

ABSTRACT
A screening of more than 1,500 drug-resistant strains of Mycobacterium tuberculosis revealed evolutionary patterns characteristic of positive selection for three alanine racemase (Alr) mutations. We investigated these mutations using molecular modeling, in vitro MIC testing, as well as direct measurements of enzymatic activity, which demonstrated that these mutations likely confer resistance to d-cycloserine.
KEYWORDS: Mycobacterium tuberculosis, cycloserine, alanine racemase
TEXT
In 2015, the Global Drug Facility declared that the cost of d-cycloserine (DCS), a group C drug to treat tuberculosis (TB), would be cut by more than half to as little as $0.19 per capsule to support the treatment of multidrug-resistant (MDR) and extensively drug-resistant (XDR) TB, which represent a major threat to public health (1). In light of this announcement, a better understanding of the resistance mechanisms to this drug is required to facilitate phenotypic as well as genotypic drug susceptibility testing (DST), both in the context of surveillance and individual patient treatment, to avoid the severe side effects of this drug (2, 3).
Studies of the mode of action of DCS in mycobacteria have produced contradictory results, with some studies pointing to alanine racemase (Alr) as the primary target and others supporting d-alanine–d-alanine ligase (DdlA) (4–9). However, molecular data from Mycobacterium tuberculosis complex (MTBC) have implicated only alr in DCS resistance, which can also be conferred by mutations in alanine dehydrogenase (ald) or a permease (cycA) gene (10, 11). Using molecular modeling, we had predicted that the alr M319T mutation observed in an XDR strain would likely confer resistance to DCS, which was subsequently confirmed by Desjardins et al. using the unrelated strain TKK_04_0105 (see Table S1 in the supplemental material) (2, 11). Desjardins et al. described a number of additional alr mutations in strains with elevated DCS MICs, including a C-to-T nucleotide change 8 bp upstream of the experimentally confirmed start codon of alr (strain TKK_02_0050 in Table S1) (11, 12). This was notable, as Merker et al. had previously reported that, compared with the susceptible parental alr wild-type strain, the acquisition of this mutation during treatment with DCS correlated with DCS resistance, which suggested that alr mutations might be both necessary and sufficient to confer DCS resistance (13).
To gain further insights into the impact of alr mutations, we first confirmed that the aforementioned alr C-8T promoter mutant that evolved during treatment correlated in MICs above the current World Health Organization (WHO)-endorsed critical concentration (CC) of 30 μg/ml using the 1% proportion method on Löwenstein-Jensen (LJ) (strains PBm0 and PBm14 in Table S1; Desjardins et al. and Merker et al. had used 10% as the critical proportion and therefore had not adhered to the current WHO recommendations [11, 13, 14]). Using the same method, we also showed that two strains with alr M319T or Y364D mutations from XDR TB patients with a treatment history with DCS had MICs above the CC (Table S1). Moreover, we observed the M319T mutation in three XDR strains (PT1, PT2, and PT5) from Lisbon, Portugal (15). Although no CC exists for MGIT 960, this mutation correlated in an MIC increase from 16 to 64 μg/ml compared with three closely related wild-type control strains (PT3, PT6, and PT7) and one more distantly related control strain (PT4), which supported the role of this mutation in DCS resistance (Fig. 1A; Table S1). In contrast, no or minimal MIC increases were recorded when testing these Portuguese strains using Sensititre MycoTB plates (Table S1) (16). Finally, a pre-XDR alr R373L mutant from a patient with DCS exposure, which also harbored a deletion in ald, tested resistant on LJ using the 1% proportion method (Tables S1 and S2).
FIG 1.

Maximum likelihood tree based on a concatenated sequence alignment of 45,740 variable sites (1,000 resamplings, general time-reversible [GTR] nucleotide substitution model) showing the alr mutants from Table S1 in the context of a globally representative reference collection of 287 MTBC strains. (A) Zoomed-in part of the overall tree (B), showing the phylogenetic relationship between the three Portuguese M319T mutants (PT1, PT5, and PT2) and the control strains (PT7, PT3, PT6, and PT4) tested in MGIT and Sensititre. The three Indian M319T, R364D, and R373G mutants that were tested with the 1% proportion LJ method in this study are underlined. The C-8T, M319T, and R364D mutations were homoplastic (i.e., they were acquired multiple times independently) and two different amino acid changes were observed at codon 373 (i.e., R373L and R373G). Thus, all mutations show evolutionary patterns of positive selection. SNPs, single-nucleotide polymorphisms; CAS, central Asian strain.
To study the importance of the C-8T, M319T, Y364D, and R373L mutations from an evolutionary perspective, we screened previously published and unpublished genomes of more than 1,500 MDR strains (mostly from Germany, eastern Europe, and Swaziland), which identified eight additional strains with mutations at these alr positions or codons (Table S1). Interrogating the genomes of these 17 strains in the context of a phylogenetically diverse reference collection that included all major MTBC lineages and species showed that the mutations had either been acquired multiple times independently and/or that different amino acid changes were present at the same codons (Fig. 1B). These mutation patterns are typically a signal of positive selection, which could have occurred in response to DCS exposure.
Molecular modeling of these coding mutations supported this hypothesis. Alr functions as a homodimer, aided by the cofactor pyridoxal 5′-phosphate (PLP) to which it is covalently bound. DCS inhibits Alr irreversibly by covalently bonding to PLP (4). We generated and analyzed a model of the complex between the M. tuberculosis Alr and DCS (AlrMtb-DCS) (Fig. S1) (4, 17). Amino acid residues 319 and 364 were located directly in the active site (Fig. S1B). M319T was positioned close enough to allow interaction with the DCS moiety, which, given the large change of the character of the side chain, could strongly affect DCS reactivity (Fig. S1C). Y364 is involved in the positioning of the phosphate moiety of PLP and thus represents a prominent active site residue in the conserved inner layer of the substrate entrance corridor of Alr (Fig. S1B) (17). A mutation to aspartic acid introduced a shorter and negatively charged side chain, which could potentially affect PLP orientation in the active site (Fig. S1C). Moreover, it could influence DCS uptake through alteration of the entrance corridor. Interestingly, M319 is located near Y364 and, as a result, it is possible that the M319T mutation could alter the interaction with Y364, thereby affecting DCS inhibition. In contrast, the R373L mutation was not directly located within the active site but near the dimer interface and close to residues M319 and D320, which play an important role in the makeup of the active site (Fig. S1B). Consequently, the replacement of arginine with the short and hydrophobic side chain of leucine might disrupt molecular interactions at the dimer interface as well as destabilize the DCS binding site (Fig. S1C).
To test these predictions experimentally, we expressed and purified the aforementioned AlrMtb-coding mutants, along with wild-type AlrMtb, and determined their half-maximal inhibitory concentration (IC50) to measure the effectiveness of inhibition by DCS (Fig. 2). The IC50 for wild-type AlrMtb was 26.4 ± 1.7 μM, which was in the range previously reported for this compound (18, 19). From our structure-based analysis, we expected the two mutations located in the active site to show the greatest effect on DCS inhibition. Indeed, the M319T mutant enzyme showed minimal inhibition by DCS, even at 1,000 μM (Fig. 2). Thus, the IC50 of this mutant could not be determined. The IC50 of the Y364D mutant showed a 50-fold increase to 1,328.0 ± 340.0 μM. The R373L mutation, which was not located directly within the active site, also showed a significant increase in resistance to DCS, with an IC50 of 712.0 ± 138.5 μM (27-fold increase).
FIG 2.
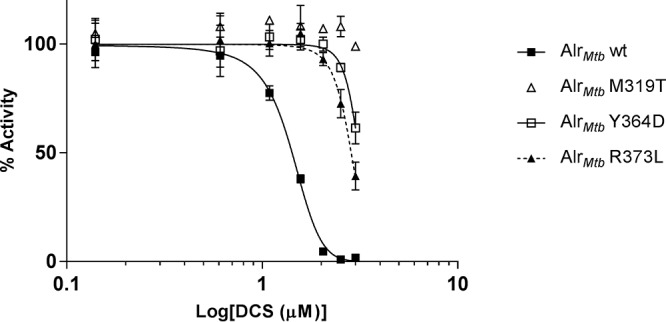
Determination of DCS IC50 for wild-type (wt) AlrMtb and the M319T, Y364D, and R373L mutants. The activity was normalized against a control with no DCS present in the assay mixture. The activity assay at each concentration was performed in triplicate, resulting in the error bars, which represent the 95% confidence interval. A variable slope model was fitted to determine the IC50s, which were 26.4 ± 1.7, 1,328.0 ± 340.0, and 712.0 ± 138.5 μM for the wild-type, Y364D, and R373L enzymes, respectively. The inhibition of M319T was too weak to allow for IC50 determination.
Taken together, these data suggested that alr mutations likely confer DCS resistance, although allelic exchange experiments are required to formally prove this (particularly for R373L, which coincided with a deletion in ald and, consequently, may not be sufficient to confer resistance on its own). Although the relationship between MICs and IC50s can be complex, the observation that MICs increased by only 4- to 16-fold versus at least 25-fold increases for IC50s supports the notion that DCS inhibits multiple targets, as noted earlier. This study should be complemented with extensive MIC testing of phylogenetically diverse pansusceptible MTBC strains to define the epidemiological cutoff value, given that it is unclear based on which evidence the current WHO CC on LJ has been set (3, 14, 20, 21). Moreover, further MIC testing of likely DCS-resistant strains is needed to investigate whether the Sensititre system is less reliable at detecting DCS resistance than are LJ and MGIT. Finally, the impact of alr mutations on resistance on terizidone remains to be investigated.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the University of Otago, Health Research Council Explorer grant and Maurice Wilkins Centre. In addition, parts of this study were supported by the European Union PathoNgenTrace project (grant FP7-278864-2) and the German Center for Infection Research (DZIF). Further funds were received from Fundação para a Ciência e a Tecnologia, Portugal, through the grants UID/Multi/04413/2013 (to M.V. and D.M.), SFRH/BPD/100688/2014 (to D.M.), and SFRH/BPD/95406/2013 (to J.P.). F.C. was supported by the Wellcome Trust 201344/Z/16/Z. T.G.C. was funded by the Medical Research Council UK (grants MR/K000551/1, MR/M01360X/1, and MR/N010469/1). Further support was received the Indian Council of Medical Research, New Delhi and Health Innovation Challenge Fund (grants HICF-T5-342 and WT098600), a parallel funding partnership between the UK Department of Health and the Wellcome Trust. C.U.K. is a research associate at Wolfson College, Cambridge, UK.
The views expressed in this publication are those of the authors and not necessarily those of the Department of Health, Public Health England, or the Wellcome Trust.
K.L.K., Y.N., and H.K.O.-R. have received funding for alanine racemase-related projects from L2 Diagnostics LLC, New Haven, CT. J.P., S.J.P., and C.U.K. have collaborated with Illumina, Inc. on a number of scientific projects. J.P. has received funding for travel and accommodation from Pacific Biosciences, Inc. and Illumina, Inc. S.J.P. has received funding for travel and accommodation from Illumina, Inc. C.U.K. is a consultant for the Foundation for Innovative New Diagnostics. The Bill & Melinda Gates Foundation and Janssen Pharmaceutica covered C.U.K.'s travel and accommodation to present at meetings. The European Society of Mycobacteriology awarded C.U.K. and M.M. the Gertrud Meissner Award, which is sponsored by Hain Lifescience.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01575-17.
REFERENCES
- 1.Stop TB Partnership. 2015. Stop TB Partnership's Global Drug Facility (GDF) achieves historic price reduction for MDR-TB drug cycloserine. Stop TB Partnership, Geneva, Switzerland: http://www.stoptb.org/news/announcements/2015/a15_006.asp. [Google Scholar]
- 2.Köser CU, Bryant JM, Becq J, Török ME, Ellington MJ, Marti-Renom MA, Carmichael AJ, Parkhill J, Smith GP, Peacock SJ. 2013. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. N Engl J Med 369:290–292. doi: 10.1056/NEJMc1215305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schön T, Miotto P, Köser CU, Viveiros M, Böttger E, Cambau E. 2017. Mycobacterium tuberculosis drug-resistance testing: challenges, recent developments and perspectives. Clin Microbiol Infect 23:154–160. doi: 10.1016/j.cmi.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 4.Fenn TD, Stamper GF, Morollo AA, Ringe D. 2003. A side reaction of alanine racemase: transamination of cycloserine. Biochemistry 42:5775–5783. doi: 10.1021/bi027022d. [DOI] [PubMed] [Google Scholar]
- 5.Milligan DL, Tran SL, Strych U, Cook GM, Krause KL. 2007. The alanine racemase of Mycobacterium smegmatis is essential for growth in the absence of d-alanine. J Bacteriol 189:8381–8386. doi: 10.1128/JB.01201-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awasthy D, Bharath S, Subbulakshmi V, Sharma U. 2012. Alanine racemase mutants of Mycobacterium tuberculosis require d-alanine for growth and are defective for survival in macrophages and mice. Microbiology 158:319–327. doi: 10.1099/mic.0.054064-0. [DOI] [PubMed] [Google Scholar]
- 7.Prosser GA, de Carvalho LP. 2013. Metabolomics reveal d-alanine:d-alanine ligase As the target of d-cycloserine in Mycobacterium tuberculosis. ACS Med Chem Lett 4:1233–1237. doi: 10.1021/ml400349n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halouska S, Fenton RJ, Zinniel DK, Marshall DD, Barletta RG, Powers R. 2014. Metabolomics analysis identifies d-alanine–d-alanine ligase as the primary lethal target of d-cycloserine in mycobacteria. J Proteome Res 13:1065–1076. doi: 10.1021/pr4010579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marshall DD, Halouska S, Zinniel DK, Fenton RJ, Kenealy K, Chahal HK, Rathnaiah G, Barletta RG, Powers R. 2017. Assessment of metabolic changes in Mycobacterium smegmatis wild-type and alr mutant strains: evidence of a new pathway of d-alanine biosynthesis. J Proteome Res 16:1270–1279. doi: 10.1021/acs.jproteome.6b00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen JM, Uplekar S, Gordon SV, Cole ST. 2012. A point mutation in cycA partially contributes to the d-cycloserine resistance trait of Mycobacterium bovis BCG vaccine strains. PLoS One 7:e43467. doi: 10.1371/journal.pone.0043467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desjardins CA, Cohen KA, Munsamy V, Abeel T, Maharaj K, Walker BJ, Shea TP, Almeida DV, Manson AL, Salazar A, Padayatchi N, O'Donnell MR, Mlisana KP, Wortman J, Birren BW, Grosset J, Earl AM, Pym AS. 2016. Genomic and functional analyses of Mycobacterium tuberculosis strains implicate ald in d-cycloserine resistance. Nat Genet 48:544–551. doi: 10.1038/ng.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strych U, Penland RL, Jimenez M, Krause KL, Benedik MJ. 2001. Characterization of the alanine racemases from two mycobacteria. FEMS Microbiol Lett 196:93–98. doi: 10.1111/j.1574-6968.2001.tb10547.x. [DOI] [PubMed] [Google Scholar]
- 13.Merker M, Kohl TA, Roetzer A, Truebe L, Richter E, Rüsch-Gerdes S, Fattorini L, Oggioni MR, Cox H, Varaine F, Niemann S. 2013. Whole genome sequencing reveals complex evolution patterns of multidrug-resistant Mycobacterium tuberculosis Beijing strains in patients. PLoS One 8:e82551. doi: 10.1371/journal.pone.0082551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.World Health Organization. 2014. Companion handbook to the WHO guidelines for the programmatic management of drug-resistant tuberculosis. World Health Organization, Geneva, Switzerland: http://apps.who.int/iris/bitstream/10665/130918/1/9789241548809_eng.pdf?ua=1&ua=1. [PubMed] [Google Scholar]
- 15.Perdigão J, Silva H, Machado D, Macedo R, Maltez F, Silva C, Jordao L, Couto I, Mallard K, Coll F, Hill-Cawthorne GA, McNerney R, Pain A, Clark TG, Viveiros M, Portugal I. 2014. Unraveling Mycobacterium tuberculosis genomic diversity and evolution in Lisbon, Portugal, a highly drug resistant setting. BMC Genomics 15:991. doi: 10.1186/1471-2164-15-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heysell SK, Pholwat S, Mpagama SG, Pazia SJ, Kumburu H, Ndusilo N, Gratz J, Houpt ER, Kibiki GS. 2015. Sensititre MycoTB plate compared to Bactec MGIT 960 for first- and second-line antituberculosis drug susceptibility testing in Tanzania: a call to operationalize MICs. Antimicrob Agents Chemother 59:7104–7108. doi: 10.1128/AAC.01117-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LeMagueres P, Im H, Ebalunode J, Strych U, Benedik MJ, Briggs JM, Kohn H, Krause KL. 2005. The 1.9 Å crystal structure of alanine racemase from Mycobacterium tuberculosis contains a conserved entryway into the active site. Biochemistry 44:1471–1481. doi: 10.1021/bi0486583. [DOI] [PubMed] [Google Scholar]
- 18.Kim MG, Strych U, Krause K, Benedik M, Kohn H. 2003. N(2)-substituted d,l-cycloserine derivatives: synthesis and evaluation as alanine racemase inhibitors. J Antibiot (Tokyo) 56:160–168. doi: 10.7164/antibiotics.56.160. [DOI] [PubMed] [Google Scholar]
- 19.Anthony KG, Strych U, Yeung KR, Shoen CS, Perez O, Krause KL, Cynamon MH, Aristoff PA, Koski RA. 2011. New classes of alanine racemase inhibitors identified by high-throughput screening show antimicrobial activity against Mycobacterium tuberculosis. PLoS One 6:e20374. doi: 10.1371/journal.pone.0020374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ängeby K, Juréen P, Kahlmeter G, Hoffner SE, Schön T. 2012. Challenging a dogma: antimicrobial susceptibility testing breakpoints for Mycobacterium tuberculosis. Bull World Health Organ 90:693–698. doi: 10.2471/BLT.11.096644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Köser CU, Feuerriegel S, Summers DK, Archer JA, Niemann S. 2012. Importance of the genetic diversity within the Mycobacterium tuberculosis complex for the development of novel antibiotics and diagnostic tests of drug resistance. Antimicrob Agents Chemother 56:6080–6087. doi: 10.1128/AAC.01641-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.