
We investigated the role of H2O2 in activating insulin-dependent changes in cardiac myocyte metabolic and inotropic pathways. The sources of insulin-dependent H2O2 generation were also studied.
Methods and results
In addition to the canonical role of insulin in modulating cardiac metabolic pathways, we found that insulin also inhibited beta adrenergic-induced increases in cardiac contractility. Catalase and NADPH oxidase (NOX) inhibitors blunted activation of insulin-responsive kinases Akt and mTOR and attenuated beta adrenergic receptor-mediated responses. These insulin responses were lost in a mouse model of type 2 diabetes, suggesting a role for these H2O2-dependent pathways in the diabetic heart. The H2O2-sensitive fluorescent biosensor HyPer revealed rapid increases in cytosolic and caveolar H2O2 concentrations in response to insulin treatment, which were blocked by NOX inhibitors and attenuated in NOX2 KO and NOX4 KO mice. In NOX2 KO cardiac myocytes, insulin-mediated phosphorylation of Akt and mTOR was blocked, while these responses were unaffected in cardiac myocytes from NOX4 KO mice. In contrast, insulin’s effects on contractility were lost in cardiac myocytes from NOX4 KO animals but were retained in NOX2 KO mice.
Conclusions
These studies identify a proximal point of bifurcation in cardiac insulin signaling through the simultaneous activation of both NOX2 and NOX4. Each NOX isoform generates H2O2 in cardiac myocytes with distinct time courses, with H2O2 derived from NOX2 augmenting Akt-dependent metabolic effects of insulin, while H2O2 from NOX4 blocks beta adrenergic increases in inotropy. These findings suggest that insulin resistance in the diabetic heart may lead to potentially deleterious potentiation of beta adrenergic responses.
Keywords: Insulin, fluorescent biosensors, redox signaling
Subject Codes: Cell Signaling/Signal Transduction, Contractile Function, Myocardial Biology, Diabetes, type II
Graphical Abstract
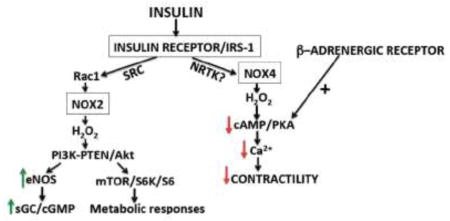
Introduction
The molecular mechanisms of insulin action have been extensively characterized in the three archetypal insulin target tissues: fat, liver, and skeletal muscle (1). In the heart, however, our understanding of insulin-modulated signaling pathways has lagged behind. It is clear that the canonical metabolic responses to insulin, such as activation of kinase Akt and its downstream targets, translocation of glucose transporters and stimulation of anabolic pathways, operate in cardiac myocytes (2). These metabolic responses to insulin serve to counterbalance some of the catabolic consequences of beta adrenergic stimulation in the heart. In addition to increasing catabolism, beta adrenergic agonists augment cardiac contractility through increases in systolic calcium cycling and phosphorylation of sarcomeric proteins by the cyclic AMP-activated protein kinase PKA (3). We have previously shown that insulin attenuates the increase in cardiac myocyte contractility elicited by beta adrenergic receptor activation (4). Despite the apparent counter-regulatory relationships between insulin and beta adrenergic signaling, the molecular mechanisms that facilitate cross-talk between the two pathways in cardiac myocytes remain incompletely understood. Here we extended our previous observations of insulin’s interaction with the beta adrenergic pathway to identify a new role for the stable reactive oxygen species (ROS) hydrogen peroxide (H2O2) produced by NADPH oxidases (NOXs), which we have discovered to control the proximal insulin receptor signaling pathways including its ability to modulate beta adrenergic responses.
For many years, ROS were studied principally in the context of pathological oxidative stress, and they have been found to instigate cellular damage in diverse chronic disease states ranging from neurodegeneration to atherosclerosis to diabetes (5–8). Yet the stable ROS H2O2 has also been identified as an important second messenger for many physiologic processes, a role termed “oxidative eustress” (6), and has been clearly implicated in insulin signaling in fat and liver (9). A causal relationship between H2O2 and metabolic responses to insulin has been described in publications dating back to the 1970s (10–12), yet many of the essential molecular details have remained unexplored. Several reversibly oxidized signaling proteins have been definitively shown to be regulated by insulin receptor activation including PTEN, PTP1-B, and CaMK (13, 14). However, the intracellular source(s) of insulin-modulated H2O2 have not been established. Thus, there is an apparent paradox between the roles of H2O2 in physiological insulin signaling and the adverse effects of ROS that underlie the oxidative stress that is associated with deranged insulin signaling in diabetes. During the development of clinical diabetes, there appears to be a transition from the salutary function of H2O2 in physiological insulin signaling to the pathological roles of ROS that are seen in the advanced diabetic state. The term “diabetic cardiomyopathy” refers to the enigmatic cardiac dysfunction that is often seen in patients with diabetes, independent of the deleterious effects of coronary atherosclerosis or hypertension on the heart (15–17). Therapies targeting this unique form of cardiac dysfunction are lacking. Since cardiac myocytes represent the most oxidatively active cells in the body in normal physiology (15), it is essential to delineate the intracellular pathways that control the fate of ROS in these cells both in the normal and diseased heart in order to understand the changes that occur with the development of diabetic cardiomyopathy.
The intracellular metabolism of ROS in cardiac myocytes involves a broad range of enzymes expressed in a diverse set of organelles. Two of the principal intracellular sources of reactive oxygen species in cardiac myocytes are the mitochondrial electron transport chain and the NOX family of NADPH oxidases (18–20). The NOX proteins are a family of membrane-bound multimeric enzymes that produce ROS using oxygen and NADPH as substrates (21–23). The different NOX isoforms differ in their tissue distribution and biological roles; both the NOX2 and NOX4 isoforms have been studied in cardiac myocytes, yet their roles in cardiac biology are controversial (24–26). While the intracellular pathways controlling receptor-dependent NOX4 activation in cardiac myocytes are incompletely understood (25, 27, 28), it is generally agreed that ROS production by NOX2 modulates signal transduction through the angiotensin II receptor in these cells (29, 30).
In contrast to angiotensin-II, beta adrenergic stimulation does not directly affect H2O2 concentrations in cardiac myocytes (29). The potential roles of insulin-modulated H2O2 in cardiac myocyte function have remained unexplored, and are the focus of the current studies. In the studies presented here, we exploited informative biosensors and genetic models to analyze insulin-modulated pathways in cardiac myocytes, and identified the proximal signaling events whereby insulin elicits cellular responses through H2O2. Evidence is presented that insulin signaling in the heart depends on H2O2 produced by two distinct NADPH oxidase isoforms: NOX2 and NOX4, leading to differential effects on distinct cardiac myocyte signaling and physiological responses. These NOX-dependent effects of insulin were lost in cardiac myocytes isolated from an in vivo model of type II diabetes. These findings identified previously unknown roles of NOX-generated H2O2 in cardiac myocyte insulin signaling that may add to our understanding of the pathophysiology of diabetic cardiomyopathy.
Materials and Methods
A detailed explanation of the methods can be found in the online data supplement.
Live-cell imaging of intracellular H2O2 in cardiac myocytes using the biosensor HyPer
Fluorescent biosensors, including the H2O2-specific biosensor HyPer and its derivatives, were expressed in adult cardiac myocytes using a recombinant adeno-associated viral vector. Over the course of these studies, HyPer3, which reportedly has a broader dynamic range than the original HyPer (50), became available during the course of these studies. As a result, HyPer3 was used as the control for the SypHer pH-control studies and for analysis of subcellular H2O2 generation. A caveolae-targeted form of HyPer was generated by fusing the N terminal sequence of bovine eNOS to the probe (producing cav-HyPer3). This strategy has been previously validated as yielding caveolar localization (51). Mito-HyPer3 was generated by replacing HyPer in mito-HyPer (containing a N-terminal double mitochondrial targeting sequence derived from subunit VIII of human cytochrome C oxidase (52)) with HyPer3. cDNA for HyPer, HyPer3, cav-HyPer3, mito-HyPer3 and SypHer2 were cloned by PCR into an AAV expression vector in which expression is driven by the CMV promoter (Stratagene). The Children’s Hospital Boston Viral Vector Core produced AAV-9 viral particles with the Rep2/Cap9 encapsulation construct. Approximately 5×1011 genome copies were suspended in 100 microliters PBS and injected retro-orbitally in 4–5 week old mice. Cardiac myocytes were isolated 4–6 weeks later and plated on laminin-coated glass dishes (Mattek) where they were maintained in culture media prior to experiments. Culture medium was replaced with a physiological balanced salt solution and the dishes were mounted in an Olympus DSU inverted fluorescence microscope equipped with an on-stage incubator at 37°C in a 4% CO2 environment. For live-cell imaging, HyPer was excited with 420 nm and 480 nm while visualized with a 40X oil immersion objective (Olympus). Images were acquired with a CCD camera (Hamamatsu) after filtering at HyPer’s emission wavelength of 530 nm. Images were analyzed post-hoc with a custom-written macro in Metamorph software. After background subtraction, ratiometric images were generated by dividing images excited with 480 nm by images excited with 420 nm. In order to mitigate artifact from any spontaneous contractions during the time course experiment, ratiometric time course traces were median filtered with a window size of 3 frames prior to statistical analysis.
Statistical analysis
All experiments were performed at least three times. Mean values for individual experiments are expressed as means ± standard error. Statistical differences were assessed by ANOVA as indicated with a Tukey’s post hoc test for multiple comparisons. Fluorescence time course traces were analyzed by two-way ANOVA with a Bonferroni post hoc correction. A p value of <0.05 was considered statistically significant.
Results
Insulin attenuates β-adrenergic induced increases in inotropy in adult cardiac myocytes
In isolated adult murine cardiac myocytes, insulin did not significantly alter basal cardiac myocyte contractility assessed by both sarcomere shortening and systolic calcium transients (Figures 1A and 1B). However, insulin pretreatment attenuated isoproterenol-induced increases in inotropy, as evidenced by a decrease in both systolic calcium transients and sarcomere shortening. To validate that these effects of insulin on cardiac myocyte contractility were mediated by the insulin receptor rather than related cell surface receptors (for example, the IGF-1 receptor), we compared insulin’s effect on cardiac myocytes from cardiac-specific insulin receptor KO (CIRKO) mice with cardiac myocytes from wild-type mice (31). In cardiac myocytes from CIRKO mice, insulin did not attenuate isoproterenol-induced increases in inotropy (Figure 1C). Additionally, in CIRKO cardiac myocytes, insulin was unable to activate the canonical anabolic signaling cascade mediated by Akt, p70S6K and ribosomal S6 (Figure 1D)
Figure 1. Effects of insulin on beta adrenergic responses in cardiac myocytes.

A) Representative transients of sarcomere length and intracellular calcium from cardiac myocytes paced at 1 Hz. The transients represent cardiac myocytes at baseline, treated with isoproterenol (Iso, 10 nM, 5 min), pretreated with insulin (Ins, 10 nM, 30 min) and treated with isoproterenol after insulin pretreatment (Iso+Ins). B) Pooled data from cardiac myocytes treated with the conditions in A. The number of cells, which were isolated from 4 mice, measured for each condition is indicated in each bar. ** indicates p<0.01 and *** Indicates p<0.001 by ANOVA C) Pooled sarcomere shortening data from cardiac myocytes isolated from CIRKO (IR−/−) mice (white bars, 4 mice) and from wild-type (IR+/+) littermate controls (black bars, 3 mice). D) Representative immunoblot of cardiac myocytes isolated from wild-type littermate controls (IR+/+, 3 mice in total) or CIRKO (IR−/−, 3 mice in total) animals simulated with insulin for the times indicated.
Cardiac myocytes were next isolated from mice fed a high fat diet for 7 weeks, a model of type II diabetes (32). Cardiac myocytes from these high fat-fed mice exhibited similar increases in contractility when treated with isoproterenol compared to mice fed a normal chow diet. However, insulin failed to attenuate isoproterenol’s positive inotropic effect in cardiac myocytes isolated from mice fed a high fat diet (Figure S1). Additionally, insulin-modulated phosphorylation responses were significantly diminished in cardiac myocytes isolated from mice fed a high fat diet (Figure S2).
Insulin-dependent phosphorylation responses in cardiac myocytes are markedly attenuated by catalase and by NADPH oxidase inhibition
A time course and dose response were characterized for insulin-dependent phosphorylation of key signaling proteins in cardiac myocytes. Insulin-dependent phosphorylation responses were sensitive to insulin at concentrations as low as 0.1 nM (Figs. S3 and S4). Insulin rapidly induced phosphorylation of the insulin receptor β at Tyr1146, Insulin Receptor Substrate 1 (IRS-1) at Tyr895 and of Akt at both Ser473 and Ser308. By contrast, the insulin-promoted phosphorylations of mTOR at Ser2448, p70S6K at Thr421 and Ser424 (Thr421/Ser424), S6 at Ser235 and Ser236 (Ser235/236) and IRS-1 at Ser636 and Ser639 (Ser636/639) were maximal only at 30 min. Subsequent analyses of the effects of inhibitors were therefore pursued 5 min after insulin addition for studies of phosphorylation of the insulin receptor, IRS-1 at Tyr895 and Akt; phosphorylation responses were analyzed after 30 min for mTOR, p70S6K, and IRS-1 at Ser636/639. Adult mouse cardiac myocytes were treated with insulin in the presence and absence of catalase, which degrades H2O2. Preincubation of cardiac myocytes with catalase blunted the subsequent insulin-promoted increases in all of these phosphorylation responses (Figures 2A and S5), implicating H2O2 as a key second messenger in cardiac myocyte insulin signaling.
Fig. 2. Effects of catalase and NOX inhibitor apocynin on insulin-modulated phosphoprotein responses.

A) Representative immunoblots for insulin signaling proteins from cardiac myocytes isolated from 5 mice total incubated overnight with PEG-catalase (PEG-cat, 200 U/mL) or vehicle, and treated with insulin (10 nM) for 5 min (Left) or for 30 min (Right). Below the immunoblots are densitometric analyses for pAkt at Ser473 and pmTOR at Ser2448 (see Supplementary figure S3 for complete densitometric analysis of all targets). B) Representative immunoblots and densitometric analyses for pAkt and pmTOR from cardiac myocytes from 6 mice pretreated with the NOX inhibitor apocynin (Apo, 500 μM) followed by insulin stimulation for 5 minutes (Akt) or 30 minutes (mTOR). (See Figure S4 for complete densitometric analysis.)
Pharmacologic inhibitors were next used to explore the involvement of NADPH oxidases in insulin-induced protein phosphorylation in cardiac myocytes. All insulin-dependent phosphorylation responses were attenuated in cells pretreated with the NOX inhibitor apocynin (Figure 2B), implicating one or more NADPH oxidase isoforms in the insulin phosphorylation response. The NOX inhibitors VAS2870 and diphenyleneiodonium (DPI) similarly attenuated insulin-promoted phosphorylation in these cells (Figures S6 and S7), strengthening the evidence for the involvement of NADPH oxidase(s) in the insulin signaling response.
NADPH oxidase inhibition prevents insulin-induced attenuation of phospholamban phosphorylation and contractility
The NOX inhibitor apocynin completely blocked insulin’s previously observed effect of attenuating the isoproterenol-stimulated increase in cardiac myocyte contractility, as measured both by sarcomere shortening and amplitude of systolic calcium transients (Figures 3A and 3B). To explore whether these effects of insulin and apocynin on contractile physiology were mediated through alterations in adrenergic signaling, phosphorylation of the PKA substrate phospholamban (33) was studied as an indicator of beta adrenergic signaling activity. Insulin treatment attenuated the increase in phospholamban phosphorylation at Ser16 seen after isoproterenol treatment, but cardiac myocytes treated with apocynin were no longer sensitive to the inhibitory effect of insulin (Figure 3C). Supporting these findings, isoproterenol induced an increase in the phosphorylation of Ser157 of VASP (vasodilator-stimulated phosphoprotein), another protein substrate of PKA (34) that was attenuated by insulin and restored with apocynin (Figure S8).
Figure 3. Effects of apocynin on contractility and beta adrenergic signaling in cardiac myocytes.

A) Representative sarcomere shortening and calcium transients from cardiac myocytes treated with insulin (10 nM, 30 min) with or without apocynin pretreatment (Apo, 30 min 500 μM) followed by stimulation with isoproterenol (10 nM, 5 min). B) Pooled data from (A). The number of cells, which were isolated from 7 mice, measured for each condition is indicated in each bar. C) Representative immunoblot for phospholamban phosphorylated at Ser16 (pPLN) from cultured cardiac myocytes isolated from 4 mice treated with isoproterenol (10 nM, 5 min) after insulin stimulation (10 nM, 30min) with or without apocynin (500 μM, 30 min) pretreatment. Pooled densitometric analyses from 4 independent experiments are shown to the right. * indicates p<0.05, **p<0.01, and ***p<0.001 compared to unstimulated cells.
Insulin elicits rapid NOX-dependent increases in intracellular H2O2
Changes in intracellular H2O2 concentrations were next assessed using the H2O2 fluorescent biosensor HyPer (35), which changes its fluorescence properties when oxidized by H2O2. HyPer was cloned into an adeno-associated viral expression vector, which was packaged into an AAV9 serotype to direct the expression of the recombinant biosensor in cardiac myocytes (36). 4–6 weeks after retro-orbital injection of the recombinant virus into mice, cardiac myocytes were isolated and imaged using live cell fluorescence microscopy. The AAV-based vector achieved strong cardiac expression (Figure 4A), enabling the real-time measurement of a rapid change in HyPer fluorescence following the addition of insulin (Figure 4B). This increase was blocked when the cardiac myocytes were pre-treated with the NOX inhibitor apocynin, once again implicating NADPH oxidase(s) as the major source of an increase in H2O2 seen in response to insulin. In order to control for HyPer’s known pH sensitivity, these experiments were repeated using a recombinant AAV9 construct expressing SypHer2, a variant of HyPer lacking one of the key cysteines oxidized by H2O2. After insulin stimulation, cardiac myocytes from mice expressing SypHer showed no increase in fluorescence (Figure S9), making a change in pH unlikely as the cause of HyPer’s fluorescence.
Figure 4. Direct imaging of insulin-promoted changes in intracellular H2O2 concentrations.

A) Representative ratiometric images of cardiac myocytes isolated from mice injected with an AAV9 viral vector carrying the H2O2 biosensor HyPer. Cardiac myocytes were pretreated with either vehicle (DMSO) or apocynin (500 μM) and stimulated with insulin (10 nM) at time=0. Ratiometric images are displayed with a color lookup table that extends linearly across increasing ratios from blue to red. B) Pooled data from HyPer-expressing cardiac myocytes stimulated with insulin at t=0 minutes with or without apocynin pretreatment. n=5 coverslips per condition with 20–40 cells per coverslip isolated from 3 mice total. *** indicates p<0.001 compared to DMSO.
Loss of NOX2 attenuates canonical insulin-mediated phosphorylation responses while loss of NOX4 prevents insulin-induced repression of beta adrenergic signaling
The previous results with pharmacological inhibitors strongly implicate H2O2 produced by NADPH oxidase(s) in the modulation of insulin signaling pathways in cardiac myocytes. The NOX2 and NOX4 isoforms have been previously identified in cardiac myocytes (23), so insulin-mediated responses in cardiac myocytes isolated from NOX2 KO and NOX4 KO mice were analyzed next. The insulin-mediated phosphorylations of kinase Akt and mTOR were significantly attenuated in cardiac myocytes isolated from NOX2 KO mice (Figure 5A). By contrast, these insulin-mediated phosphorylation responses were entirely unaffected in cardiac myocytes isolated from NOX4 KO mice (Figure 5B). In contrast, insulin-promoted attenuation of isoproterenol-stimulated cardiac contractility was retained in cardiac myocytes from NOX2 KO mice, yet was entirely absent in cardiac myocytes from the NOX4 KO mice (Figure 6A)- the opposite of what was observed for insulin-dependent Akt and mTOR phosphorylation responses. Consistent with the effects seen on cardiac myocyte contractility, insulin-induced attenuation of the isoproterenol-stimulated increase in phospholamban phosphorylation was retained in cardiac myocytes isolated from the NOX2 KO mice and was absent in cardiac myocytes from the NOX4 KO mice (Figure 6B). In order to assess the potential effect of genetic ablation of NOX2 and NOX4 on the expression of other NOX-associated gene products, qPCR was performed on cardiac myocytes from NOX2 KO and NOX4 KO mice. No significant alterations in expression of Nox1, gp91phox (Nox2), Nox4, Rac1 or p22phox were observed (Figure S10).
Figure 5. Insulin-modulated phosphoprotein responses in cardiac myocytes deficient for NOX2 or NOX4.

A) Representative immunoblots for cardiac myocytes from wild-type (n=4) mice and NOX2 KO (n=4) mice treated with insulin (1 nM) for the indicated times (Left). Densitometric analyses for pAkt and pmTOR from at least three independent experiments are shown to the right. B) Representative immunoblots for cardiac myocytes from NOX4 KO (n=4) mice with wild-type control cardiac myocytes (n=4) treated with insulin (1 nM) for the indicated times (Left). Densitometric analyses from at least three independent experiments for the pAkt and pmTOR are shown to the right. * indicates p<0.05, **p<0.01, and ***p<0.001.
Figure 6. Contractility and beta adrenergic signaling responses to insulin and isoproterenol in NOX2 KO and NOX4 KO cardiac myocytes.

A) Pooled data from sarcomere shortening transients in cardiac myocytes isolated from NOX2 KO (n=4) mice, NOX4 KO (n=5) mice, or wild-type controls (n=7 mice). Cardiac myocytes were stimulated with isoproterenol (10 nM, 5 min) with or without insulin pretreatment (10 nM, 30 min). (B) Representative immunoblots of NOX2 or NOX4 KO cardiac myocytes treated with isoproterenol with or without insulin pretreatment (right side of the blots). wild-type cardiac myocyte lysates were loaded on the left side of the gel. Below the representative blots are the densitometric analyses of pooled data from at least three independent experiments. * indicates p<0.05, **p<0.01, and ***p<0.001.
NOX2 KO and NOX4 KO cardiac myocytes demonstrate attenuated H2O2 production with distinct time courses
Having observed differential roles of NOX2 and NOX4 in insulin-modulated cardiac myocyte responses (Figures 5 and 6), we performed a temporal analysis of H2O2 production in cardiac myocytes from NOX2 KO and NOX4 KO mice. NOX2 KO and NOX4 KO mice were injected with the AAV9-HyPer biosensor, and live cell imaging was performed on isolated cardiac myocytes to quantitate the HyPer response in real time following addition of insulin (as in Figure 4). While there remains a significant insulin-stimulated increase in H2O2 in cardiac myocytes from both the NOX2 KO and NOX4 KO mice, there were striking differences in the temporal profile of the HyPer response that distinguished cardiac myocytes from these NOX knockout mice (Figure 7A). Cardiac myocytes from NOX2 KO mice showed a marked attenuation in the insulin-stimulated H2O2 response, and sustained a modest yet statistically significant production of H2O2 when stimulated with insulin. Most notably, these NOX2 KO cardiac myocytes did not show the initial rapid phase of H2O2 production that was seen in cardiac myocytes form wild-type mice. In contrast, cardiac myocytes from NOX4 KO mice maintained the early phase of production of H2O2, which eventually reached concentrations of H2O2 that were similar to those observed in cardiac myocytes from NOX2 KO mice. These results imply that insulin activates both NOX2 and NOX4 in cardiac myocytes, with NOX2 modulating the initial rapid increase in H2O2 generation, and NOX4 producing a more modest yet stable H2O2 response following insulin stimulation.
Figure 7. Insulin-induced changes in intracellular H2O2 in NOX2 and NOX4 KO cardiac myocytes and calveolar and mitochondrial subcellular compartments.

A) Pooled data from cardiac myocytes isolated from wild-type (n=163 cells from 5 coverslips from 2 mice), NOX2 (n= 109 cells from 4 coverslips from 2 mice) or NOX4 (n=111 cells from 4 coverslips from 2 mice) mice, expressing HyPer, and stimulated with insulin (10 nM) at t=0. * indicates p<0.05 comparing NOX4 KO to wild-type, and # indicates p<0.05 comparing NOX2 KO to wild-type by 2-way ANOVA. B) Pooled data from cardiac myocytes expressing HyPer3 (n=58 cells from 4 coverslips from 1 mouse), mito-HyPer3 (n=98 cells from 5 coverslips from 2 mice) or cav-HyPer3 (n=99 cells from 6 coverslips from 2 mice) treated with insulin at time t=0. * indicates p<0.05 comparing mito-HyPer3 to HyPer3.
Insulin stimulates an increase of H2O2 in caveolae and a decrease in mitochondrial matrix H2O2
Because NOXs have been reported to be localized to specific subcellular compartments, changes in local H2O2 concentrations were next explored using differentially targeted variants of HyPer in order to identify the compartment(s) of H2O2 production in response to insulin. Cardiac myocytes expressing HyPer constructs targeted to the caveolae, mitochondria or cytosol were stimulated with insulin while local H2O2 was measured by real time fluorescence microscopy. Insulin stimulation triggered a rapid increase in the HyPer fluorescence ratio in the caveolar compartment, while a consistent and significant decrease in the HyPer signal was observed in the mitochondrial matrix (Figure 7B).
Discussion
Differential insulin signaling via NOX2 and NOX4
These studies have exploited novel biosensors, physiological approaches, biochemical analyses, and cellular imaging methods to identify a central role for hydrogen peroxide (H2O2) in the modulation of insulin signaling pathways in the heart. A role for H2O2 was suggested by the observation that catalase (which degrades H2O2) and the NADPH oxidase (NOX) inhibitor apocynin block insulin-modulated physiological and phosphorylation responses (Figures 1–3). Changes in intracellular H2O2 in response to insulin were then directly documented using cell imaging techniques (Figure 4). Recombinant cardiotropic AAV9 viruses expressing variants of the H2O2 biosensor HyPer were developed and injected into mice to infect cardiac tissues. Cardiac myocytes from these mice were imaged to quantitate intracellular H2O2 levels in real time by analyzing the HyPer fluorescence ratio. We found that insulin stimulation elicited a rapid increase in intracellular H2O2 levels. These findings using the H2O2 biosensor HyPer, in combination with our observations using catalase (Figure 2), lead us to conclude that the effects of insulin are specifically mediated by H2O2, as distinguished from other hydroperoxides, hydroxyl radicals, or protonated superoxide.
The insulin-modulated H2O2 response was markedly attenuated by NADPH oxidase inhibitors (Figure 4). Previous studies have shown that cardiac myocytes express both NOX2 and NOX4 (37), but there are currently no optimal isoform-specific NOX inhibitors (37). In order to identify the NADPH oxidase isoforms that modulate the insulin H2O2 response, insulin-induced phosphorylation and physiological responses were explored in cardiac myocytes isolated from NOX2 KO and NOX4 KO mouse lines. Insulin-promoted phosphorylation of Akt, mTOR and other “classical” insulin-responsive phosphoproteins were attenuated in the NOX2 knockout, but were retained in cardiac myocytes from NOX4 KO mice (Figure 5). In contrast, responses that involve the insulin-dependent attenuation of beta–adrenergic receptor-mediated responses (cardiac myocyte contractility and phospholamban phosphorylation) were lost in the NOX4 KO mice, yet were retained in cardiac myocytes from NOX2 KO mice (Figure 6). These findings indicate that both NOX2 and NOX4 are involved in the H2O2-mediated insulin response, and suggest that different H2O2 signaling pathways are activated downstream of NOX2 vs. NOX4. Insulin signaling therefore appears to trigger a bifurcating response, wherein two distinct NADPH oxidase isoforms are activated contemporaneously, but control divergent distal signaling processes.
H2O2 responses in NOX2 KO and NOX4 KO were studied in mice infected with the H2O2-sensitive AAV9-HyPer biosensor, and the insulin-stimulated increase in H2O2 was attenuated but not completely abolished in cardiac myocytes isolated from either NOX2 KO or NOX4 KO mice compared to wild-type cardiac myocytes (Figure 7A). These findings provide additional evidence that the insulin-dependent H2O2 response involves both the NOX2 and NOX4 isoforms. These two NOX isoforms appear to be activated by insulin yet have distinct temporal profiles: the NOX2 isoform mediates a rapid increase in intracellular H2O2 that peaks at approximately 2–3 minutes following addition of insulin, while the NOX4 isoform has a slower but more sustained H2O2 response (Figure 7A). H2O2 generated by NOX2 and NOX4 thus appear to modulate pathways in cardiac myocytes that are both temporally and functionally distinct.
NOX2 regulation of insulin-modulated phosphorylation pathways
The role of NOX2 in the heart is incompletely understood; most studies have focused on a deleterious role for NOX2, although a protective role for NOX2 in ischemic preconditioning has also been reported (38). NOX2 is the NOX isoform that has been most clearly linked to receptor-mediated activation of ROS synthesis (39): angiotensin II acts through the AT-1 receptor to promote the rapid activation of NOX2 by enhancing the binding of the small GTPase Rac1, an essential component of the active NOX2 complex. Yet the role of NOX2 in insulin signal transduction remains almost entirely unexplored, with a single previous report in cultured myotubes finding that knockdown of p47phox (a component of the NOX2 complex) attenuates insulin-induced ROS production (40). Supporting a role for NOX2-derived H2O2 in insulin signal transduction in metabolic tissues, the NOX2 knockout mouse has been reported to have elevated serum glucose concentrations and lower body weights (41). However, given that studies in other cell types have indicated that NOX4 rather than NOX2 amplifies insulin signaling (42), we were surprised to find a similar role fulfilled by NOX2 in cardiac myocytes. These differences highlight the different roles that signaling proteins may play in various cell types. Our studies have used immunochemical approaches to identify multiple phosphoproteins that are dynamically modulated by insulin-regulated H2O2, but of course many other protein kinases/phosphoprotein phosphatases still remain to be studied in these cells. It is plausible that these other phosphorylation pathways may be differentially regulated by H2O2-dependent signals. It is also important to note that our data indicate that NOX-derived H2O2 augments insulin signaling, but is not essential to activate the canonical insulin-modulated kinase cascade in cardiac myocytes. This is consistent with previous studies of insulin-regulated redox-sensitive signaling proteins such as PTP-1B, which is a plausible candidate for transducing the H2O2–dependent insulin responses that have been documented in the present studies.
The enigma of NOX4 activation
Unlike NOX2, the rapid receptor-dependent activation of NOX4 has not been extensively characterized; only a single report has linked NOX4 to insulin receptor signaling in adipocytes, but by unknown mechanisms (42). Indeed, the entirely unaffected Akt and mTOR responses observed in NOX4 KO cardiac myocytes was unexpected given the previous findings in adipocytes. The role of NOX4 in the heart is controversial: some studies ascribe a deleterious role of NOX4 in heart failure (26, 27) while other studies find a protective role for the enzyme in various heart failure models (25, 37). None of these previous studies of NOX4 explored a role in cardiac insulin signaling. Most previous reports postulate that cellular regulation of NOX4 activity is achieved by transcriptional activation, leading to changes in NOX4 protein abundance that occur over many hours (43). However, the time course that we have observed for the insulin-dependent H2O2 response is not compatible with genetic induction of NOX4, and indicates that activation of NOX4 likely occurs at the post-translational level. The study of post-translational modifications of NOX4 remains a nascent field with limited biochemical tools available and only one phosphorylation site (Tyr566) having being recently described as modifying enzyme activity (44). Another important question prompted by these studies is the mechanism by which NOX4 attenuates beta adrenergic signaling. We have recently shown that insulin’s effects on inotropy may be mediated through a pathway involving the GPCR kinase GRK2, the β2 adrenergic receptor, and the phosphodiesterase isoform PDE-4D (45). It has also been reported that activation of eNOS by exogenous H2O2 can lead to the S-nitrosylation and activation of GRK2 in the heart (46), suggesting another mechanism whereby NOX4 may modulate contractility. It is also plausible that NOX4-derived H2O2 may directly activate the cyclic GMP-dependent protein kinase (47). Clearly, much remains to be learned about the pathway(s) whereby NOX4 modulates cyclic nucleotide signaling in cardiac myocytes and other cell types.
The specificity with which various NOX isoforms are localized to different subcellular compartments remains controversial. NOX4 has been reported in the mitochondrial matrix, nucleus, plasma membrane and endoplasmic reticulum of various cell types (26–28) while NOX2 has been reported in the plasma membrane and endoplasmic reticulum (22). We used differentially targeted HyPer3 constructs to probe H2O2 dynamics in both caveolae and mitochondria in cardiac myocytes isolated from mice infected with these recombinant AAV9 viruses. We were intrigued to find that insulin promoted a striking increase in H2O2 levels in caveolae, to a magnitude similar to that observed in the cytosol (Figure 7B). By contrast, in the mitochondrial matrix, insulin promoted a significant decrease in the HyPer ratio. The marked insulin-dependent decrease in the HyPer ratio within the cardiac myocyte mitochondrial matrix is likely to be multifactorial, possibly reflecting the effects of insulin on metabolic switching and/or pH changes in the mitochondria in these cells; additional experiments (beyond the scope of these studies) are needed to define the biochemical basis for this observation.
Insulin, diabetes, and control of cardiac function
These studies extend our understanding of the role of insulin in control of cardiac myocyte physiology. While insulin appears to have no substantive direct effect on basal cardiac contractility, insulin markedly attenuates the positive inotropic response to beta adrenergic receptor activation. This inhibitory effect of insulin is lost both in cardiac myocytes isolated from NOX4 KO mice and as a consequence of high fat feeding (Figure S1). The relevance of these responses in the intact animal will need to be tested in future in vivo studies. Unfortunately, assessment of cardiac function and insulin signaling in response to both isoproterenol and insulin infusion in intact animals would be complicated by the counter-regulatory physiologic responses that would be elicited by any changes in inotropy, as well as changes the changes in serum glucose that would be a consequence of systemic insulin administration.
These findings may represent an important step towards understanding the pathophysiology of diabetic cardiomyopathy, a disease in which diabetics develop cardiac dysfunction independently of coexisting vascular disease and hypertension. The results provide clear evidence of cardiac insulin resistance in a mouse model of type II diabetes, in which there is marked attenuation of insulin signaling and physiological responses in cardiac myocytes isolated from mice fed a high fat diet (Figure 1). We acknowledge that these observations stand independently of the NOX2 and NOX4 pathways described in the rest of these studies. A thorough examination of these two key redox pathways in the context of high fat diet will be needed to ascertain whether insulin resistance is mediated in part by altered activity of NOX2 or NOX4. Regardless of whether alterations occur at the level of H2O2 production or more proximally, the insulin-resistant cardiac myocytes isolated from mice fed a high fat diet appear to lose a key physiological counter-regulatory force on adrenergic signaling. Excessive adrenergic stimulation is a known driving force for heart failure and cardiac dysfunction (48). Insulin’s physiological inhibitory effect on beta adrenergic signaling through NOX4 may therefore be cardioprotective, a hypothesis that will need to be tested in an in vivo setting. In such a model, the insulin-resistant heart loses a protective effect of insulin, allowing for excessive beta adrenergic stimulation. The restoration of insulin’s physiologic and potentially cardioprotective “beta blocking” effects by increasing flux through NOX4 may represent a promising therapeutic target in diabetic cardiomyopathy and heart failure.
Supplementary Material
highlighs.
Insulin signaling in cardiac myocytes is modulated by hydrogen peroxide derived from two distinct NADPH oxidase isoforms, Nox2 and Nox4.
Nox2 couples insulin to “canonical” insulin-modulated phosphorylation pathways in cardiac myocytes involving kinase Akt and mTor.
By contrast, Nox4 modulates insulin-dependent attenuation of beta-adrenergic pathways by suppressing cAMP/PKA-dependent physiological responses- revealing a “beta blocker-like” effect of insulin in cardiac myocytes.
In an animal model of type two diabetes, these insulin-dependent hydrogen peroxide-mediated responses are abrogated, suggesting an involvement of insulin-modulated oxidant pathways in the pathogenesis of diabetic cardiomyopathy.
Acknowledgments
These studies were supported by NIH grants PO1-HL48743 and RO1-HL46457 (to TM) and R01-DK092065, R01-HL127764, and R01-HL112413 (to EDA).
Abbreviations
- ROS
Reactive oxygen species
- PKA
Protein kinase A
- NOX
NADPH oxidase
- PTP1B
Protein tyrosine phosphatase 1B
- CaMK
Calmodulin Kinase
- mTOR
Mammalian target of rapamycin
- AAV
Adeno-associated virus
- CIRKO
Cardiac insulin receptor knockout
- IGF-1
Insulin-related growth factor 1
- eNOS
endothelial nitric oxide synthase
- GRK2
G protein receptor kinase 2
- PDE-4D
Phosphodiesterase 4 D
Footnotes
Disclosures: The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Boucher J, Kleinridders A, Ronald Kahn C. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harbor Perspectives in Biology. 2014;6(1) doi: 10.1101/cshperspect.a009191. pii: a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertrand L, Horman S, Beauloye C, Vanoverschelde JL. Insulin signalling in the heart. Cardiovascular Research. 2008;79(2):238–248. doi: 10.1093/cvr/cvn093. [DOI] [PubMed] [Google Scholar]
- 3.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415(6868):206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 4.Fu Q, Xu B, Liu Y, Parikh D, Li J, Li Y, Zhang Y, Riehle C, Zhu Y, Rawlings T, Shi Q, Clark RB, Chen X, Abel ED, Xiang YK. Insulin inhibits cardiac contractility by inducing a Gi-biased β2-adrenergic signaling in hearts. Diabetes. 2014;63(8):2676–89. doi: 10.2337/db13-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. Journal of Biological Chemistry. 2014;289:8735–41. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biology. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leopold JA, Loscalzo J. Oxidative enzymopathies and vascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(7):1332–1340. doi: 10.1161/01.ATV.0000163846.51473.09. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Wang W, Li L, Perry G, Lee H, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2014;1842(8):1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spencer NY, Yan Z, Boudreau RL, Zhang Y, Luo M, Li Q, Tian X, Shah aM, Davisson RL, Davidson B, Banfi B, Engelhardt JF. Control of hepatic nuclear superoxide production by glucose 6-phosphate dehydrogenase and NADPH oxidase-4. Journal of Biological Chemistry. 2011;286(11):8977–8987. doi: 10.1074/jbc.M110.193821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.May JM, de Haën C. The insulin-like effect of hydrogen peroxide on pathways of lipid synthesis in rat adipocytes. Journal of Biological Chemistry. 1979;254(18):9017–9021. [PubMed] [Google Scholar]
- 11.May JM, de Haën C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. Journal of Biological Chemistry. 1979;254(7):2214–2220. [PubMed] [Google Scholar]
- 12.Sartoretto JL, Kalwa H, Shiroto T, Sartoretto SM, Pluth MD, Lippard SJ, Michel T. Role of Ca2+ in the Control of H2O2-modulated phosphorylation pathways leading to eNOS activation in cardiac myocytes. PLoS ONE. 2012;7(9):e44627. doi: 10.1371/journal.pone.0044627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal. 2005;7(5–6):560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- 14.Anderson ME. Oxidant stress promotes disease by activating CaMKII. Journal of Molecular and Cellular Cardiology. 2015;89:160–167. doi: 10.1016/j.yjmcc.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murarka S, Movahed MR. Diabetic cardiomyopathy. Journal of Cardiac Failure. 2010;16(12):971–979. doi: 10.1016/j.cardfail.2010.07.249. [DOI] [PubMed] [Google Scholar]
- 16.Pappachan JM, Varughese GI, Sriraman R, Arunagirinathan Joseph Pappachan GM. Diabetic cardiomyopathy: Pathophysiology, diagnostic evaluation and management. World J Diabetes. 2013;4(45):177–189. doi: 10.4239/wjd.v4.i5.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dandamudi S, Slusser J, Mahoney DW, Redfield MM, Rodeheffer RJ, Chen HH. The prevalence of diabetic cardiomyopathy: A population-based study in Olmsted County, Minnesota. Journal of Cardiac Failure. 2014;20(5):304–309. doi: 10.1016/j.cardfail.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature Reviews: Molecular Cell Biology. 2014;15(6):411–21. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 19.Finkel T. Signal transduction by reactive oxygen species. Journal of Cell Biology. 2011;194(1):7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circulation Research. 2015;116(3):531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxidants & Redox Signaling. 2009;11(6):1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J-M, Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. Journal of Biological Chemistry. 2002;277(22):19952–60. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- 23.Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circulation Research. 2012;110(10):1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreira LF, Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Radical Biology and Medicine. 2016;98:1–11. doi: 10.1016/j.freeradbiomed.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, Brewer AC, Schröder K, Santos CXC, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(42):18121–6. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J. Activation of Nox4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the PERK/eIF-2α/ATF4 pathway. Circulation Research. 2013;113(11):1253–1264. doi: 10.1161/CIRCRESAHA.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circulation Research. 2010;106(7):1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie L-H, Tian B, Sadoshima J. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circulation Research. 2013;112(4):651–63. doi: 10.1161/CIRCRESAHA.112.279760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sartoretto JL, Kalwa H, Pluth MD, Lippard SJ, Michel T, Romero N, Michel T. Hydrogen peroxide differentially modulates cardiac myocyte nitric oxide synthesis. Proceedings of the National Academy of Sciences. 2011;528(38):61–78. doi: 10.1073/pnas.1111331108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zablocki D, Sadoshima J. Solving the cardiac hypertrophy riddle: The angiotensin II-mechanical stress connection. Circulation Research. 2013;113(11):1192–1195. doi: 10.1161/CIRCRESAHA.113.302501. [DOI] [PubMed] [Google Scholar]
- 31.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Ronald Kahn C, Dale Abel E. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. Journal of Clinical Investigation. 2002;109(5):629–639. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagao M, Asai A, Sugihara H, Oikawa S. Fat intake and the development of type 2 diabetes. Endocrine Journal. 2015;60(9):1065–75. doi: 10.1507/endocrj.EJ15-0055. [DOI] [PubMed] [Google Scholar]
- 33.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phopholamban/SERCA2a regulatome. Circulation Research. 2012;110(12):1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sartoretto JL, Jin BY, Bauer M, Gertler FB, Liao R, Michel T. Regulation of VASP phosphorylation in cardiac myocytes: differential regulation by cyclic nucleotides and modulation of protein expression in diabetic and hypertrophic heart. American Journal of Physiology. Heart and Circulatory Physiology. 2009;297(5):H1697–H1710. doi: 10.1152/ajpheart.00595.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bilan DS, Belousov VV. HyPer family probes: state of the art. Antioxidants & Redox Signaling. 2015;24(3):731–51. doi: 10.1089/ars.2015.6586. [DOI] [PubMed] [Google Scholar]
- 36.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy: The Journal of the American Society of Gene Therapy. 2008;16(6):1073–80. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxidants & Redox Signaling. 2012;18(9):120827084821005. doi: 10.1089/ars.2012.4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsushima S, Tsutsui H, Sadoshima J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends in Cardiovascular Medicine. 2014;24(5):202–205. doi: 10.1016/j.tcm.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M, Prosser BL, Bamboye Ma, Gondim ANS, Santos CX, Martin D, Ghigo A, Perino A, Brewer AC, Ward CW, Hirsch E, Lederer WJ, Shah AM. Contractile function during angiotensin-II activation. Journal of the American College of Cardiology. 2015;66(3):261–272. doi: 10.1016/j.jacc.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Espinosa A, García A, Härtel S, Hidalgo C, Jaimovich E. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. Journal of Biological Chemistry. 2009;284(4):2568–2575. doi: 10.1074/jbc.M804249200. [DOI] [PubMed] [Google Scholar]
- 41.De Figueiredo ASP, Salmon AB, Bruno F, Jimenez F, Martinez HG, Halade GV, Ahuja S, Clark RA, DeFronzo RA, Abboud HE, El Jamali A. Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. Journal of Biological Chemistry. 2015;290(21):13427–13439. doi: 10.1074/jbc.M114.626077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth D, Goldstein BJ, Lambeth JD. The NAD(P)H oxidase homolog nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Molecular and Cellular Biology. 2004;24(5):1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen F, Haigh S, Barman S, Fulton DJR. From form to function: The role of Nox4 in the cardiovascular system. Frontiers in Physiology. 2012;3:412. doi: 10.3389/fphys.2012.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsushima S, Kuroda J, Zhai P, Liu T, Ikeda S, Nagarajan N, Oka SI, Yokota T, Kinugawa S, Hsu CP, Li H, Tsutsui H, Sadoshima J. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. Journal of Clinical Investigation. 2016;126(9):3403–3416. doi: 10.1172/JCI85624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Q, Liu Y, Fu Q, Xu B, Zhang Y, Kim S, Tan R, Barbagallo F, West T, Anderson E, Wei W, Abel DD, Xiang YK. Inhibiting insulin-mediated β2AR activation prevents diabetes-associated cardiac dysfunction. Circulation. 2016;135(1):73–88. doi: 10.1161/CIRCULATIONAHA.116.022281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang ZM, Gao E, Fonseca FV, Hayashi H, Shang X, Hoffman NE, Chuprun JK, Tian X, Tilley DG, Madesh M, Lefer DJ, Stamler JS, Koch WJ. Convergence of G protein-coupled receptor and S-nitrosylation signaling determines the outcome to cardiac ischemic injury. Science Signaling. 2013;6(299):ra95. doi: 10.1126/scisignal.2004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgoyne JR. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science. 2007;317:1393–1398. doi: 10.1126/science.1144318. [DOI] [PubMed] [Google Scholar]
- 48.Lachowska K, Gruchała M, Narkiewicz K, Hering D. Sympathetic activation in chronic heart failure: Potential benefits of interventional therapies. Current Hypertension Reports. 2016;18(7):1–11. doi: 10.1007/s11906-016-0660-7. [DOI] [PubMed] [Google Scholar]
- 49.Sartoretto JL, Kalwa H, Romero N, Michel T. In vivo imaging of nitric oxide and hydrogen peroxide in cardiac myocytes. Methods in Enzymology. 2013;528:61–78. doi: 10.1016/B978-0-12-405881-1.00004-5. [DOI] [PubMed] [Google Scholar]
- 50.Bilan DS, Pase L, Joosen L, Gorokhovatsky AY, Ermakova YG, Gadella TWJ, Grabher C, Schultz C, Lukyanov S, Belousov VV. HyPer-3: A genetically encoded H2O2 probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chemical Biology. 2013;8(3):535–542. doi: 10.1021/cb300625g. [DOI] [PubMed] [Google Scholar]
- 51.Kalwa H, Sartoretto JL, Martinelli R, Romero N, Steinhorn BS, Tao M, Ozaki CK, Carman CV, Michel T. Central role for hydrogen peroxide in P2Y1 ADP receptor-mediated cellular responses in vascular endothelium. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(9):3383–8. doi: 10.1073/pnas.1320854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rizzuto R, Nakase H, Darras B, Francke U, Fabrizi GM, Mengel T, Walsh F, Kadenbach B, DiMauro S, Schon EA. A gene specifying subunit VIII of human cytochrome c oxidase is localized to chromosome 11 and is expressed in both muscle and non-muscle tissues. Journal of Biological Chemistry. 1989;264(18):10595–10600. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.