

Abstract
Background
While pediatric low-grade glioma/glioneuronal tumors (LGG/LGGNTs) are considered slow-growing, indolent tumors with excellent long-term prognosis, mortality due to the disease is not unknown. Few studies have addressed the cause of death in this population.
Methods
Retrospective review of clinicopathologic and radiologic data for children 21 years or younger with LGG/LGGNT who died at St. Jude Children’s Research Hospital between April 1985 and June 2015. Our primary objective was to determine the causes and timing of mortality in affected children.
Results
For the 87 eligible patients, median age at diagnosis was 7.7 years (range, 0.21–21 years), median age at death was 14.26 years (range, 0.58–32 years), and median time to death from diagnosis was 4.02 years (range, 0.21–24 years). Midbrain/thalamus was the most common tumor location (n=34), followed by suprasellar/hypothalamic (n=18) and cerebrocortical (n=13). Astrocytoma not otherwise specified (n=24), pilocytic astrocytoma (n=23), and fibrillary astrocytoma (n=11) were the predominant histologic diagnoses. Causes of death included progressive primary disease (PD) (n=43), progression of primary disease with histological features of a high-grade glioma at progression or at autopsy (PD-HGG) (n=15), second cancer (n=3), suicide (n=4), and vehicular accident (n=3). Among the 15 patients with PD-HGG, 12 received radiation therapy before histologic confirmation of progression.
Conclusions
PD and PD-HGG contributed to 66% of the mortality in our patient cohort. Early psychological intervention should be included as part of the multidisciplinary management approach of children with LGG/LGGNT to reduce the risk of suicide in vulnerable subjects.
Keywords: low-grade glioma, children, suicide, violent deaths, progressive disease
INTRODUCTION
Pediatric low-grade gliomas (LGGs) and low-grade glioneuronal tumors (LGGNTs) (WHO Grade I & II tumors) generally have excellent outcomes with prolonged survival, especially when a gross-total resection (GTR) of the tumor is achieved.1–5 LGG/LGGNTs are often viewed as a chronic disease with an indolent course, in contrast to some other central nervous system (CNS) malignancies [e.g., embryonal tumors or high-grade gliomas (HGGs)]. Children with LGG/LGGNT often require multiple treatment regimens to halt tumor progression.6, 7 A vast majority survive into adulthood, but a small proportion ultimately succumb. The reported rate of mortality in children with LGG/LGGNT is 5% to 18%.1, 2, 4, 8, 9 The causes of mortality include, but are not limited to, progression of primary disease (PD), pathologic progression to HGG, and treatment-associated complications.8–10 Barring a few studies, there is a scarcity of literature on the causes of death of these children, and the clinicopathologic variables that may predict a fatal outcome.8, 9 In an attempt to systematically delineate the causes and timing of mortality in this population, we undertook a retrospective study of children with LGG/LGGNT who died during the course of their treatment or follow-up at St. Jude Children’s Research Hospital (St. Jude).
METHODS
Study Design
We conducted a retrospective review of the medical records of all children with a WHO Grade I or II LGG/LGGNT, who were 21 years or younger at diagnosis and had died during the course of their treatment or follow-up at St. Jude. The study period was April 1985 to June 2015. The date of first presentation at St. Jude was considered the study entry point. Patients were included in the study if they had a histologic confirmation of a WHO Grade I or II LGG/LGGNT or in the presence of corroborative tumor-related symptoms, an imaging finding compatible with WHO Grade I or II LGG/LGGNT. An example of the latter is visual impairment in children with optic nerve lesion, in whom the diagnosis of LGG is often made without biopsy. The data collected included patient demographics, tumor location and histology, metastatic status, number and type of treatments received, the cause of mortality, and time to death from diagnosis. This study was approved by the St. Jude Institutional Review Board.
The date of first imaging, when available, was considered the date of diagnosis. If that data was not available, then the date of first biopsy or tumor resection and histologic confirmation of LGG/LGGNT was considered the date of diagnosis. The cause of death was ascertained from the medical record or death certificate. The treatments administered were categorized as surgical resection, chemotherapy, or radiation therapy (RT).
Surgical Outcomes
The extent of surgical resection was determined by the operating surgeon, imaging results, or both. The categories of surgical resection were defined as follows: gross-total resection (GTR), when no tumor remained visible in the surgical field; near-total resection (NTR), when more than 90% of the tumor was removed; sub-total resection (STR), when 50% to 90% of the tumor was removed: and partial resection (PR), when less than 50% of the tumor was removed. When the surgical intent was to obtain tumor tissue for diagnosis without any effort to resect the tumor, it was termed biopsy only.
Other Treatments
Chemotherapy regimens varied, depending on the availability of ongoing clinical trials or non-protocol treatment plans, from single-agent therapy (e.g., parenteral cyclophosphamide or oral temozolamide) to the more traditional combination chemotherapy (e.g., vincristine/carboplatin). RT was either focal, whole-brain, or craniospinal irradiation. When more than one treatment modality was used at diagnosis or each progression, it was counted as a single regimen.
Causes of and Time to Death
Time to death was calculated as the period from the time of diagnosis to that of mortality. The causes of mortality were classified as follows: PD, progression of primary disease with histological features of a high-grade glioma at progression or autopsy (PD-HGG), second malignancy (i.e., when the second tumor was noncontiguous in location and/or histologically different from the primary LGG/LGGNT), therapy-induced or tumor location–associated complications, and miscellaneous causes. A diagnosis of PD-HGG was made based on clinical suspicion of progression and when corroborating histologic confirmation of LGG/LGGNT at diagnosis and that of an HGG at progression or on autopsy was available.
Statistical Methods
Descriptive statistics were used to designate the distributions of variables of interest. The results are presented as median, range, and percentages.
RESULTS
Patient Characteristics
Of the 727 patients with LGG/LGGNT treated at our institution during the study period, 87 (12%) died during treatment or follow-up. Among the 727 patients, 370 patients had their domiciliary residence in Memphis and adjoining cities, for which St. Jude is the primary tertiary hospital of care. The remaining 357 patients were referred from other institutions to St. Jude. Among those who died from the disease, 42 patients were from the Memphis geographic location while 45 were referred to St. Jude for care. All 87 subjects were included in this study. The median age of the cohort at diagnosis was 7.7 years (range, 0.21–21 years). There was a predominance of male patients in our study, with a male-to-female ratio of 1.7:1 (Table 1). Two-thirds of our patients (n=59) identified as white race; about one-third identified as black race (n=24); one patient identified as mixed race; and three identified as other. Three patients had a diagnosis of neurofibromatosis type 1 (NF-1).
TABLE 1.
Patient characteristics (N = 87)
| Characteristic | n (%) |
|---|---|
| Sex | |
| Female | 32 (37) |
| Male | 55 (63) |
| Race | |
| Black | 24 (27.5) |
| White | 59 (68) |
| Mixed | 1 (1) |
| Other | 3 (3.5) |
| Tumor location | |
| Midbrain/thalamus | 34 (39) |
| Suprasellar/hypothalamus | 18 (21) |
| Cerebrocortical | 13 (15) |
| Brain stem | 12 (14) |
| Tectum | 3 (3) |
| Cerebellum | 2 (2) |
| Cervicomedullary | 1 (1) |
| Spine | 4 (5) |
| Histology | |
| Astrocytoma, NOS | 24 (28) |
| Pilocytic astrocytoma | 23 (26) |
| Fibrillary astrocytoma | 11 (13) |
| Oligodendroglioma/oligoastrocytoma | 7 (8) |
| Pleomorphic xanthoastrocytoma | 6 (7) |
| Ganglioglioma | 4 (4.5) |
| Glioma, NOS | 4 (4.5) |
| Desmoplastic infantile ganglioglioma | 1 (1) |
| Glioneural tumor | 1 (1) |
| Not available | 6 (7) |
| Macroscopic metastasis (M2/3) | |
| At Diagnosis | |
| Yes | 7 (8) |
| No | 52 (60) |
| Not available | 28 (32) |
| During the course of illness | |
| Yes | 30 (34) |
| No | 57 (66) |
| Neurofibromatosis-1 | 3 (4) |
Abbreviation: NOS, not otherwise specified
Tumor Location, Histology, and Metastasis
The midbrain and/or thalamus was the most common location of the tumor, being found in 39% (n=34) of the cases. This was followed by the suprasellar/hypothalamic area in 21% (n=18) of patients and cerebrocortical and brain stem distribution in approximately 15% (n=13) and 14% (n=12), respectively (Table 1, Fig. 1). Other primary tumors were located in the tectum (n=3), cerebellum (n=2), cervicomedullary (n=1), and spine (n=4). Histopathology data was available for 93% (n=81) of the patients, and all tissue samples were reviewed at our institution, except for that of one patient who had PD-HGG and for whom the high-grade pathology was unavailable for review. The three most common diagnoses included astrocytoma not otherwise specified (28%, n=24), pilocytic astrocytoma (26%, n=23), and diffuse fibrillary astrocytoma (13%, n=11). Remaining tumors included oligodendroglioma/oligoastrocytoma (n=7), pleomorphic xanthoastrocytoma (n=6), ganglioglioma (n=4), glioma not otherwise specified (n=4), desmoplastic infantile ganglioglioma (n=1) and glioneural tumor (n=1).
Figure. 1.
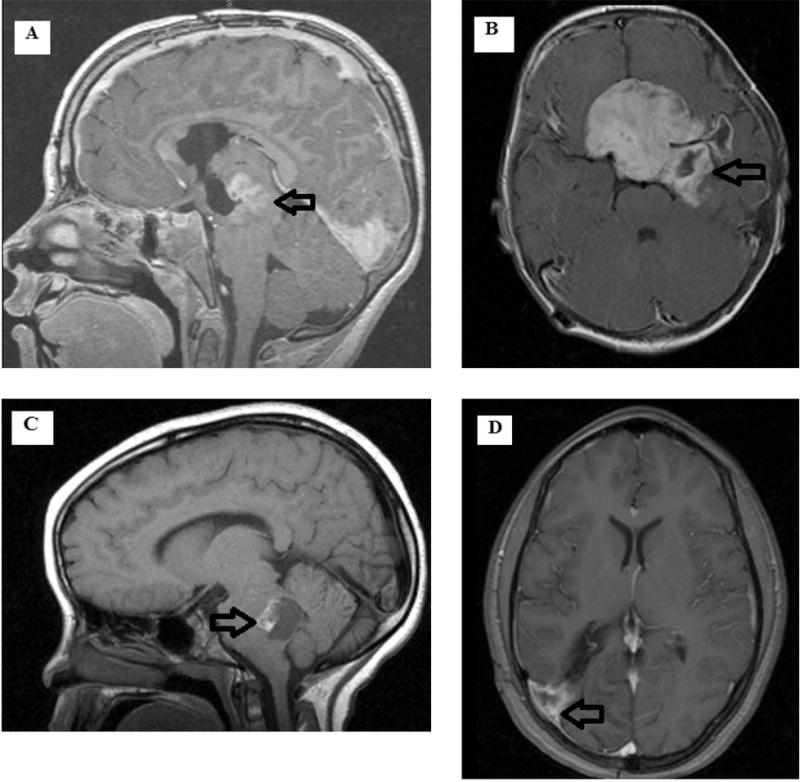
Representative images of the most common locations of LGG/LGGNT in the study cohort. (A) Sagittal T1 contrast image of a thalamic/midbrain tumor. (B) Axial T1 contrast image of a suprasellar/hypothalamic tumor. (C) Saggital T1 contrast image of an exophytic pontomedullary (brain stem) tumor. (D) Axial T1 contrast image of a right parieto-occipital tumor. Arrows note tumor location.
Histopathology was unavailable for review in six patients: two with optic pathway gliomas, two with pontomedullary tumors, one with a tectal tumor, and one with a thalamic/midbrain tumor. The diagnosis of a LGG/LGGNT was made in these patients based on the long duration of symptoms secondary to the location of the tumor and magnetic resonance imaging (MRI) features. None of these patients underwent surgical intervention. Metastatic imaging work-ups were available for 59(68%) patients at diagnosis, 7 of who had evidence of dissemination. This contrasts with a finding of 19 patients with metastatic disease at diagnosis in a larger cohort of 599 patients with LGG, alive or dead, reported from our institution by Chamdine et al.11 All patients had at least one imaging examination (either computed tomography or MRI) to investigate for dissemination. Thirty (34%) patients had imaging evidence of disease spread to a site distant from the primary tumor location during the course of their illness (Table 1).
Causes of Mortality and Time to Death
PD refractory to treatment was the most common cause of death in our study population; 43 (49%) children succumbed to disease. PD-HGG was the second most common cause of death, occurring in 15 (17%) patients. All 15 patients had a histologic confirmation of a HGG, with tissue samples available at diagnosis and progression or autopsy. Glioblastoma was the most common diagnosis at the time of progression or on autopsy in these 15 patients (Table 2).
TABLE 2.
Descriptions of 15 patients with PD-HGG
| Patient # |
Age at diagnosis (years) |
Site of disease |
Method of obtaining initial tissue sample |
Site of initial tissue sampling |
Initial histopathology |
Radiation therapy prior to repeat sampling Yes/No/Type |
Method of obtaining repeat tissue sample |
Site of repeat tissue sample |
Repeat histopathology |
Time from initial histologic diagnosis to MT/second HGG (years) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.2 | Midbrain/Thalamus | Biopsy | Thalamus | Astrocytoma, NOS | Yes/Focal | STR | Midbrain, Thalamus | GB | 11.9 |
| 2 | 0.8 | Midbrain/Thalamus | Biopsy | Midbrain | Fibrillary astrocytoma | No | Biopsy | Thalamus | GB | 6.5 |
| 3 | 4.8 | Optic pathway/ Cerebellum | STR | Cerebellum | Pilocytic astrocytoma | Yes/Focal | Autopsy | Cerebellum | GB | 5.5 |
| 4 | 5.2 | Midbrain/Thalamus | Biopsy | Thalamus | Astrocytoma, NOS | Yes/Focal | Autopsy | Midbrain/brain stem | GB | 10.7 |
| 5 | 6.4 | Midbrain/Thalamus | Biopsy | Thalamus | Oligodendroglioma | Yes/CSI | Autopsy | Thalamus | Anaplastic oligodendroglioma | 1.3 |
| 6 | 7.7 | Midbrain/Thalamus | NTR | Thalamus | Glioma, NOS | Yes/Focal | PR | Left CP angle lesion | Glioma, High Grade | 4.7 |
| 7 | 8.8 | Midbrain/ Thalamus | Biopsy | Thalamus | Fibrillary astrocytoma | Yes/Focal | PR | Thalamus | GB | 1.3 |
| 8 | 12.3 | Hemispherical | GTR | Right parieto-occipital cortex | PXA | No | GTR | Right parietal lobe | Anaplastic PXA | 1.9 |
| 9 | 13.3 | Midbrain/Thalamus | STR | Thalamus | Pilocytic astrocytoma | Yes/Focal | Biopsy | Frontal lobe | Glioma, High Grade | 1.7 |
| 10 | 16.8 | Hemispherical | PR | Right frontal lobe | Oligoastrocytoma | Yes/Focal | GTR | Right frontal lobe | GB | 2 |
| 11 | 16.3 | Midbrain/Thalamus | GTR | Thalamus | Ganglioglioma | No | Biopsy | Thalamus | Astrocytoma, High Grade | 7.4 |
| 12 | 21 | Midbrain/Thalamus | Biopsy | Right midbrain | Pilocytic astrocytoma | Yes/Focal | Autopsy | Midbrain | GB | 9.3 |
|
| ||||||||||
| Likely second HGG | ||||||||||
|
| ||||||||||
| 13 | 2.9 | Midbrain/Thalamus | Biopsy | Right Thalamus | Fibrillary astrocytoma | Yes/Focal | PR | Right basal ganglia, temporal lobe | GB | 6.7 |
| 14 | 8.1 | Midbrain/Thalamus | Biopsy | Midbrain | Pilocytic astrocytoma | Yes/Focal | Biopsy | Right cerebellar hemisphere | GB | 22.9 |
| 15 | 21 | Midbrain/Thalamus | Biopsy | Midbrain | Astrocytoma, NOS | Yes/Focal | Biopsy | Right middle CP | Glioma, High Grade | 17.9 |
Abbreviations: PD-HGG, progressive disease with high-grade glioma at progression; CP, cerebellar peduncle ;GB, glioblastoma; GTR, gross-total resection; HGG, high-grade glioma; NOS, not otherwise specified; MT, malignant transformation; NTR, near-total resection; PR, partial resection; PXA, pleomorphic xanthoastrocytoma; STR, subtotal resection
We hypothesized that some tumors with PD-HGG were second malignancies. The images and tissue-sampling sites at diagnosis and at progression/ autopsy were further analyzed for these 15 patients. We found that in 12/15 patients the location of the primary tumor and tumor at progression were unequivocally identical or contiguous (Table 2). The same was true for the site of tissue sampling at diagnosis and progression or autopsy. However, in three patients (13, 14, and 15; Table 2), the sites of sampling the primary tumor and that of progression were noncontiguous, albeit very close to each other, raising the possibility of those tumors being second malignancies. (Fig. 2).
Figure. 2.
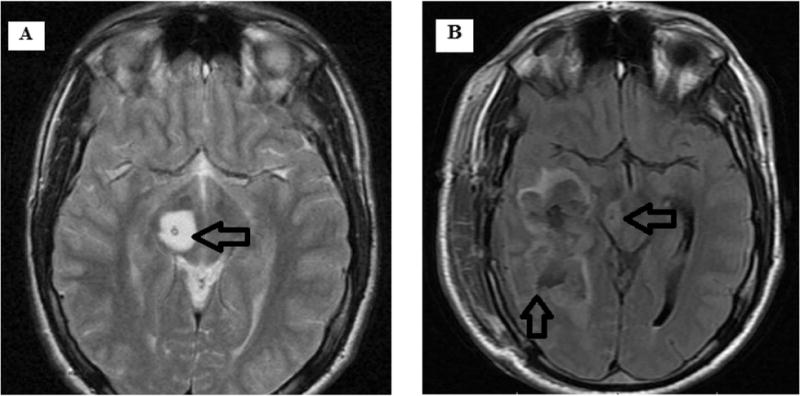
Magnetic resonance imaging of patient #13. (A) Axial T2-weighted image of the right thalamic tumor that was present at diagnosis. (B) Axial T2-weighted image of a noncontiguous but closely located high-grade glial neoplasm that was detected at follow-up. This patient received focal radiation therapy to the primary tumor during the period between diagnosis and histological confirmation of a high-grade glioma at progression.
Three patients died of second malignancy. Two of the three patients had NF-1: malignant peripheral nerve sheath tumor developed in one, and medulloblastoma developed in the other. The third patient suffered refractory T-cell acute lymphoblastic leukemia. The other tumor-related causes of death included shunt infection and sepsis (n=3) and respiratory compromise due to a brainstem location of the tumor (n=3). Two patients committed suicide; one patient’s reason for the act was anxiety and apprehension about PD. Miscellaneous causes of death included aspiration pneumonia and adult respiratory distress syndrome (n=2), stroke (n=2), renal toxicity due to chemotherapy (n=1), endocardial fibrosis (n=1), tumor hemorrhage (n=1), and pontine demyelination (n=1). Two patients died in motor vehicle accidents.
The cause of death was obtained from death certificates for eight patients and included suicide (n=2), motor vehicle accident (n=1), hydrocephalus due to shunt failure (n=1), and primary disease (n=4). The precise details of the events leading to death were lacking in these cases; hence, we listed them separately. The four patients whose deaths were documented as due to primary disease were not included in the analysis of time to death for those with PD due to a lack of sufficient information pertaining to their deaths.
The median age at death for the cohort was 14.26 years (range, 0.58–32 years), and the median time to death from diagnosis was 4.02 years (range, 0.21–24 years). For those who died of combined causes of PD and PD-HGG, when analyzed separately from the cohort, the median time to death was slightly shorter at 2.67 years (range, 0.35–24 years) (Table 3). Approximately three-fourths of those who died of PD did so within 5 years of diagnosis. Notably, seven patients died of PD, and five died of PD-HGG more than 10 years from diagnosis. (Table 4).
TABLE 3.
Descriptive statistics of patient ages
| Study subjects | Median Age (years) |
Range (years) |
|---|---|---|
| Entire cohort (N=87) | ||
| Age at diagnosis | 7.7 | 0.21–21 |
| Age at death | 14.26 | 0.58–32 |
| Time to death from diagnosis | 4.02 | 0.21–24 |
| PD+PD-HGG (n=58) | ||
| Age at diagnosis | 8.09 | 0.21–21 |
| Age at death | 14.17 | 4.74–32 |
| Time to death from diagnosis | 2.67 | 0.35–24 |
Abbreviations: HGG, high-grade glioma; PD, progressive disease; PD-HGG, progressive disease with high-grade glioma at progression
TABLE 4.
Time to death due to PD or PD-HGG
| Cause of death (n) | Median time to death from diagnosis Years (range) | Time to death from diagnosis (years) |
n (%) |
|---|---|---|---|
| PD (43) | <2 | 22 (51) | |
| 1.98 (0.36–16.82) | 2 – <5 | 8 (19) | |
| 5 – <10 | 6 (14) | ||
| 10 – <20 | 7 (16) | ||
| PD-HGG (15) | <2 | 2 (13.5) | |
| 2 – <5 | 3 (20) | ||
| 6.74 (0.68–23.97) | 5 – <10 | 5 (33) | |
| 10 – <20 | 3 (20) | ||
| >20 | 2 (13.5) |
Abbreviations: HGG, high-grade glioma; PD, progressive disease; PD-HGG, progressive disease with high-grade glioma at progression
Treatment Details
Our patients underwent surgical resection, chemotherapy, RT, or a combination thereof. The median number of treatments received was two (range, 0–6), and 20 (23%) patients received four or more therapies. Although 12 of 15 patients who had PD-HGG received RT during the course of their illness, four had confirmation of HGG only upon autopsy. Therefore, these four patients may have already progressed to HGG before receiving RT. In the remaining eight patients, RT was administered at variable times before histologic confirmation of HGG. Of the remaining 72 patients, 45 received RT but had no histologically documented HGG.
DISCUSSION
Despite its overall good prognosis, LGG/LGGNT in children can result in mortality, either due to tumor-related causes or complications arising from therapeutic intervention(s).1, 4, 8, 9 In this single-institution retrospective review of all LGGs/LGGNTs treated during a 30-year period, the mortality rate was 12%, with a median time to death from diagnosis of 4.02 years (range, 0.21–24 years). Contrary to a finding in the population-based study of children with LGG by Krishnatry et al,8 death due to treatment-resistant PD was the most common cause of death in our cohort. Chemotherapy regimens in the current study varied, depending on the year of diagnosis. Children diagnosed after the late 1990s were more likely to have received vincristine and carboplatin as the first-line therapy, and those who were treated during the earlier years received the regimen included in institutional clinical trials prevailing at the time of diagnosis. Among those who died of PD, two-thirds had tumors that arose in areas of the CNS considered difficult or improbable to completely resect (e.g., midbrain, thalamus, suprasellar chiasmatic area). Attempted tumor resection in those areas has a high likelihood of permanent neurologic sequelae. This finding mirrors the previous reports of less favorable outcomes of patients with tumors located in the suprasellar chiasmatic, hypothalamic, or other midline locations.8, 12–15
More than half of the patients in our study who died of PD did so within 2 years of diagnosis. Thirty one of the 43 children who died of PD had undergone only a biopsy for histologic confirmation of LGG at diagnosis. Biopsies sometimes miss areas of high-grade tumor due to the limited availability of tissue sample. However, we thought it was necessary to understand the cause of death in patients with biopsy-only evidence of LGG/LGGNT, because in a substantial proportion of patients this is the only way to determine the histology and grade of tumor, as a more extensive resection is not possible due to the tumor’s location. Hence, we included these patients in our study to determine whether their causes of death were similar to those in previous studies of LGG outcome.4, 14 This raises an important concern that clinicians face––tissue samples can miss high-grade areas of the tumor, resulting in the clinician erroneously classifying the tumor as low grade. More importantly, this does not exclude the likelihood that some tumors are true LGG/LGGNTs with inherently aggressive behavior. In a recent study by Ryall et al,15 the investigators examined the prognostic impact of H3K27M and MAPK-pathway mutations in pediatric thalamic glioma. Children with LGG who harbored the H3K27M mutation had a worse prognosis than did those with wild-type H3, and all patients harboring the mutation succumbed to their disease.16 Therefore, it is vital that subjects who die of early progression of LGG/LGGNT not be excluded from the analysis, as the information obtained from those cases will help educate families about possible outcomes. Metabolic imaging [e.g., positron-emission tomography (PET), methionine-PET scans, and MR spectroscopy] are increasingly demonstrating their utility in delineating high-grade areas of tumor. This is proving beneficial to optimizing biopsy specimens and enhancing the diagnostic yield with better delineation of the grade of a tumor.17–19 When available, these approaches should be considered before obtaining a biopsy sample.
Among those in our cohort who died of PD, 16% did so more than 10 years after diagnosis. This contrasts with results from the study by Krishnatry et al, who found no deaths due to PD at more than 5 years from diagnosis and at a median follow-up of 12.73 years (range, 0.02–33.33 years).8 Our study included patients treated at a tertiary referral institution; thus, their risk factors may have differed from those in the population-based study by Krishnatry et al.8 Our results highlight the importance of long-term surveillance of patients with LGG/LGGNT beyond the traditional 5-year period. The natural history of pediatric LGGs includes a slowing and sometimes an arrest of tumor growth with time.3, 20–23 This has been the basis for using less aggressive therapies and deferring RT in children with LGG in the hope of avoiding potential long-term toxicities. However, it is important not to lose sight of the finding that late progression of the disease and mortality are possible, thus necessitating prolonged follow-up of these patients well into adulthood.
Malignant transformation (MT) of LGG to a high-grade tumor is a well-described phenomenon in children and adults.10, 24–27 However, the true incidence of this in children remains to be elucidated. In our study, 15 patients had documented HGGs, either at progression or autopsy. All of them had a previous histologic confirmation of LGG/LGGNT by biopsy (n=9), GTR/NTR (n=3), or STR/PR (n=3). Glioblastoma was the most common finding at progression; it was present in 9/15 cases. Only one patient had cancer predisposition in the form of NF-1. In a recent study by Mistry et al, the incidence of MT was 2.6%,27 and Broniscer et al demonstrated a 6.7% ± 3.9% rate of MT of grade II astrocytoma.10 In our study cohort, we can only hypothesize that among the 15 children with PD-HGG, those with progression at the primary site may have had either a MT or a molecularly high-grade tumor from the outset despite a benign histology, and those with progression at a noncontiguous site had a second malignancy. Due to the lack of molecular data, we are unable to differentiate between these entities with certainty. As demonstrated by Ryall et al,16 the presence of the H3K27M mutation in midline LGGs results in an adverse outcome. This clinical conundrum reinforces the need to obtain molecular data, including the detection of the H3K27M mutation, in tumors arising from the midline and thus are misdiagnosed as a low-grade tumor due to a nonrepresentative tumor specimen. Of note, 12/15 of our study subjects with PD-HGG had a midline tumor.
Among the 15 patients with HGG at progression, 12 had received RT at variable times before the diagnosis of HGG. Four patients had histologic confirmation of HGG only at autopsy. Therefore, some may have progressed to an HGG before administration of RT. It is tempting to infer from these data that the RT may have transformed at least some LGGs. However, we did not intend to study the incidence of MT or second HGG; thus, a larger analysis of patients with LGG who received different treatment modalities is desirable to determine not only the true incidence of MT but also the role of radiation in MT of LGG/LGGNT. MT of a LGG can arise even in the absence of exposure to RT, and additional molecular features of the tumor may cause progression to a more aggressive tumor.4, 10, 24, 28 Recent studies by Bandopadhyaya et al and Krishnatry et al demonstrated increased risk of tumor- and nontumor-related deaths in children with LGG who received RT.2, 8 These findings further reinforce the need for caution using RT in children with LGG/LGGNT and due consideration must be given for using potentially less toxic therapies, like molecularly targeted agents.
In our study, seven deaths (4 suicides and 3 motor vehicle accidents) were seemingly unrelated to the tumor. A deeper interrogation of the reasons underlying these violent deaths revealed anxiety related to the possibility of tumor recurrence in one patient as the reason for a fatal self-inflicted gunshot injury. Although this patient had no evidence of disease recurrence on imaging obtained periodically and was receiving counseling, his fear of being unable to cope with a recurrence drove him to his death. The second suicidal patient had intractable headache refractory to multiple medications. That patient had a ventricular peritoneal shunt for a tectal tumor and did not have any evidence of hydrocephalus as the cause of headache. The reason for suicide was undetermined in the remaining two patients, neither of whom had any evidence of disease on their last surveillance imaging. Brinkman et al found an approximately 12% incidence of suicidal ideation among a group of pediatric brain tumor survivors, the majority of whom were LGG survivors.29 Similarly, Gunnes et al found an increased hazard ratio for suicide in survivors of childhood cancers, including those with CNS tumors.30 A review by Shah et al demonstrated a higher incidence of psychiatric problems in childhood brain tumor survivors than in the general population.31 These studies and the four suicides in our study underscore the importance of early recognition and prompt intervention of any psychological dysfunction in children with LGG/LGGNT.
One of our patients who died in a motor vehicle accident was hit by a drunk driver. We did not have details of the circumstances leading to the deaths of the other two patients. However, one had a history of left-sided superior visual field defects and well-controlled generalized seizures, both of which could potentially result in fatal injuries should the subject be driving a car without appropriate clearance from medical providers. These deaths highlight the importance of multidisciplinary care of these patients to help them reintegrate into society after completing treatment. At St. Jude, patients with limited vision, for example, are sent early to a low-vision clinic and rehabilitation services.32
In the current era of genomic technological advances, characterizing the tumor’s genetic alterations at diagnosis and progression is invaluable not only for understanding the biology of the tumors but also for prognostication and exploring therapeutic options targeting those changes. Recent studies have demonstrated the role of the MAPK pathway in the pathogenesis of pediatric LGG/LGGNT33–37 and molecular changes occurring in childhood HGG.10, 27. Additionally, reports have suggested that LGG/LGGNTs with the KIAA1549–BRAF fusion have a better prognosis in comparison to the fusion negative cases38, 39 and that the presence of BRAFV600E mutation in ganglioglioma is associated with shorter recurrence-free survival in children.40 Therefore, we must identify the patients who are at the greatest risk of PD and dying of their disease, so that effective therapeutic strategies can be formulated. Our retrospective study is limited, however, by the absence of this molecular data.
CONCLUSIONS
Mortality and its causes in children with LGG/LGGNT is an inadequately studied phenomenon in an otherwise extensively researched pediatric brain tumor. Despite the excellent overall survival, a significant number of affected children die, primarily from PD and in some cases, many years after diagnosis. Children with LGG/LGGNT warrant continued (and in a select few, long-term) monitoring and surveillance. Our findings help to better delineate these causes and plan therapeutic interventions, including early psychological evaluation and support for vulnerable children and multidisciplinary care for children with LGG/LGGNT. Molecular characterization of the tumor is emerging as an essential component of the investigative work up of these patients.
Acknowledgments
The authors gratefully acknowledge Dr. David W. Ellison for his critical review of the manuscript and invaluable suggestions and Dr. Angela McArthur for her scientific editing of the manuscript.
Funding: This work was supported by grant CA21765 from the National Institutes of Health and by ALSAC.
Abbreviations
- LGG
Low-grade glioma
- LGGNTs
Low-grade glioneuronal tumors
- HGG
High-grade glioma
- PD
Progressive disease
- PD-HGG
Progressive disease with high-grade glioma at progression
- RT
Radiation therapy
- GTR
Gross total resection
- NTR
Near total resection
- STR
Subtotal resection
- CSI
Cranio-spinal irradiation
- NF-1
Neurofibromatosis type I
- PXA
Pleomorphic xanthoastrocytoma
- PET
Positron-emission tomography
- MT
Malignant transformation
Footnotes
Author contributions: Santhosh A. Upadhyaya and Ibrahim Qaddoumi designed the study, collected data, analyzed the data, and wrote the manuscript. Yahya Ghazwani helped collect the data and reviewed the manuscript. Shengjie Wu analyzed the data and conducted statistical analysis. Alberto Broniscer, Frederick. A. Boop, and Amar Gajjar cared for the patients, analyzed the data, and reviewed the manuscript.
Conflict of interest: The authors do not have any conflicts of interest to disclose.
References
- 1.Gajjar A, Sanford RA, Heideman R, et al. Low-grade astrocytoma: a decade of experience at St. Jude Children’s Research Hospital. J Clin Oncol. 1997;15:2792–2799. doi: 10.1200/JCO.1997.15.8.2792. [DOI] [PubMed] [Google Scholar]
- 2.Bandopadhayay P, Bergthold G, London WB, et al. Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: an analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr Blood Cancer. 2014;61:1173–1179. doi: 10.1002/pbc.24958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong GT, Conklin HM, Huang S, et al. Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro Oncol. 2011;13:223–234. doi: 10.1093/neuonc/noq178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gnekow AK, Falkenstein F, von Hornstein S, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. Neuro Oncol. 2012;14:1265–1284. doi: 10.1093/neuonc/nos202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh KS, Hung J, Robertson PL, et al. Outcomes of multidisciplinary management in pediatric low-grade gliomas. Int J Radiat Oncol Biol Phys. 2011;81:e481–488. doi: 10.1016/j.ijrobp.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 6.Perilongo G. Considerations on the role of chemotherapy and modern radiotherapy in the treatment of childhood low grade glioma. J Neurooncol. 2005;75:301–307. doi: 10.1007/s11060-005-6754-8. [DOI] [PubMed] [Google Scholar]
- 7.Chalil A, Ramaswamy V. Low Grade Gliomas in Children. J Child Neurol. 2016;31:517–522. doi: 10.1177/0883073815599259. [DOI] [PubMed] [Google Scholar]
- 8.Krishnatry R, Zhukova N, Guerreiro Stucklin AS, et al. Clinical and treatment factors determining long-term outcomes for adult survivors of childhood low-grade glioma: A population-based study. Cancer. 2016;122:1261–1269. doi: 10.1002/cncr.29907. [DOI] [PubMed] [Google Scholar]
- 9.Rakotonjanahary J, De Carli E, Delion M, et al. Mortality in Children with Optic Pathway Glioma Treated with Up-Front BB-SFOP Chemotherapy. PLoS One. 2015;10:e0127676. doi: 10.1371/journal.pone.0127676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broniscer A, Baker SJ, West AN, et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol. 2007;25:682–689. doi: 10.1200/JCO.2006.06.8213. [DOI] [PubMed] [Google Scholar]
- 11.Chamdine O, Broniscer A, Wu S, Gajjar A, Qaddoumi I. Metastatic Low-Grade Gliomas in Children: 20 Years’ Experience at St. Jude Children’s Research Hospital. Pediatr Blood Cancer. 2016;63:62–70. doi: 10.1002/pbc.25731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters O, Gnekow AK, Rating D, Wolff JE. Impact of location on outcome in children with low-grade oligodendroglioma. Pediatr Blood Cancer. 2004;43:250–256. doi: 10.1002/pbc.20111. [DOI] [PubMed] [Google Scholar]
- 13.Stokland T, Liu JF, Ironside JW, et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702) Neuro Oncol. 2010;12:1257–1268. doi: 10.1093/neuonc/noq092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ater JL, Zhou T, Holmes E, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the Children’s Oncology Group. J Clin Oncol. 2012;30:2641–2647. doi: 10.1200/JCO.2011.36.6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broadway SJ, Ogg RJ, Scoggins MA, Sanford R, Patay Z, Boop FA. Surgical management of tumors producing the thalamopeduncular syndrome of childhood. Journal of neurosurgery Pediatrics. 2011;7:589–595. doi: 10.3171/2011.4.PEDS119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryall S, Krishnatry R, Arnoldo A, et al. Targeted detection of genetic alterations reveal the prognostic impact of H3K27M and MAPK pathway aberrations in paediatric thalamic glioma. Acta Neuropathol Commun. 2016;4:93. doi: 10.1186/s40478-016-0353-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guzman-De-Villoria JA, Mateos-Perez JM, Fernandez-Garcia P, Castro E, Desco M. Added value of advanced over conventional magnetic resonance imaging in grading gliomas and other primary brain tumors. Cancer imaging : the official publication of the International Cancer Imaging Society. 2014;14:35. doi: 10.1186/s40644-014-0035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Messing-Junger AM, Floeth FW, Pauleit D, et al. Multimodal target point assessment for stereotactic biopsy in children with diffuse bithalamic astrocytomas. Childs Nerv Syst. 2002;18:445–449. doi: 10.1007/s00381-002-0644-6. [DOI] [PubMed] [Google Scholar]
- 19.Morana G, Piccardo A, Milanaccio C, et al. Value of 18F-3,4-dihydroxyphenylalanine PET/MR image fusion in pediatric supratentorial infiltrative astrocytomas: a prospective pilot study. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2014;55:718–723. doi: 10.2967/jnumed.113.125500. [DOI] [PubMed] [Google Scholar]
- 20.Gunny RS, Hayward RD, Phipps KP, Harding BN, Saunders DE. Spontaneous regression of residual low-grade cerebellar pilocytic astrocytomas in children. Pediatr Radiol. 2005;35:1086–1091. doi: 10.1007/s00247-005-1546-z. [DOI] [PubMed] [Google Scholar]
- 21.Loh JK, Lieu AS, Chai CY, et al. Arrested growth and spontaneous tumor regression of partially resected low-grade cerebellar astrocytomas in children. Childs Nerv Syst. 2013;29:2051–2055. doi: 10.1007/s00381-013-2113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ogiwara H, Bowman RM, Tomita T. Long-term follow-up of pediatric benign cerebellar astrocytomas. Neurosurgery. 2012;70:40–47. doi: 10.1227/NEU.0b013e31822ff0ed. discussion 47–48. [DOI] [PubMed] [Google Scholar]
- 23.Tabori U, Vukovic B, Zielenska M, et al. The role of telomere maintenance in the spontaneous growth arrest of pediatric low-grade gliomas. Neoplasia. 2006;8:136–142. doi: 10.1593/neo.05715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krieger MD, Gonzalez-Gomez I, Levy ML, McComb JG. Recurrence patterns and anaplastic change in a long-term study of pilocytic astrocytomas. Pediatr Neurosurg. 1997;27:1–11. doi: 10.1159/000121218. [DOI] [PubMed] [Google Scholar]
- 25.Marton E, Feletti A, Orvieto E, Longatti P. Malignant progression in pleomorphic xanthoastrocytoma: personal experience and review of the literature. J Neurol Sci. 2007;252:144–153. doi: 10.1016/j.jns.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 26.McCormack BM, Miller DC, Budzilovich GN, Voorhees GJ, Ransohoff J. Treatment and survival of low-grade astrocytoma in adults–1977–1988. Neurosurgery. 1992;31:636–642. doi: 10.1227/00006123-199210000-00004. discussion 642. [DOI] [PubMed] [Google Scholar]
- 27.Mistry M, Zhukova N, Merico D, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol. 2015;33:1015–1022. doi: 10.1200/JCO.2014.58.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rumana CS, Valadka AB. Radiation therapy and malignant degeneration of benign supratentorial gangliogliomas. Neurosurgery. 1998;42:1038–1043. doi: 10.1097/00006123-199805000-00049. [DOI] [PubMed] [Google Scholar]
- 29.Brinkman TM, Liptak CC, Delaney BL, Chordas CA, Muriel AC, Manley PE. Suicide ideation in pediatric and adult survivors of childhood brain tumors. J Neurooncol. 2013;113:425–432. doi: 10.1007/s11060-013-1130-6. [DOI] [PubMed] [Google Scholar]
- 30.Gunnes MW, Lie RT, Bjorge T, et al. Suicide and violent deaths in survivors of cancer in childhood, adolescence and young adulthood-A national cohort study. International journal of cancer. 2016 doi: 10.1002/ijc.30474. [DOI] [PubMed] [Google Scholar]
- 31.Shah SS, Dellarole A, Peterson EC, et al. Long-term psychiatric outcomes in pediatric brain tumor survivors. Childs Nerv Syst. 2015;31:653–663. doi: 10.1007/s00381-015-2669-7. [DOI] [PubMed] [Google Scholar]
- 32.Sparrow J, Brennan R, Mao S, et al. Participation in an occupational therapy referral program for children with retinoblastoma. J Pediatr Rehabil Med. 2016;9:117–124. doi: 10.3233/PRM-160372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cin H, Meyer C, Herr R, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol. 2011;121:763–774. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- 34.Jones DT, Gronych J, Lichter P, Witt O, Pfister SM. MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci. 2012;69:1799–1811. doi: 10.1007/s00018-011-0898-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Penman CL, Faulkner C, Lowis SP, Kurian KM. Current Understanding of BRAF Alterations in Diagnosis, Prognosis, and Therapeutic Targeting in Pediatric Low-Grade Gliomas. Front Oncol. 2015;5:54. doi: 10.3389/fonc.2015.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qaddoumi I, Orisme W, Wen J, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131:833–845. doi: 10.1007/s00401-016-1539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45:602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Becker AP, Scapulatempo-Neto C, Carloni AC, et al. KIAA1549: BRAF Gene Fusion and FGFR1 Hotspot Mutations Are Prognostic Factors in Pilocytic Astrocytomas. J Neuropathol Exp Neurol. 2015;74:743–754. doi: 10.1097/NEN.0000000000000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hawkins C, Walker E, Mohamed N, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res. 2011;17:4790–4798. doi: 10.1158/1078-0432.CCR-11-0034. [DOI] [PubMed] [Google Scholar]
- 40.Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol. 2013;125:901–910. doi: 10.1007/s00401-013-1120-y. [DOI] [PubMed] [Google Scholar]