

Abstract
BACKGROUND
Valproic acid (VPA) has demonstrated beneficial effects in preclinical models of cancer, neurologic diseases, and traumatic injuries. The purpose of this trial (Clinicaltrials.gov NCT0195156) was to characterize the single-dose plasma pharmacokinetics of 15 to 150 mg/kg of intravenous VPA, and to determine the maximum tolerated dose (MTD) of intravenous VPA above the previously established MTD of 60 mg/kg.
METHODS
Healthy male and female subjects aged 18–65 and with BMI between 18 kg/m2 and 30 kg/m2 were enrolled into this double-blinded, placebo-controlled, dose-escalation trial. Subjects were randomized 3:1 to intravenous VPA or placebo infused over an hour. Serial plasma samples over 72 hours were assayed by liquid chromatography tandem mass spectrometry and analyzed by non-compartmental and population pharmacokinetic methods. The final model was used to derive individual Bayesian estimates of exposure and classification and regression tree analyses were used to identify Cmax and AUC breakpoints associated with adverse events. Safety monitoring included a thorough assessment of laboratory tests, hemodynamic parameters, cardiac rhythm monitoring, and cognitive testing.
RESULTS
Fifty-nine healthy subjects (mean: 30±12 years) were enrolled, and 44 received VPA. The MTD was 140 mg/kg with a mean (SD) Cmax of 1271 (178) mg/L compared to 248 (25.4) mg/L at the 60 mg/kg dose. A two-compartment model without system parameter covariates adequately characterized VPA pharmacokinetics. The number of adverse events correlated with the dosage (R2= 0.56). Headache and nausea lasting greater than 12 hours were the key dose limiting toxicities. Transient hypoacusis (n=19), chills (n=18), and headaches (n=16) were common with Cmax and AUC breakpoints of >276 mg/L and >3873 h•mg/L predictive of transient hypoacusis.
CONCLUSIONS
The MTD of intravenous VPA is 140 mg/kg in healthy subjects. These results support further evaluation of high dose VPA in trauma patients with life-threatening injuries.
Keywords: Valproic acid, Valproate, Trauma, Cancer, Brain Injury, Maximum Tolerated Dose
I. INTRODUCTION
The FDA originally approved valproic acid (VPA) in 1978 for the treatment of epilepsy, an indication for which it is routinely used today. Evidence supports multiple mechanisms of action that may explain VPA’s antiepileptic effects, including gamma-Aminobutyric acid (GABA)ergic potentiation, glutamate and N-methyl-D-aspartate receptor (NMDA) inhibition, and blockage of voltage-gated sodium channels [1]. More recently, VPA has been identified as a non-selective histone deacetylase (HDAC) inhibitor [2]. Acetylation and deacetylation of histone proteins plays a central role in the regulation of gene expression. By increasing histone protein acetylation, HDAC inhibitors relax chromatin conformation and enhance transcriptional activity [3]. HDAC inhibitors have also been shown to increase acetylation of non-histone proteins, the regulatory scope of which has been compared to other major posttranslational modifications like phosphorylation [4].
As the scientific community’s understanding of acetylation expands, agents that modify the acetylome have garnered more attention. Intensive research is now being conducted in the fields of oncology, neurology, and rheumatology, where VPA and other HDAC inhibitors have shown promise treating conditions like leukemia, multiple sclerosis, and arthritis [5–7]. Our group has focused on the use of VPA in the setting of traumatic injuries, where preclinical studies have demonstrated improved outcomes both in-vitro and in-vivo [8]. The use of VPA to treat injury is a novel concept and represents an opportunity to improve outcomes in trauma, which is the number one cause of death and disability in people under age 46 in the United States [9]. In this population, hemorrhage has been identified as the leading cause of preventable death. Our ability to treat hemorrhage, however, is limited, and requires rapid access to surgical care and blood products [10–12]. In large animal models of traumatic injury and hemorrhage, we have shown that intravenously administered VPA can serve as a bridge to definitive care. In swine subjected to hemorrhage and polytrauma, treatment with VPA decreased mortality by 50% and did so in the absence of isotonic fluid resuscitation [13, 14]. Improved outcomes have also been validated in more complex models of injury, including those combining hemorrhagic shock with sepsis [15–17] or traumatic brain injury (TBI) [18–20].
In preclinical studies of traumatic injury, intravenous VPA has been administered as a single dose ranging from 150 to 400 mg/kg administered over a period of 90 to 180 minutes. Based on simple allometry, the estimated human equivalent dose is 140 to 360 mg/kg. This is significantly higher than previously established maximum tolerated single intravenous doses of 60 and 75 mg/kg [21, 22]. Given these differences in dosing, we conducted an FDA approved, phase 1, single dose, double-blind, placebo-controlled trial to determine the safety and tolerability of intravenous VPA in healthy human subjects (Clinicaltrials.gov NCT0195156). Data generated from pharmacokinetic, pharmacodynamic, and safety profile testing was used to determine the maximum tolerated dose of VPA for use in a phase Ib trial studying patients in hemorrhagic shock. This is the first study to evaluate high dose intravenous VPA in healthy adult volunteers, and provides safety and pharmacokinetic insights that will serve to benefit investigators seeking to evaluate this compound for a broad range of indications.
II. METHODS
a. Patients
Healthy male and female subjects aged 18–65 and with BMI between 18 kg/m2 and 30 kg/m2 were eligible for the study. Medical history, physical examination, electrocardiogram (ECG), and routine laboratory tests (blood chemistry, hematology, and urinalysis) in addition to hepatitis B and C and HIV screening were performed prior enrollment to rule out any clinically significant medical conditions. Female subjects were required to be surgically sterilized or postmenopausal. Pregnancy was ruled out with a serum pregnancy test. Major exclusion criteria included: use of prescription or non-prescription drugs, herbs, or dietary supplements within 14 days of the first dose of study medication; abstinence of caffeine from Day -1 to Day 4; any history of Valproic Acid use; current alcohol, tobacco, or illicit drug use or past alcohol or illicit drug abuse; hypertension (sitting blood pressure > 140 (systolic) or > 90 (diastolic) on two evaluations at least 10 minutes apart); vaccination, treatment with an investigational drug, or blood donation within 30 days of the first dose of the study medication; febrile illness within 5 days of the first dose of the study medication; inadequate venous access; or inability to be confined to the clinical research facility as required by the protocol. For a detailed explanation of eligibility criteria please see the complete study protocol in the supplemental material.
b. Study design
VPA was studied using a double-blinded, placebo-controlled, single dose-escalation trial design. In each 8-person cohort, subjects were randomized 3:1 to receive one dose of intravenous VPA or placebo infused over a period of 60 minutes. Patients were monitored for adverse events and blood was drawn at timed intervals to evaluate VPA’s pharmacokinetic (PK), pharmacodynamic (PD), and safety profile. VPA dosing was based on manufacturer’s recommendations and previous clinical trials. The formulation of VPA used was Depacon®, the sodium salt of valproic acid (sodium 2-propylpentanoate; Abbvie, Chicago, Illinois). Per manufacturer’s recommendations, the starting dose of VPA for the treatment of seizures is 15 mg/kg/day and the maximum dose is 60 mg/kg/day. A dose escalation trial in which cancer patients received a single dose of intravenous VPA daily for five days found the maximum tolerated dose to be 60 mg/kg/day [21]. The starting dose for this single administration study in healthy subjects was 15 mg/kg, with subsequent doses increasing to 30, 60, 90, 120, 130, 140 and 150 mg/kg. VPA (100 mg/mL) was diluted with saline to a total volume of 300 mL and prepared within 24 hours of study drug administration. Placebo consisted of normal saline and was identical in appearance to the study drug. All patients, clinicians, and data collectors were blinded to subject assignments for the duration of the study.
Toxicity was monitored and documented using the Medical Dictionary for Regulatory Activities [23]. Adverse events were categorized as mild, moderate, or severe based on predefined definitions and were determined based on subject symptoms, physical exam findings, vital signs, clinical laboratory parameters, ECG, and Abbreviated Mental Test [24] and Mini Mental State Exam [25] scores. Complete definitions of mild, moderate, and severe adverse events are described in the complete study protocol (supplemental material). Briefly, a moderate adverse event was defined as an event of sufficient severity to require more than one dose of treatment to alleviate symptoms and/or an event that takes longer than 12 hours to resolve. Additional physiologic and biochemical criteria are included in the study protocol. A serious adverse event was defined as an event that results in death, life threatening illness, hospitalization, persistent or significant disability, or a congenital anomaly. Study investigators determined the relationship of adverse events to VPA, which were definitely, probably, possibly, unlikely, or definitely not related to the study drug. Dose limiting toxicities (DLT) were defined as any definite, probable, or possible drug-related moderate or higher adverse event. Dose escalation occurred when less than two subjects in any group experienced DLTs. The study’s SRC and independent DSMB approved each dose escalation. Depending on the results of preceding dose groups VPA dosing could be adjusted in the event that a DLT was observed in two or more subjects in any cohort. This included the opportunity for dose de-escalation. Changes to study drug dosing required approval by the DSMB.
Subjects were admitted to the clinical research unit for 5 days (1 day prior to drug administration and 4 days after) and returned for a final follow-up 14±2 days after discharge. While admitted, vital signs and physical examination were performed at regular intervals. Cognitive testing using the AMT and the MMSE were performed immediately before drug administration and at 1, 4, 12, and 24 hours after. A score of <7 on the AMT and <26 on the MMSE were considered moderate adverse events. PK and clinical safety labs, including complete blood count, comprehensive metabolic panel, magnesium, phosphorus, lipase, amylase, prothrombin time, and partial thromboplastin time, were drawn immediately before drug infusion and at 55 minutes, 2, 4, 8, 16, 24 36, 60, and 72 hours after the initiation of drug infusion. PD labs were drawn immediately before drug infusion and at 2, 4, 8, 16, 24, 48, and 72 hours after the initiation of drug infusion. PD evaluation was specific to VPA’s mechanism of action as an HDAC inhibitor and included determination of histone 3 acetylation using Western Blot and evaluation of differentially expressed proteins in peripheral blood mononuclear cells using protein mass spectrometry. Initial PD results are published separately [26].
c. Bioanalysis
Briefly, plasma samples were assayed for VPA using a liquid chromatography mass spectrometry method (LC-MS). An ABI-3200 Qtrap mass spectrometer with electrospray ionization probe was interfaced with an Agilent 1200 series high performance LC (HPLC) system for sample analysis. The Analyst Software Version 1.4.2 package supplied by Applied Bio-systems (MDS SCIEX) was used to control the LC–MS/MS system, as well as for data acquisition and processing. For complete details regarding plasma analysis please see the supplemental material.
d. Pharmacokinetic Analyses
The PK data analyses were performed in three stages, including exploratory analyses of concentration-time profiles, non-compartmental analyses, and population pharmacokinetic analyses. Concentration-time profiles were visualized using STATA/IC version 14 (Stata Corp, College Station, Texas, USA). Non-compartmental analysis was performed using Phoenix/WINNONLIN 6.3 (Pharsight, St. Louis, Missouri, USA) to derive initial PK system parameter estimates for population analyses. Population pharmacokinetic analyses were performed using PMetrics® version 1.5.0 (LAPK, Los Angeles, California, USA) with an iterated two-stage Bayesian algorithm evoked through R. One, two, and three compartment linear models with zero-order rates of input were tested. This was followed by non-linear model such as Michaelis-Menten and a parallel linear/MM model. Model discrimination was performed based on goodness of fit plots of the population observed versus predicted, individual observed versus predicted, comparisons of inter-individual variability, and ε-shrinkage. The Cmax was the plasma concentration at the end of a 1-hour infusion. The final model was utilized to generate empiric Bayesian estimates of system parameters, AUC24, AUCinf, and C24.
e. Pharmacodynamic Safety Analyses
Assessment of exposure-safety relationships were based on repeated measured values and subject-reported adverse events. The variables analyzed included vital signs (heart rate, temperature, blood pressure, and respiratory rate), laboratory parameters (hematology, chemistry), organ specific laboratory measures (liver transaminases, pancreatic enzymes), and coagulation markers over time. A longitudinal approach using multi-level regression analysis was used to compare the effects of dose-level on temporal changes in continuous measured values. The subject-reported adverse events were binary coded in to six groups that included presence or absence of neurological changes, hearing changes, visual changes, nausea, vomiting, and chills. The effect of VPA dose on these adverse events was tested using Fisher’s exact test to identify significant relationships (p<0.05). Classification and auto regression tree analysis were used to identify breakpoints for dose (mg/kg), dose (mg), Cmax, and AUCinf associated with subject-reported adverse events. All statistical analyses were executed using STATA/IC version 14 (Stata Corp, College Station, Texas, USA).
f. Study approval
The study was approved by the University of Michigan IRBMED and conducted under a FDA investigational new drug application (IND 113,010). Participants gave written informed consent prior to inclusion in the study. The trial was registered on ClinicalTrials.gov (NCT0195156).
III. RESULTS
a. Safety
Fifty-nine healthy subjects (5 female, mean age 30.2 ± 11.7 years, range 18–60 years old) were enrolled for a single, predefined dose of VPA (Figure 1). Subjects were Caucasian (n = 47, 80%), African-American (n =10, 17%), and Asian (n = 2, 3%). Forty-four subjects (75%) received VPA and 15 received placebo (25%). No dose-limiting toxicities (DLTs) or serious adverse events (AEs) were observed in subjects enrolled in dose cohorts up to 120 mg/kg (15, 30, 60, 90, 120 mg/kg). Two of three subjects in the 150 mg/kg dose cohort experienced at least one moderate adverse event. These included headache and nausea lasting greater than 12 hours that did not resolve following administration of acetaminophen or ondasetron, respectively. Both subjects’ symptoms resolved without further intervention and within 24 hours of VPA infusion.
Figure 1.
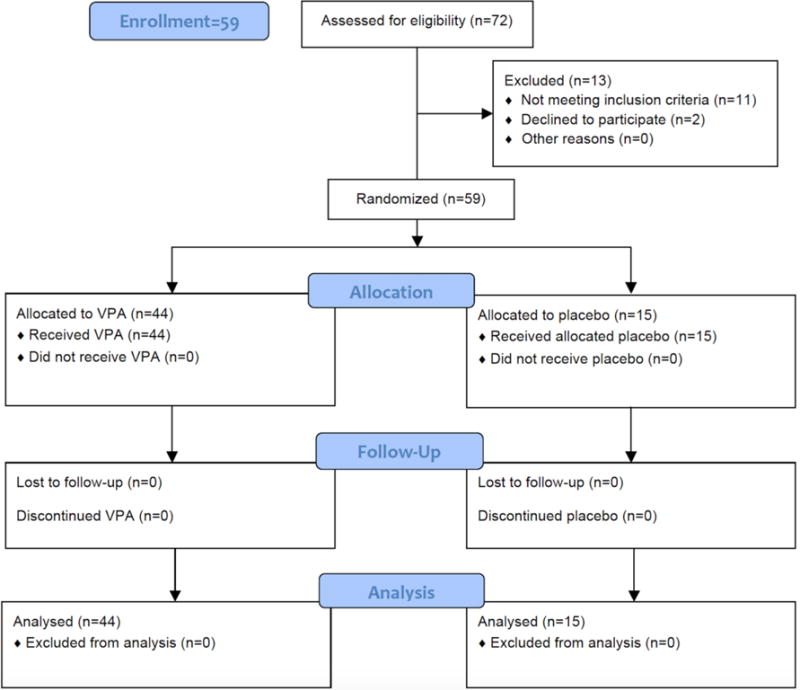
Screening and enrollment flow chart
Given these findings, an FDA and IRB approved amendment was made to the protocol to allow for de-escalation of dosing to better determine the maximum tolerated dose (MTD) of VPA. With the guidance of the Safety Review Committee (SRC) and Data and Safety Monitoring Board (DSMB), doses of 130 and 140 mg/kg were evaluated. Ultimately, the MTD single intravenous dose of VPA was defined as 140 mg/kg. A total of 127 AEs were observed in all cohorts and are summarized in Table 1. The most common AEs included hypoacusis (n = 19 subjects), chills (n = 18), headache (n = 16), tinnitus (n = 15), and nausea (n = 10), all of which were determined “likely” related to the study drug. Overall, 43 of 59 subjects (73%) experienced at least one adverse event. Many subjects developed more than one adverse event. No significant drug-related abnormalities were seen in other safety measures, including clinical safety labs (complete blood count, comprehensive metabolic panel, magnesium, phosphorus, lipase, amylase, prothrombin time, and partial thromboplastin time), ECG parameters, and cognitive testing (Abbreviated Mental Test and Mini Mental State Exam). There was no evidence of complications related to VPA’s Black Box warnings (hepatotoxicity, pancreatitis, and teratogenicity). All subjects completed the study procedures per protocol guidelines (supplemental material).
Table 1.
Number of subject-reported adverse events by dosing cohorts, grouped by category
| Category | Neurological | Hearing | Visual | Gastrointestinal | Chills | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Dose (mg/kg) | Headache | Feeling abnormal | Fatigue | Dizziness | Hypoacusis | Tinnitus | Blurred vision | Nausea | Vomiting | Chills |
| 15 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 30 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 60 | 1 | 0 | 0 | 0 | 1 | 2 | 0 | 1 | 1 | 0 |
| 90 | 2 | 1 | 1 | 0 | 3 | 0 | 1 | 1 | 0 | 2 |
| 120 | 3 | 1 | 2 | 2 | 3 | 4 | 2 | 2 | 1 | 2 |
| 130 | 3 | 2 | 4 | 1 | 4 | 4 | 1 | 3 | 1 | 6 |
| 140 | 5 | 0 | 2 | 3 | 6 | 4 | 2 | 2 | 1 | 7 |
| 150 | 2 | 0 | 0 | 2 | 2 | 1 | 0 | 1 | 1 | 1 |
|
| ||||||||||
| Total | 16 | 4 | 9 | 8 | 19 | 15 | 6 | 10 | 5 | 18 |
Data are presented as number of self-reported adverse events (AE) in each dose category. Many subjects reported more than one AE
b. Pharmacokinetic Analysis
The mean concentration-time profiles are illustrated in Figure 2 based on dose-level. The 140 mg/kg dose cohort had the highest concentrations with a mean maximum plasma concentration (Cmax) of 1,271 mg/L. Evaluation of the logarithm transformed concentration-time data revealed that the profile declined in a biphasic manner. Consistent with this observation, a two-compartment model was identified as the optimal model predictive of VPA concentration-time profiles using lowest Akaike’s information criterion (AIC). Comparisons of the observed versus predicted plots for the population and individual model are illustrated in the supplemental material, which shows the two-compartment model to be optimal (Figure S1). A summary of system parameters generated using alternate linear PK models is detailed in Table 2. The mean volume of the central compartment (Vc) was estimated to be 11 L with a mean VPA clearance (CL) of 1.16 L/h. A summary of the final model generated by individual estimates of systemic exposure is provided in Table 3. As shown, trends toward shortening half-life were observed at higher dosing levels based on a mg/kg basis. Non-linear models of clearance (CL) were tested but did not improve the model fit relative to the two-compartment linear model. Differences in exposure by dosing group could, however, be explained by actual differences in body weight in dosing groups. For example, the concentrations observed in the 150 mg/kg group were lower than the 140 mg/kg group due to a lower median body weight of 64 kg and 78 kg, respectively, leading to lower absolute doses. Figure 3 illustrates good relationships between the observed AUCinf, Cmax values, and absolute doses relative to dose (based on weight). Consistent with these findings, the inter-individual variability in system parameters such as CL and Vc were not explained by body weight alone (Figure S2). Regression of AUCinf to VPA absolute dose (Figures 2b) shows a proportionate relationship (slope = 0.927, near unity with no constant), whereas the administered dose nearly approximates the AUCinf. Similarly, regression of Cmax to VPA absolute dose (Figures 2d) shows a proportionate relationship (slope = 0.0847, with no constant) whereas one eleventh of the administered dose (consistent with Vc estimate) approximates the Cmax.
Figure 2.
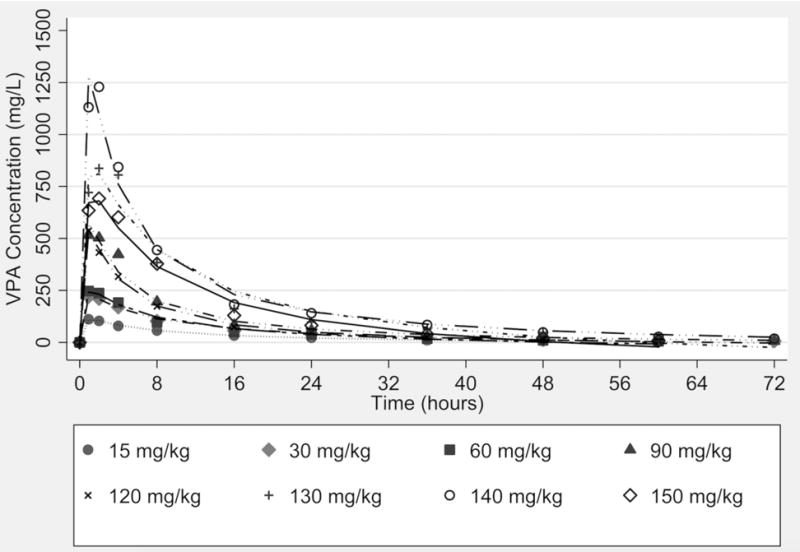
Observed mean concentration-time profile of valproic acid (VPA) by dose administration group.
Table 2.
Pharmacokinetic system parameters based on 1-, 2-, and 3-compartment models
| Model | CL (L/h) | Vc (L) | Q1 (L/h) | Vp1 (L) | Q2 (L/h) | Vp2 (L) | AIC |
|---|---|---|---|---|---|---|---|
| 1-Compartment | 1.25 (0.51) | 19.5 (5.57) | NA | NA | NA | NA | 94.85 |
| 2-Compartment | 1.16 (0.48) | 11.1 (2.62) | 0.64 (0.32) | 7.70 (3.28) | NA | NA | 91.03 |
| 3-Compartment | 1.16 (0.49) | 10.8 (2.74) | 0.62 (0.29) | 7.63 (3.29) | 3.48(1.47) | 0.32(0.06) | 96.14 |
CL, clearance; Vc, volume of distribution of the central compartment; Q1, intercompartmental clearance between central and first peripheral compartment; Vp1, volume of distribution of the first peripheral compartment; Q2, intercompartmental clearance between central and second peripheral compartment; Vp2, volume of distribution of the second peripheral compartment; NA, not applicable. Values reported as mean (standard deviation).
Table 3.
Weight, absolute dosage, and exposure parameters of dosing cohorts
| Dose (mg/kg) | Weight (kg) | Dose (mg) | Cmax (mg/L) | C24 (mg/L) | Half-life (h) | AUC24 (h•mg/L) | AUCinf (h•mg/L) |
|---|---|---|---|---|---|---|---|
| 15 | 78.8 (14.3) | 1182 (214) | 112 (11.8) | 22.6 (5.64) | 12.1 (1.8) | 1184 (152) | 1714 (340) |
| 30 | 71.7 (9.6) | 2151 (288) | 235 (21.1) | 39.5 (11.8) | 12.1 (3.4) | 2205 (311) | 3386 (1033) |
| 60 | 66.8 (6.9) | 4009 (412) | 248 (25.4) | 39.8 (2.83) | 11.3 (2.1) | 2241 (174) | 3043 (377) |
| 90 | 77.4 (14.7) | 6966 (1322) | 541 (62.3) | 56.9 (25.4) | 8.9 (1.5) | 4626 (1044) | 5812 (1424) |
| 120 | 74.9 (12.2) | 8990 (1467) | 537 (70.7) | 49.7 (19.7) | 9.0 (2.1) | 3843 (723) | 4983 (1332) |
| 130 | 82.1 (9.5) | 10667 (1231) | 855 (90.1) | 97.2 (11.9) | 8.8 (2.3) | 7745 (1404) | 11273 (1658) |
| 140 | 77.9 (6.8) | 10899 (955) | 1271 (178) | 140 (19.3) | 9.7 (1.0) | 9870 (898) | 13520 (1621) |
| 150 | 64.2 (4.4) | 9630 (658) | 694 (62.9) | 81.4 (34.7) | 7.9 (3.7) | 6911 (571) | 8521 (226) |
Values reported as mean (standard deviation).
Figure 3. AUCinf and Cmax of weight-based and absolute doses of VPA.
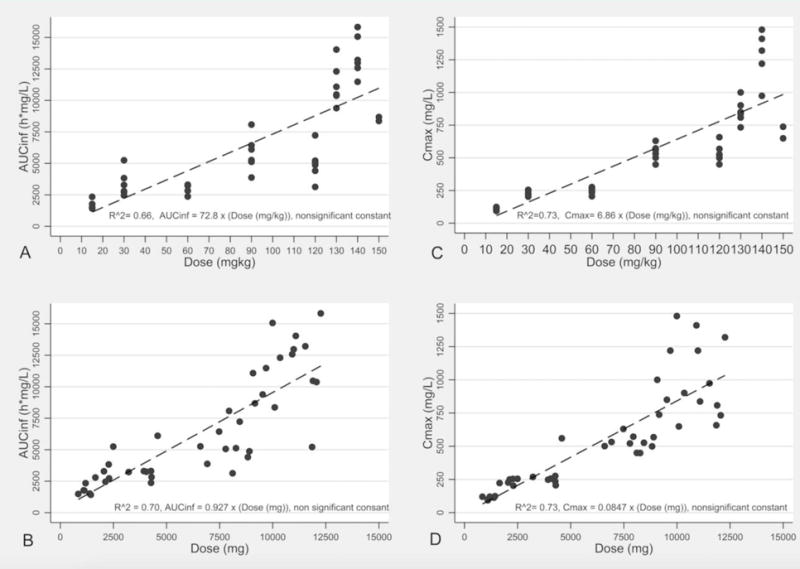
Scatter and linear regression fit plot (with equation) of the relationship between area under the concentration-time curve from time zero to infinity (AUCinf) over weight-based dose (A) and absolute dose (B), and the maximum concentration (Cmax) over weight-based dose (C) and absolute dose (D).
c. Pharmacodynamic Safety Analysis
Comparisons of continuous measured variables over time demonstrated no significant relationship to dose-level except for heart rate. A transient elevation in the heart rate was observed near the end of VPA infusion (Figure S3) and a significant correlation (p < 0.001, R2 = 0.35) was observed between tachycardia (≥100 beats per minute, BPM) and the dose (Figure S4). The predicted change was approximately one BPM per 1000 mg of VPA above the predicted 10 BPM increase associated with the placebo. A strong correlation (R2 = 0.56) was also observed between the number of subject-reported adverse events and the dose-level (Figure 4). A summary of the breakpoint values for Cmax and AUCinf and the doses associated with these adverse events are provided in Table 4. As demonstrated, the risk for selected self-reported adverse events is dose/exposure dependent.
Figure 4. Adverse events and VPA dose.
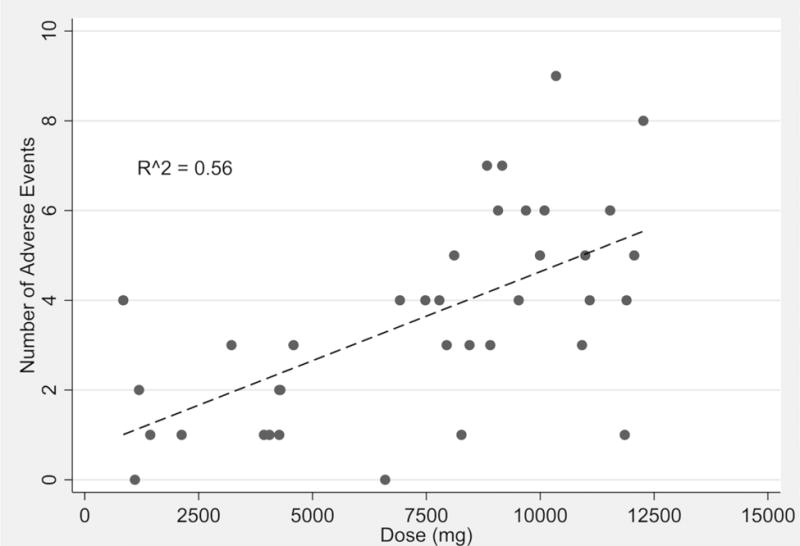
Scatter and linear regression fit plot of the number of subject-reported adverse events over VPA dose (mg).
Table 4.
Breakpoints identified for the occurrence of adverse events.
| Adverse Events | Cmax | AUCinf | Dose |
|---|---|---|---|
| Neurological | >256 mg/L (1.54) | NA | NA |
| Hearing | >276 mg/L (1.79) | >3873 h mg/L (1.77) | >3222 mg (1.70) |
| Chills | >738 mg/L (3.35) | >8680 h mg/L (3.35) | >6597 mg (2.24) |
Cmax, maximum concentration; AUCinf, area under the concentration time from zero to infinity. Predicted relative hazard ratio (RHR) is listed in parenthesis.
d. Pharmacodynamic Effect Analysis
Pharmacodynamic evaluation was specific to VPA’s mechanism of action as an HDAC inhibitor and included determination of histone 3 acetylation using Western Blot and protein mass spectrometry. Protein mass spectrometry was also used to evaluate differentially expressed proteins in peripheral blood mononuclear cells. These findings are published separately [26].
IV. DISCUSSION
We have demonstrated that the maximum tolerated single intravenous dose of VPA in healthy humans is 140 mg/kg when infused over a 1-hour period. This is significantly higher than both the maximum recommended oral dose (60 mg/kg/day) and previously established maximum intravenous dose (75 mg) [22]. Adverse events were mild and no drug-related abnormalities were seen in other safety measures including clinical labs, ECG, and cognitive testing. Pharmacokinetic analysis demonstrated a two-compartment model predictive of VPA concentration-time profiles, with a strong correlation observed between the number of reported adverse events and the dose-level.
In this dose escalation trial involving healthy humans AEs related to VPA administration were mild. The most common side effects associated with treatment included hypoacusis, chills, headache, tinnitus, and nausea. Subjects tolerated these symptoms well, with all events resolving within 12 hours of infusion. The DLTs experienced by two patients in the 150 mg/kg cohort included headache lasting greater than 12 hours that was not relieved with acetaminophen and nausea lasting greater than 12 hours that was not relieved with odansetron. For both subjects, symptoms resolved without further intervention within 24 hours of the infusion. These findings suggest that in the setting of cancer, traumatic injury, or other conditions in which VPA is utilized, the mild side effects associated with high dose intravenous VPA are manageable. While neurovestibular side effects were prevalent, there was no significant difference between subjects’ scores on the Abbreviated Mental Test and Mini Mental State Exam before and after VPA administration. Clinical safety labs were also unremarkable. Given black box warnings for hepatoxicity and pancreatitis, data related to organ function was collected. Liver function tests, amylase, and lipase were unchanged following a single dose of VPA, and no patients developed clinical evidence of organ injury. Vital signs were also monitored closely but were largely unaffected by VPA administration. The exception was mildly elevated heart rate, which is discussed above.
To date, two previous trials have evaluated high dose intravenous VPA in human subjects. In patients with progressing solid tumors, Atmaca et al [21] established 60 mg/kg/day to be the MTD of VPA. Daily doses of VPA were divided in half and administered intravenously twice daily over a one-hour period for 5 days. In another trial, Munster et al [22] studied VPA in combination with epirubicin treatment for patients with advanced solid tumor malignancies. In the original study design, intravenous VPA was used as a loading dose before treatment with 5 oral doses every 12 hours. Due to DLTs in patients receiving loading doses of 60 mg/kg or 75 mg/kg of intravenous VPA, the intravenous loading dose was changed to an oral administered dose. Once the transition was made to completely oral dosing the MTD of VPA was determined to be 140 mg/kg [22]. DLTs in these studies included somnolence, confusion, and dizziness – findings that were also identified in our study population. In contrast to our study, subjects in these trials also developed hematologic disturbances (i.e. neutropenia), physiologic changes (i.e. fevers), and ECG changes (i.e. QTc prolongation), which we did not identify in healthy volunteers. Important differences between these trials and ours include the patient population studied, the method by which VPA was administered, and the fact that all patients included in this study were not currently taking any other pharmacologic agents.
Although oral dosing can result in high plasma concentrations of VPA, we sought to evaluate intravenously administered VPA. The rationale for this decision was related to our anticipated use of VPA in traumatized patients, who may not be able to have orally administered agents. While the package insert for Depacon® state that intravenous and oral administration of VPA are equivalent, we found that the peak plasma concentrations of VPA in our study was significantly higher than those identified in patients given the same dose of oral VPA (140 mg/kg dose, IV dosing (current study): 1,000–1,250 μg/mL vs. oral dosing [22]: 300–400 μg/mL). In addition, we were able to show that a two-compartment linear model adequately explains the concentration-time data across a 14-fold range of absolute doses (846 mg to 12,264 mg). This population model explained 85.6% of the inter-individual variability without a covariate structure—implying that predictable exposure should be expected in future clinical trials involving high doses of VPA.
The 2-compartment model developed was also able to identify pharmacodynamics breakpoints predictive of drug safety, and demonstrated a higher threshold for neurologic toxicities than previously established. In regards to VPA metabolism, intravenous VPA has a clearance value of approximately 1 L/h. This has practical implications for future dose selection, as an AUC breakpoint predictive of therapeutic effect will approximate the AUC. Prior work from investigators at the Kentucky poison control center provides additional insight into VPA toxicity. In this series, 133 patients with acute VPA ingestion and VPA serum concentrations greater than 100 mg/L were evaluated [27]. The mean (range) Cmax VPA concentration was 378 (110–1840) mg/L. Two patients had a fatal outcome, and a Cmax concentration greater than 850 mg/L was found to be predictive of serious adverse events such as coma and respiratory depression. However, these evaluated Cmax values were measurements taken on average 7.4 hours after ingestion. As such, the true Cmax would expected to be much higher, which would be more consistent with our findings.
Despite improved outcomes in large animal models of hemorrhage and TBI, VPA’s protective mechanism of action is not well understood. However, recent work from our lab has yielded exciting clues. As a part of this trial, peripheral blood mononuclear cells were collected from subjects for proteomic analysis. In total, 140 differentially expressed proteins were identified. Functional annotation revealed alteration in proteins related to cell death and survival, fatty acid metabolism, neurologic disorders, and cellular component organization. Full results from this analysis have been published separately[26], and additional work is currently being conducted to better understand these findings.
We have also identified specific transcriptional programs in the central nervous system that are induced by VPA. Using a variety sources, including published studies, public and commercial databases, and animal experiments, we found that VPA may be acting through regulatory pathways that enhance neurogenesis and reduce gliogenesis. Genes which encode transcription factors that specify neuronal cell fate, including MEF2D, MYT1L, NEUROD1, PAX6 and TBR1, and their target genes, are up-regulated by VPA. In fact, NEUROD1 has regulatory interactions with 38% of the genes regulated by VPA in a swine model of TBI and hemorrhagic shock. VPA was also found to repress genes responsible for oligodendrogenesis, angiogenesis, and endothelial cell proliferation (unpublished data)[28]. In a separate study, we used immunofluoroscopy to show that VPA protects the integrity of the blood brain barrier in swine subjected to TBI and hemorrhagic shock. Animals treated with VPA had increased expression of tight-junction and basement membrane associated proteins, including zona occludin-1, laminin, and claudin-5. Similar outcomes were achieved in vitro, where monolayers of endothelial cells subjected to anoxia and treated with VPA had significantly decreased permeability relative to anoxic controls [29].
With such broad clinical application, the MTD of intravenous VPA is an important discovery. However, there are limitations to this trial. First, the majority of subjects in this study were young, healthy males. This is in part explained by the exclusion of women of childbearing age due to VPA’s known teratogenicity. VPA’s pharmacokinetic and pharmacodynamic profiles may be altered in different patient populations. For this reason, the safety and tolerability of VPA is currently being evaluated in patients in hemorrhagic shock. Second, this study involved a single dose of intravenous VPA. For the majority of established clinical applications like epilepsy and bipolar disorder VPA is taken on a daily basis, by mouth, and at relatively low doses. Whether the effects of VPA are optimized following a single intravenous dose is not known. We are currently in the process of developing further large animals studies to determine the optimal dose, timing, and administration strategy (e.g. split dosing, multiple dosing) of VPA in the setting of trauma. Finally, a phase I trial such as this does not establish the efficacy of VPA treatment. This study represents phase Ia of a two-part phase I trial (NCT01951560). Phase II and III trials to determine whether VPA can improve outcomes in trauma patients have already been approved for funding by the US Department of Defense.
In conclusion, this study shows that a single intravenous dose of VPA as high as 140 mg/kg is well tolerated in healthy subjects. This is significantly higher than the previously established MTD and has important implications for the treatment of a variety of disease states, including hemorrhagic shock. While additional studies are needed to determine the efficacy of high dose VPA the pharmacokinetic model developed as part of this study will help guide the design of future clinical trials.
Supplementary Material
KEY POINTS.
Valproic acid is a non-selective histone deacetylase inhibitor with important pharmacologic effects extending beyond the treatment of epilepsy that supports repurposing of this compound for novel therapeutic applications in cancer, neurologic diseases, and traumatic injuries.
The current study shows that the maximum tolerated dose of valproic acid in healthy humans is 140 mg/kg. While above current regulatory approved doses the pharmacokinetic profile achieved in this study supports the use of high dose intravenous valproic acid in clinical translational studies of trauma patients with life-threatening injuries.
Acknowledgments
This work was funded by the Department of Defense Office of Naval Research (N000141310071) and the National Institutes of Health Center for Advancing Translational Sciences (UL1TR000433). The Michigan Institute for Clinical Health Research (MICHR) provided guidance, data design, and clinical research space. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, or the US Department of Defense.
Footnotes
CLINICAL TRIAL: NCT0195156
Conflicts of interest:
There are no conflicts to declare.
Ethical approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
References
- 1.Perucca E. Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs. 2002;16(10):695–714. doi: 10.2165/00023210-200216100-00004. [DOI] [PubMed] [Google Scholar]
- 2.Gottlicher M. Valproic acid: an old drug newly discovered as inhibitor of histone deacetylases. Ann Hematol. 2004;83(Suppl 1):S91–2. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- 3.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389(6649):349–52. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 4.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science (New York, NY) 2009;325(5942):834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 5.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. The Journal of clinical investigation. 2014;124(1):30–9. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nature reviews Drug discovery. 2008;7(10):854–68. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 7.Toussirot E, Khan KA, Herbein G. Histone deacetylase inhibitors: a new and promising drug class for the treatment of arthritis? Clinical epigenetics. 2010;1(1–2):3–6. doi: 10.1007/s13148-010-0002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halaweish I, Nikolian V, Georgoff P, Li Y, Alam HB. Creating a “Prosurvival Phenotype” Through Histone Deacetylase Inhibition: Past, Present, and Future. Shock. 2015;44(Suppl 1):6–16. doi: 10.1097/SHK.0000000000000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. Web based Injury Statistics Query and Reporting System (WISQARS) [online]. Availabe at http://www.cdc.gov/injury/wisqars/index.html. Accessed June 20, 2016.
- 10.Shackford SR, Mackersie RC, Holbrook TL, Davis JW, Hollingsworth-Fridlund P, Hoyt DB, Wolf PL. The epidemiology of traumatic death. A population-based analysis. Arch Surg. 1993;128(5):571–5. doi: 10.1001/archsurg.1993.01420170107016. [DOI] [PubMed] [Google Scholar]
- 11.Holcomb JB, Tilley BC, Baraniuk S, Fox EE, Wade CE, Podbielski JM, del Junco DJ, Brasel KJ, Bulger EM, Callcut RA, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA. 2015;313(5):471–82. doi: 10.1001/jama.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pons PT, Jerome J, McMullen J, Manson J, Robinson J, Chapleau W. The Hartford Consensus on Active Shooters: Implementing the Continuum of Prehospital Trauma Response. J Emerg Med. 2015;49(6):878–85. doi: 10.1016/j.jemermed.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 13.Alam HB, Shuja F, Butt MU, Duggan M, Li Y, Zacharias N, Fukudome EY, Liu B, Demoya M, Velmahos GC. Surviving blood loss without blood transfusion in a swine poly-trauma model. Surgery. 2009;146(2):325–33. doi: 10.1016/j.surg.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Fukudome EY, Kochanek AR, Li Y, Smith EJ, Liu B, Kheirbek T, Lu J, Kim K, Hamwi K, Velmahos GC, et al. Pharmacologic resuscitation promotes survival and attenuates hemorrhage-induced activation of extracellular signal-regulated kinase 1/2. J Surg Res. 2010;163(1):118–26. doi: 10.1016/j.jss.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Liu B, Fukudome EY, Kochanek AR, Finkelstein RA, Chong W, Jin G, Lu J, deMoya MA, Velmahos GC, et al. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery. 2010;148(2):246–54. doi: 10.1016/j.surg.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dekker SE, Bambakidis T, Sillesen M, Liu B, Johnson CN, Jin G, Li Y, Alam HB. Effect of pharmacologic resuscitation on the brain gene expression profiles in a swine model of traumatic brain injury and hemorrhage. J Trauma Acute Care Surg. 2014;77(6):906–12. doi: 10.1097/TA.0000000000000345. discussion 12. [DOI] [PubMed] [Google Scholar]
- 17.Zhao T, Li Y, Liu B, Liu Z, Chong W, Duan X, Deperalta DK, Velmahos GC, Alam HB. Novel pharmacologic treatment attenuates septic shock and improves long-term survival. Surgery. 2013;154(2):206–13. doi: 10.1016/j.surg.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halaweish I, Bambakidis T, Chang Z, Wei H, Liu B, Li Y, Bonthrone T, Srinivasan A, Bonham T, Chtraklin K, et al. Addition of low-dose valproic acid to saline resuscitation provides neuroprotection and improves long-term outcomes in a large animal model of combined traumatic brain injury and hemorrhagic shock. J Trauma Acute Care Surg. 2015;79(6):911–9. doi: 10.1097/TA.0000000000000789. [DOI] [PubMed] [Google Scholar]
- 19.Jepsen CH, deMoya MA, Perner A, Sillesen M, Ostrowski SR, Alam HB, Johansson PI. Effect of valproic acid and injury on lesion size and endothelial glycocalyx shedding in a rodent model of isolated traumatic brain injury. J Trauma Acute Care Surg. 2014;77(2):292–7. doi: 10.1097/TA.0000000000000333. [DOI] [PubMed] [Google Scholar]
- 20.Jin G, Duggan M, Imam A, Demoya MA, Sillesen M, Hwabejire J, Jepsen CH, Liu B, Mejaddam AY, Lu J, et al. Pharmacologic resuscitation for hemorrhagic shock combined with traumatic brain injury. J Trauma Acute Care Surg. 2012;73(6):1461–70. doi: 10.1097/TA.0b013e3182782641. [DOI] [PubMed] [Google Scholar]
- 21.Atmaca A, Al-Batran SE, Maurer A, Neumann A, Heinzel T, Hentsch B, Schwarz SE, Hovelmann S, Gottlicher M, Knuth A, et al. Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. Br J Cancer. 2007;97(2):177–82. doi: 10.1038/sj.bjc.6603851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munster P, Marchion D, Bicaku E, Schmitt M, Lee JH, DeConti R, Simon G, Fishman M, Minton S, Garrett C, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25(15):1979–85. doi: 10.1200/JCO.2006.08.6165. [DOI] [PubMed] [Google Scholar]
- 23.Brown E. Medical Dictionary for Regulatory Activities (MedDRA®) In: Mann RD, Andrews EB, editors. Pharmacovigilance. Second. John Wiley & Sons, Ltd; Chichester, UK: 2006. [DOI] [Google Scholar]
- 24.Abbreviated Mental Test Score (AMTS) Occas Pap R Coll Gen Pract. 2002;(82):48. [Google Scholar]
- 25.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 26.Georgoff P, Halaweish I, Nikolian V, Higgins G, Bonham T, Tafatia C, Remmer H, Menon R, Liu B, Li Y, et al. Alterations in the human proteome following administration of valproic acid. J Trauma Acute Care Surg. 2016 doi: 10.1097/TA.0000000000001249. [DOI] [PubMed] [Google Scholar]
- 27.Spiller HA, Krenzelok EP, Klein-Schwartz W, Winter ML, Weber JA, Sollee DR, Bangh SA, Griffith JR. Multicenter case series of valproic acid ingestion: serum concentrations and toxicity. J Toxicol Clin Toxicol. 2000;38(7):755–60. doi: 10.1081/clt-100102388. [DOI] [PubMed] [Google Scholar]
- 28.Higgins GGP, Nikolian V. Network reconstruction reveals that valproic acid activates neurogenic transcriptional progams in adult brain following traumatic injury. 2016 doi: 10.1007/s11095-017-2130-6. Submitted for publication November 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikolian VDS, Bamdakidis T, et al. Pharmacologic Resuscitation Decreases Blood-Brain Barrier Permeability in a Porcine Model of Traumatic Brain Injury and Hemorrhagic Shock. American College of Surgeons; Washington, DC: 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.